
Abstract
Background:
Mechanisms of cortical plasticity have been recently investigated in Alzheimer’s disease (AD) patients with transcranial magnetic stimulation protocols showing a clear impairment of long-term potentiation (LTP) cortical-like plasticity mechanisms.
Objective:
We aimed to investigate mechanisms of cortico-cortical spike-timing dependent plasticity (STDP) in AD patients investigating the connections between posterior parietal cortex (PPC) and primary motor cortex (M1).
Methods:
We used a cortico-cortical paired associative stimulation (cc-PAS) protocol to repeatedly activate the connection between PPC and M1 of the left-dominant hemisphere in a sample of fifteen AD patients and ten age-matched healthy subjects. PPC transcranial magnetic stimulation preceded (ccPAS +5) or followed M1 stimulation (ccPAS – 5) by 5 ms. Motor-evoked potentials (MEPs) were collected to assess the time course of the after effects of cc-PAS protocol measuring MEP amplitude as index of cortico-cortical associative plasticity.
Results:
In healthy subjects, ccPAS – 5 protocol induced the expected long-lasting increase of MEP amplitude compatible with LTP-like cortical plasticity while PAS +5 protocol induced the opposite effect. AD patients did not show any significant modification of the amplitude of MEP after both ccPAS protocols.
Conclusions:
Our study shows that in AD patients the time-locked activation of human cortico-cortical connections is not able to form STDP, reflecting an impairment of a multi-factor plasticity process.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative process leading inescapably to dementia. Neuropathological hallmarks of AD are the deposition in several brain areas of aggregates of misfolded proteins such as amyloid plaques and neurofibrillary tangles that in turn affect primarily synaptic terminals inducing subsequently neuronal loss [1, 2]. This structural synaptic remodeling produces an engulfment of synaptic activity recorded with electrophysiological tools in vitro as imbalances of the physiological forms of long-term modifications [3].
AD typically presents with difficulty in encoding new information, affecting the hippocampal-type episodic memory [4]. This memory loss has been referred not only to local damage of the hippocampus, but also to a dysfunction of large-scale networks underlying memory processes. Indeed, in physiological conditions, the continuous refinement of synaptic connections between pyramidal cells in cortical neuronal networks has been linked to memory formation [5]. Important to this end, the efficiency of neuronal communication in encoding new stimuli is guaranteed by a precise, millisecond-scale timing of action potentials (APs) generated in pre- and postsynaptic pyramidal cells [6–11]. This spike timing-dependent plasticity (STDP), theorized by Hebb in 1949, requires that neurons near-simultaneously activated increase their efficiency if the synapse constantly assists the postsynaptic target neuron to generate APs. In in vitro studies, the repeated coupling of presynaptic and post synaptic APs results in a long-term potentiation (LTP) of synaptic efficacy, a neurophysiological effect collectively referred as the cellular correlate of learning and memory [12]. Typically, LTP is induced when presynaptic activity occurs just before postsynaptic spiking in the target cell. Conversely, long-term depression (LTD) is usually induced when the postsynaptic cell fires before the presynaptic input.
In the last decade, mechanisms of cortical plasticity have been investigated in humans by applying means of transcranial magnetic stimulation (TMS) [13, 14]. Mechanisms of cortical plasticity have been widely investigated in AD patients with TMS protocols such as theta burst stimulation (TