
Abstract
Background:
Although inflammation has been implicated in the pathogenesis of Alzheimer’s disease, the effects of systemic inflammation on brain amyloid deposition remain unclear.
Objective:
We examined the association of midlife and late-life systemic inflammation with late-life brain amyloid levels in a community sample of non-demented older adults from the Atherosclerosis Risk in Communities (ARIC) – PET Study.
Methods:
339 non-demented participants (age: 75 [SD 5]) were recruited from the ARIC Study to undergo florbetapir PET (amyloid) imaging. Blood levels of high sensitivity C-reactive protein (CRP), a marker of systemic inflammation, were measured 22 years (Visit 2), 16 years (Visit 4), and up to 2 years before PET imaging (Visit 5). Elevated brain amyloid deposition (standardized uptake value ratio >1.2) was the primary outcome.
Results:
Our primary analyses found no association of midlife and late-life CRP with late-life brain amyloid levels. However, in secondary stratified analyses, we found that higher midlife (Visit 2) CRP was associated with elevated amyloid among males (OR 1.65, 95% CI: 1.13–2.42), and among white (OR 1.33, 95% CI: 1.02–1.75), but not African American, participants (p-interactions<0.05). Among male participants, those who maintained high CRP levels (≥3 mg/L) throughout mid- and late-life were most likely to have elevated brain amyloid (OR, 8.81; 95% CI: 1.23, 62.91).
Conclusions:
Although our primary analysis does not support an association between systemic inflammation and brain amyloid deposition, we found evidence for sex- and race-dependent associations. However, findings from subgroup analyses should be interpreted with caution.
INTRODUCTION
The amyloid cascade hypothesis proposes that the accumulation of amyloid-β (Aβ) or Aβ oligomers and the deposition of cortical Aβ plaques are key causative processes in the pathophysiology of Alzheimer’s disease (AD) [1]. Considerable evidence also suggests that inflammation has a central role in driving AD pathogenesis [2]. For example, polymorphisms in genes involved in innate immune functioning [3, 4] are known to confer risk for late-onset AD and increased Aβ deposition [5]. Several studies suggest that inflammation within the central nervous system (CNS) may co-occur with the deposition of Aβ. For example, recent work has demonstrated that 18 kDa translocator protein (TSPO), a marker of microglial activation, is elevated among individuals with mild cognitive impairment (MCI) [6] and AD [7], especially in brain regions with high levels of amyloid deposition. Furthermore, pathological hallmarks of microglial activation have been found to co-localize with early Aβ deposits [8] and correlate positively with clinical severity [9], suggesting an association between neuroinflammation and amyloid deposition that may promote later neurodegenerative processes. On the other hand, preclinical and clinical trial data have demonstrated that stimulation of the cell-mediated immune response through active and passive immunotherapies can promote the phagocytosis and clearance of Aβ by activated microglial [10, 11], effectively reducing the burden of brain amyloid (albeit without any clear improvement in clinical symptoms) [12].
Although a relationship between neuroinflammation and Aβ has been consistently demonstrated, it is less clear how inflammation outside the CNS (i.e., systemic inflammation) might influence cortical Aβ deposition. Based on animal studies which have demonstrated that repeated peripheral immune challenges enhance cerebral Aβ production in mice [6–8], it has been postulated that systemic inflammation may promote the accumulation and deposition of Aβ in the brains of elderly individuals through neuroinflammatory processes. Although systemic inflammation has been associated with cognitive decline [9], dementia risk [10], and MRI-defined neurodegenerative changes [11, 12], it remains unclear whether systemic inflammation during late-life, or during the preceding decades, is associated with greater cortical Aβ deposition in older adults.
In this study, we examined the association between circulating levels of high-sensitivity C-reactive protein (CRP), a non-specific marker of systemic inflammation, and brain amyloid deposition, as measured by positron emission tomography (PET), within a biracial group of non-demented older adults in the Atherosclerosis Risk in Communities (ARIC) Study. CRP was measured 22, 16, and up to 2 years before PET imaging, allowing us to test the hypothesis that individuals with elevated systemic inflammation during midlife and those with chronically elevated systemic inflammation are more likely to have elevated cortical Aβ deposition in later life. In light of evidence that race and sex influence brain amyloid deposition [13], circulating inflammatory marker levels [14], peripheral immune signaling [15, 16], and potentially, AD pathogenesis [17, 18], we examined whether race and sex modified the relationship between systemic inflammation and cortical Aβ deposition.
METHODS
Participants
Participants included in the ARIC-PET study were recruited from an ongoing ARIC Neurocognitive Study (ARIC-NCS), an ancillary to the larger ARIC study [19]. ARIC is an ongoing population-based study, which at its onset (Visit 1; 1987–1989) included 15,792 participants from four sites across the U.S.: Washington County, MD; Forsyth County, NC; northwestern suburbs of Minneapolis, MN; and Jackson, MS (African Americans only). After the baseline visit, participants were brought back every three years for three additional visits until Visit 4 (1996–98; Fig. 1A). Participants were then invited back for a fifth visit approximately 15 years later (Visit 5; 2011–2013). At Visit 5, nearly 2,000 of 6,538 participants were selected to undergo a brain MRI, including a group of participants with cognitive impairment as well as an age-stratified sample without impairment as described previously [20]. Among these, participants without dementia, heavy current alcohol use, renal dysfunction (creatinine >2 mg/dL), or prolonged QT-c interval (>450 ms) at three ARIC sites (Forsyth County, NC; Jackson, MS; and Washington County, MD) were recruited to take part in ARIC-PET.

A) Study design and flowchart. B) Hypothetical 21-year C-reactive protein patterns. Participants were assigned to one of five categories based on blood C-reactive protein levels (<3 or≥3mg/L) at Visits 2, 4, and 5. The dotted line indicates approximate C-reactive protein level at each visit.
Of the 346 participants who completed a PET scan, we excluded one participant due to an adjudicated dementia diagnosis (based on expert committee diagnosis [21]), two for race that was non-white or non-African American, and 13 for missing APOE ɛ4 genotype. The ARIC study protocols were approved by the Institutional Review Boards at each participating center. All participants gave written informed consent at each study visit.
Inflammatory markers
We measured high-sensitivity CRP levels from blood that was collected at Visits 2, 4, and 5, and stored at – 70°C. We used the immunoturbidimetric assay to measure Visit 2 CRP levels (mg/L) in 2011–13 (storage time: 21.2 years [0.98 SD]) using the Roche Modular P chemistry analyzer (Roche Diagnostics, Indianapolis, IN). Visit 4 CRP levels (mg/L) were measured in 2008 (approximate storage time: 10.8 years [0.81 SD]) using the nephelometric method on the Siemens Dade Behring BN II analyzer (Siemens Healthcare Diagnostics, Deerfield IL). Visit 5 CRP levels (mg/L) were measured in 2011–13 (storage time: 24.1 days [42.03 SD]) using an immunoturbidimetric assay on the Beckman Coulter Olympus AU400e analyzer (Beckman Coulter Inc., Brea, CA). Differences in CRP measurement based on laboratories, assay methods, instruments, specimen type, and time of measurement were not large enough to warrant re-calibration (bias <10%) [22]. Although CRP levels are stable across prolonged storage times (e.g., >14 years) [23, 24], coefficients of variation may be higher in samples stored for extended periods [23].
To define longitudinal patterns of inflammation, participants were first categorized as having “low” or “high” CRP levels at each visit using a cut-off of 3 mg/L, above which is suggestive of ongoing systemic inflammation [25–27]. Using this “low” versus “high” CRP dichotomization, participants were categorized into one of five categories based on their pattern of CRP levels over three Visits (Fig. 1B). Stable low: low CRP levels (<3 mg/L) at Visits 2, 4, and 5 Ascending: low CRP at Visit 2 and high CRP (≥3 mg/L) at Visit 5 Descending: high CRP at Visit 2 and low CRP at Visit 5 Stable high: high CRP at Visits 2, 4, and 5 Variable: other CRP patterns (i.e., low-high-low and high-low-high)
Each of these predefined groups represents a longitudinal pattern of age-related inflammation that has been described previously in the aging literature (e.g., [28]). While it is clear that many individuals maintain low levels of inflammation as they age, studies have identified a distinct subset of aging individuals more prone to age-related diseases who experience chronic inflammation [29–31]. Other age-related inflammatory phenotypes have also been described, including a subset of individuals who transition from a non-inflammatory to an inflammatory state in older adulthood [29, 33] and, conversely, individuals who demonstrate significant reductions in inflammation with increasing age [29, 32]. Additionally, several studies have defined a subgroup of aging adults with abnormally high variability in inflammatory markers over time [34, 35].
Brain MRI and PET imaging
3T MRI scans were obtained and analyzed at the ARIC MRI Reading Center (Mayo Clinic) using methods described previously [20]. All florbetapir PET scans were performed within one year of the brain MRI. Magnetization-prepared rapid gradient echo (MP-RAGE) sequences were used for coregistration of PET images. Images were acquired between 50 and 70 min after intravenous injection of the florbetapir isotope for a 20-min uptake scan (4 X 5 min). Images were reviewed at the PET image analysis center (Johns Hopkins) for image quality before standardized uptake value ratios (SUVRs) were quantified. Thirty-four regions of interest (ROIs) were manually drawn and applied to the SUVR images in the standard Montreal Neurologic Institute (MNI) space. Additional details of PET image processing have been described previously [13]. A global measure of cortical florbetapir uptake was calculated based on a volume-dependent weighted average of the orbitofrontal, prefrontal, and superior frontal cortices; the lateral temporal, parietal, and occipital lobes; the precuneus, the anterior cingulate, and the posterior cingulate. An automated area of cerebellar gray matter was used as a reference [36]. Using methods described previously, high SUVR was defined as a value above 1.2, the sample median [13, 37]. Global SUVR was dichotomized at the sample median, rather than used as a continuous parameter, because of the highly skewed distribution of the SUVR data.
Assessment of covariates
Participants provided information on race, sex, and years of education at Visit 1. Information about previous diagnoses of chronic inflammatory conditions (i.e., arthritis, gout) and long-term use of anti-inflammatory medication (e.g., nonsteroidal anti-inflammatory drugs [NSAID], arthritis medication) was assessed via self-report at Visit 5. APOE ɛ4 genotype was assessed using a TaqMan assay (Applied Biosystems, Foster City, CA). All other variables were assessed concurrently with the assessment of CRP (i.e., Visits 2, 4, and 5). Alcohol use and cigarette smoking status (current/former/never) were determined by self-report. Body mass index (BMI, kg/m2) was calculated using height and weight. Total cholesterol was measured using the enzymatic method [38]. The Friedewald equation was used to measure low-density lipoprotein (LDL) cholesterol; high-density lipoprotein (HDL) cholesterol was measured after precipitation of non-HDL lipoprotein [39].
Coronary heart disease was adjudicated based on medical record evidence or self-report of previous myocardial infarction, coronary artery bypass graft or angioplasty, or myocardial infarction determined by ECG. Hypertension was defined as systolic blood pressure >140 mm Hg, diastolic blood pressure >90 mm Hg, or use of hypertensive medication. Diabetes was defined as either fasting glucose≥126 mg/dl or non-fasting glucose of≥200 mg/dl, current use of diabetes medication, or self-report of physician-diagnosed diabetes.
Statistical analysis
Participant characteristics are displayed stratified according to sex. Group differences were assessed using t-tests and chi-square tests. Associations between CRP and elevated brain amyloid were first assessed graphically using locally weighted scatter plot smoothing (LOWESS) curves (Supplementary Figures 1–3) to check for nonlinearity. We then examined the association of CRP at Visits 2, 4, and 5 with late-life brain amyloid (SUVR) using logistic regression models. CRP values were log transformed to correct for skewness. As appropriate, we included linear and cubic spline terms to model nonlinear associations. Time-varying covariates from the visit concurrent with CRP assessment were incorporated into the models. To examine how 21-year CRP patterns (Fig. 1B) relate to late-life brain amyloid, we used covariate-adjusted binary logistic regression. Using the stable low group as the reference, we examined the association between ascending, descending, stable high, and variable 21-year CRP patterns and elevated brain amyloid. For this analysis, we incorporated covariates from the visit concurrent with baseline CRP assessment (Visit 2). To address potential bias related use of multiple CRP assay methods, we repeated these analyses using an alternative ranking-based approach to classify participants into longitudinal CRP categories (described in the Supplementary Methods). We adjusted all analyses for a set of demographic, physiological, and clinical variables shown to be jointly associated with inflammation and AD-related outcomes (i.e., potential confounders): age, sex, race-center (Washington County, white; Forsyth County, white; Forsyth County, African American; and Jackson, African American), education, APOE ɛ4 status (defined as 0, 1, or 2 ɛ4 alleles), vascular risk factors and disease (i.e., BMI, total cholesterol, HDL, hypertension, diabetes, coronary heart disease, cigarette smoking, and alcohol use), inflammatory disease, and anti-inflammatory and cholesterol lowering medication use. Because we found evidence for effect modification by both race and sex (p-interactions <0.05; Supplementary Figures 4–6), race- and sex-stratified models are reported. To reduce potential confounding related to CRP storage time, we repeated primary analyses after adjusting for the duration of CRP storage. A two-sided p value < 0.05 was used as the cutoff for statistical significance. Analyses were conducted using Stata Version 14 (StataCorp, College Station, Tex., USA).
RESULTS
We included 330 participants in the analytic sample. Visit 2, Visit 4, and Visit 5 CRP levels were collected 22.3 years (SD 0.9), 16.2 years (SD 0.9), and 1.5 years (SD 0.6) before the amyloid PET scan, respectively. PET imaging was obtained within one year of Visit 5 MRI scans. Forty-two percent (n = 140) of participants were African American, and 56% (n = 186) were women. Participant characteristics for the full sample and sex-stratified samples are displayed in Table 1 (race-stratified characteristics are presented in Supplementary Table 1). Within both ARIC-PET and the full ARIC sample (see Supplementary Table 2), median CRP levels increased during midlife, and decreased slightly from middle- to late-life. Women tended to have higher CRP levels; however, these differences were statistically significant only in midlife. Compared to white participants, African Americans had higher CRP levels during middle- and late-life; race differences were statistically significant only within the full ARIC Cohort. Thirty-one percent (n = 96), 39% (n = 126), and 36% (n = 120) of participants had high (≥3 mg/L) CRP at Visits 2, 4, and 5, respectively. Twenty-seven percent (n = 90) of participants met criteria for MCI at the time of PET imaging based on expert committee diagnosis [21], whereas all other participants were adjudicated as cognitively normal.
Baseline (Visit 2; 1990–92) participant characteristics
Values are displayed as means (SD) for continuous variables, and column percentages for categorical variables unless otherwise specified. aAssessed at Visit 5 (2011–13). bDifference between female and male participants statistically significant (p < 0.05).
Mid- and late-life CRP levels and florbetapir PET
Visit 2 (1990–1992) CRP
We found no association between Visit 2 CRP and elevated brain amyloid in our primary analysis of the total sample. However, we found an interaction between CRP level, sex (p-interaction = 0.025), and race (p-interaction = 0.045) on brain amyloid (see Fig. 2 and Supplementary Figure 4). Sex-stratified analyses revealed that among male participants, each one SD increase in Visit 2 CRP was associated with 65% greater odds (95% CI: 1.13 to 2.42) of elevated late-life brain amyloid after adjusting for demographic variables, APOE ɛ4 status, cardiovascular risk factors and comorbid disease. However, we found no association between Visit 2 CRP and late-life brain amyloid among female participants. In race-stratified analyses, we found that each one SD increase in midlife CRP was associated with 33% greater odds (95% confidence interval [CI]: 1.02 to 1.75) of elevated brain amyloid in late life among white participants after adjusting for potentially confounding variables (Fig. 3). In contrast, we found no association among African American participants. The 3-way interaction between (race x sex x CRP) on brain amyloid was non-significant.

The adjusted odds ratio (OR) of having a standardized uptake value ratio (SUVR) >1.2 per each one standard deviation increase in C-reactive protein at Visits 2, 4, and 5. Models were adjusted for demographic characteristics, APOE ɛ4 status, anti-inflammatory and cholesterol lowering medication use, cardiovascular risk factors, and comorbid disease at the time of C-reactive protein measurement. *Sex interaction was statistically significant (p-interaction = 0.025).
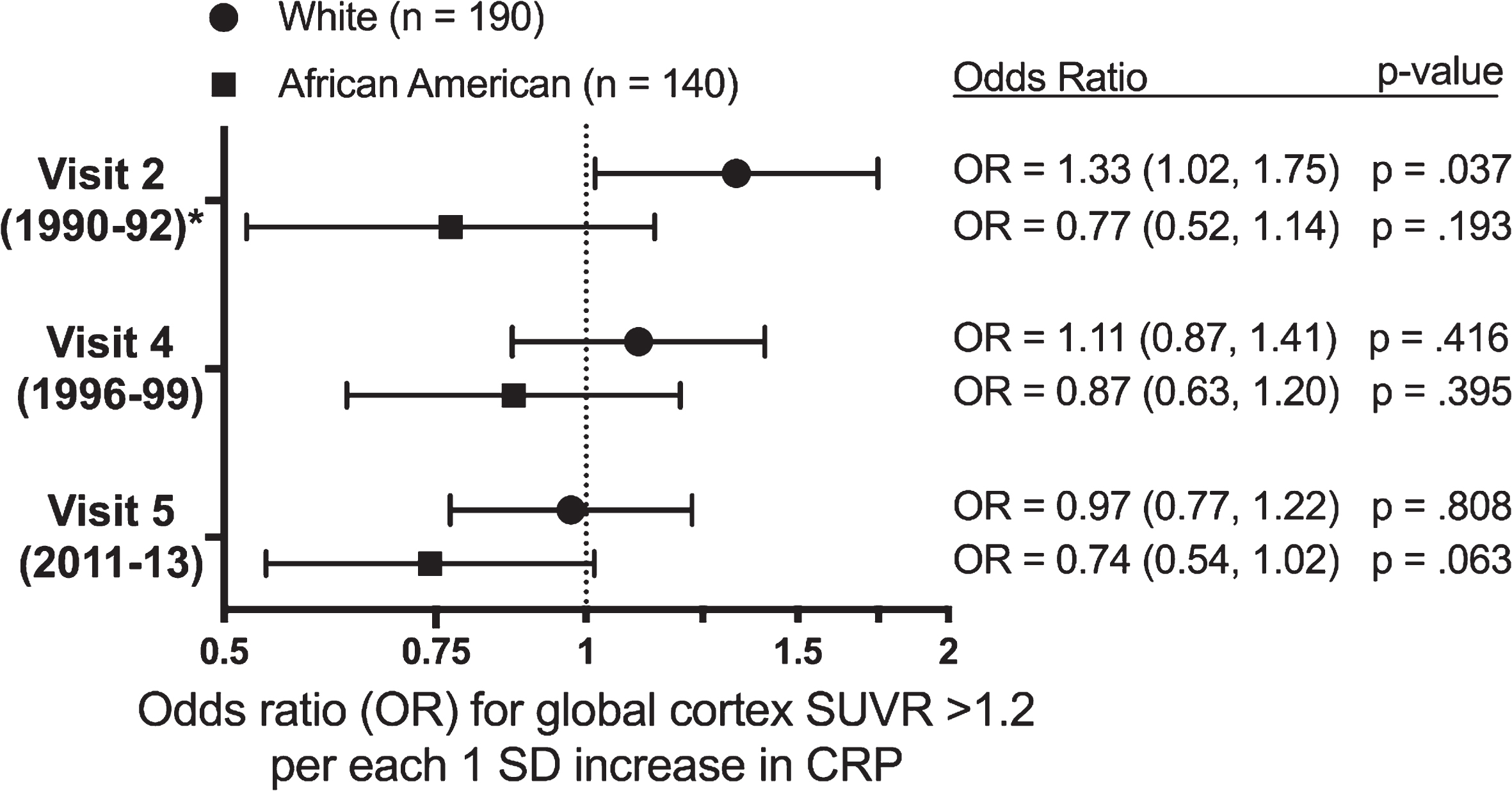
The adjusted odds ratio (OR) of having a standardized uptake value ratio (SUVR) >1.2 per each one standard deviation increase in C-reactive protein at Visits 2, 4, and 5. Models were adjusted for demographic characteristics, APOE ɛ4 status, anti-inflammatory and cholesterol lowering medication use, cardiovascular risk factors, and comorbid disease at the time of C-reactive protein measurement. *Race interaction was statistically significant (p-interaction = 0.045).
We repeated regression analyses after including a linear spline term with a knot at CRP levels of 3 mg/L based on evidence of a change in slope (Supplementary Figure 1). Among male participants and white participants, we found the incremental increase in odds of elevated brain amyloid per unit increase in CRP to be greater for CRP values at a lower range (CRP < 3 mg/L) (Supplementary Table 3).
Visit 4 (1996–1999) CRP
We found no association between Visit 4 CRP and elevated brain amyloid in the total sample after adjusting for demographic variables, APOE ɛ4 status, cardiovascular risk factors, and comorbid disease (Fig. 2). Race and sex interactions terms were not statically significant. No significant associations were found between CRP and brain amyloid deposition in sex- or race-stratified analyses (Figs. 2 and 3).
Visit 5 (2011–2013) CRP
Visit 5 CRP was not associated with brain amyloid in our primary analysis (Fig. 2); interactions by race and sex were non-significant. However, there was a non-significant association between higher CRP levels at Visit 5 and a reduced odds of elevated brain amyloid in the total sample and among African Americans in race-stratified analyses (Fig. 3). We found no association between Visit 5 CRP and brain amyloid in sex-stratified analyses. These findings were largely unchanged in regression models that included cubic and linear spline terms to model the nonlinear associations observed among African American and Female participants (Supplementary Figure 3). The results of Visit 2, Visit 4, and Visit 5 CRP analyses were similar after additionally adjusting for differences in CRP storage time.
21-year CRP pattern and florbetapir PET
The odds of elevated brain amyloid among participants with ascending, descending, stable elevated, and variable 21-year patterns of CRP did not differ from that of the stable low group in our analysis of the total sample (Supplementary Table 4). However, there was a significant interaction by sex for the association of descending CRP (p-interaction = 0.035) and stable elevated CRP (p-interaction = 0.023) with brain amyloid. In a sex-stratified analysis, we found that male participants with stable elevated CRP demonstrated significantly greater odds of elevated brain amyloid (OR, 8.81; 95% CI: 1.23, 62.91, p = 0.030) compared to male participants with stable low CRP (Fig. 4); however, this estimate was imprecise due to small sample size. We found no association among female participants. These results were similar when 21-year CRP patterns were categorized using a ranking-based approach (Supplementary Table 5).
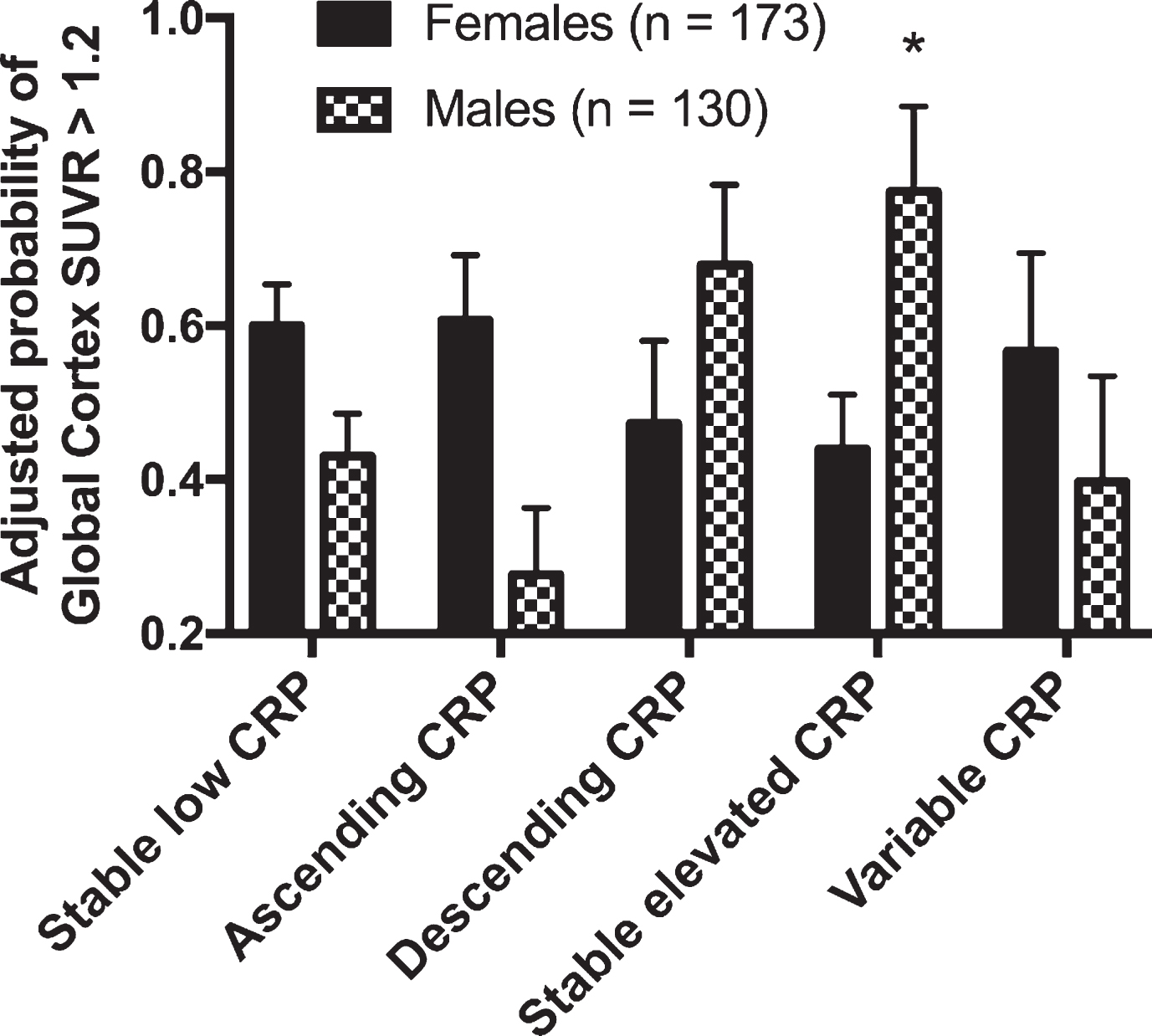
The adjusted probability of having a standardized uptake value ratio (SUVR) >1.2 according to the 21-year pattern of past C-reactive protein levels. Results are stratified by sex. Values were estimated using logistic regression models adjusted for demographic variables, APOE ɛ4 status, anti-inflammatory and cholesterol lowering medication use, Visit 2 cardiovascular risk factors, and comorbid disease. *p < 0.05 compared to the Stable Low referent group.
DISCUSSION
In this sample of non-demented older adults, we found no association between higher levels of systemic inflammation and elevated brain amyloid in our primary analyses. However, our secondary analyses indicate that midlife systemic inflammation may be associated with a higher burden of amyloid decades later within a subset of the population. Specifically, among male participants, a higher level of CRP during midlife was associated with elevated brain amyloid in late-life. We found a similar association among white, but not African American, participants when analyses were stratified by race. However, these associations were not observed when CRP was measured later in life. In our longitudinal analysis of CRP, males who demonstrated persistent systemic inflammation over 21 years spanning middle- to late-adulthood had approximately double the risk of elevated brain amyloid compared to males who maintained low CRP levels. However, this finding was not observed among females. Importantly, each of these associations occurred independently of potentially confounding demographic characteristics, vascular risk factors, and comorbid disease. Together, our findings suggest sex- and race-specific heterogeneity of the relationship between systemic inflammation and amyloid deposition.
The current understanding of the relationship between systemic inflammation and brain amyloid deposition in humans is limited. Our findings highlight the dynamic nature of this relationship and suggest that the effect of systemic inflammation on brain amyloid may be age, sex, and race dependent. Brain amyloid deposition in persons with preclinical AD typically begins during midlife, two or more decades before the disease is clinically manifested [40]. Thus, our finding that a higher level of CRP during midlife, but not late-life, is associated with elevated brain amyloid among a subset of older adults suggests that systemic inflammation may be most etiologically relevant in middle adulthood, during what is likely the early stages of brain amyloid deposition. The significance of this midlife exposure period was recently highlighted in another ARIC-PET study, which demonstrated that midlife, but not late-life, vascular risk factors are associated with brain amyloid deposition in older adults [37]. Midlife, but not late-life, systemic inflammation may be associated with an increased duration of exposure to the potentially pathogenic effects of sustained proinflammatory signaling. Previous theories have posited that individuals with chronic systemic inflammation may be at greatest risk for adverse neurologic outcomes [41, 42]. Our finding that male participants with persistently elevated mid- to late-life CRP levels demonstrate greater odds of elevated brain amyloid is consistent with this notion.
In what is, to the best of our knowledge, the only previous study to examine the association between circulating immune markers and amyloid deposition measured using PET, a greater percentage of CD8+ T cells was found in the blood of non-demented older adults with elevated brain amyloid [43]. Although these findings suggest that the adaptive immune response is heightened in individuals with greater levels of brain amyloid, the small sample size and cross-sectional study design make it difficult to draw strong conclusions. CSF studies have provided additional evidence for an association between systemic inflammation and molecular biomarkers of AD. For example, plasma and CSF levels of soluble tumor necrosis factor receptor-1 and -2 correlated strongly with CSF levels of Aβ40, tau, and β-site APP – cleaving enzyme 1 (BACE1) activity in a cross-sectional study of non-demented older adults [44]. However, in other studies which have included middle-aged and older adults, circulating levels of inflammatory proteins (e.g., Alpha-2 macroglobulin, interleukin 6, interleukin 8) were more strongly associated with CSF markers of neurodegeneration and neurofibrillary tangle formation (tau and phosphorylated tau) than with markers of Aβ deposition (Aβ42) [45–47]. Although these results provide additional support for the relationship between systemic inflammatory markers and AD pathology, the degree to which inflammation is associated with amyloid deposition versus tau pathology or neurodegeneration more generally is still unclear.
Our findings which indicate that systemic inflammation, when elevated in midlife or elevated chronically, is associated with higher brain amyloid levels among men, are in line with previous studies, which have found stronger associations between circulating inflammatory markers and indicators of neurodegeneration among male participants [44, 49]. These sex differences may be explained by several factors, including sex differences in monocyte and lymphocyte function [50], sexual dimorphism in inflammatory gene expression [15, 51], and a hormonal influence on the peripheral immune signaling [52]. Race also appeared to modify the association of midlife CRP with late-life brain amyloid in the current study. Race differences in physiological levels of CRP and the regulation of inflammatory signaling pathways have been described [16] and may account for discordant findings. The observed racial differences may also be the results of confounding due to dissimilar sample characteristics or environmental effects related to differences in geographic location. Compared to white participants, African American participants had a higher burden of chronic disease and greater physiological heterogeneity. Relatedly, the majority of African American participants included in this study (96%) were located at the Jackson, MS site, whereas white participants were located in Washington County, MD or Forsyth County, NC. Lastly, race differences may also result from a survival bias, as we have found that African Americans (particularly African Americans with high CRP) are more likely to die or drop out of the study before Visit 5 (Supplementary Table 6).
The associations between midlife CRP level and late-life brain amyloid deposition support theories which have identified systemic inflammation as a potential driver of Aβ accumulation. However, there are other plausible explanations for the current findings. For example, it is possible that as Aβ begins to accumulate in the brain, it initiates a compensatory response within the central nervous system that, in turn, activates the peripheral immune system. Aβ may induce a low-grade immune response in the periphery as it leaves CSF and interstitial fluid through lymphatic drainage, where it can then enter peripheral circulation [43, 53]. Alternatively, systemic inflammation may be an epiphenomenon of another biological process, not accounted for in the current study, which itself promotes brain amyloid deposition. Although causality cannot be assumed from the current findings, support for a causal link between systemic inflammation and Aβ deposition has emerged from translational and experimental studies [6–8]. For example, previous work using animal models has demonstrated that systemic inflammation can trigger a neuroinflammatory response, which, in turn, promotes AβPP expression and the intracellular accumulation of Aβ [7].
Strengths of the current study include the prospective study design, serial measurement of CRP, extended follow-up period, use of a relatively large population-based sample, the inclusion of a large number of African American participants, and careful measurement and adjustment for potentially confounding variables. However, the current results must be interpreted within the context of several limitations. First, although CRP is widely used as a non-specific marker of systemic inflammation, without a broader assay of inflammatory cytokines and chemokines, our ability to understand the more nuanced associations between pro- and anti-inflammatory signaling and brain amyloid deposition may be limited. Second, given the 15-year duration between Visits 4 and 5, there is an increased likelihood that relevant changes in CRP were unmeasured. Therefore, the characterization of CRP patterns over this period may be vulnerable to misclassification. Future studies will benefit from a more frequent longitudinal assessment of blood inflammatory markers. Third, the current study may have been underpowered to detect some of the more modest associations in our primary and subgroup analyses, and the analysis of multiple race- and sex-stratified subgroups increases the probability of type 1 error. Although the moderating effects of sex and race have been demonstrated in similar studies, findings from our subgroup analyses should be interpreted with caution. Lastly, the current findings may be subject to selection bias resulting from differential attrition, as participants with the highest levels of midlife systemic inflammation were more likely to die or dropout before Visit 5 (Supplementary Table 6). The exclusion of participants with dementia from PET analyses may have similarly resulted in an omission of participants with highest levels of systemic inflammation and brain amyloid, biasing results toward the null.
Despite these limitations, the current study provides insight into the relationship between systemic inflammation during middle- and late-life and a central component of AD pathology. However, our findings, which suggest sex- and race-specific associations between CRP and brain amyloid, should be interpreted with caution, as future studies will be needed to corroborate or disconfirm these results. While there is biological plausibility and previous support for our sex-specific findings, the explanations underlying racial differences are more likely related to non-biological factors. Together, these findings highlight the need to further understand how sex and race may influence the role of immune functioning in AD pathogenesis.
Footnotes
ACKNOWLEDGMENTS
The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). Neurocognitive data is collected by U01 HL096812, HL096814, HL096899, HL096902, HL096917 from the National Heart, Lung, and Blood Institute and the National Institute of Neurological Disorders and Stroke, with previous brain MRI examinations funded by R01-HL70825 from the National Heart, Lung, and Blood Institute. This study was also supported by contracts T32 AG027668 (Dr. Walker) and K24 AG052573 (Dr. Gottesman) from the National Institute on Aging. The ARIC-PET study is funded by the National Institute on Aging (R01AG040282). Dr. Selvin was supported by NIH/NIDDK grants K24DK106414 and R01DK089174. Dr. Gross was supported by K01- AG050699 from the National Institute on Aging. Avid Radiopharmaceuticals provided the florbetapir isotope for the study, but had no role in the study design or interpretation of results. Reagents for the C-reactive protein assays were donated by Roche Diagnostics Corp. The sponsors had no role in the design and conduct of the study; collection management, analysis and interpretation of the data; or preparation review, or approval of the manuscript. The authors thank the staff and participants of the ARIC study for their important contributions.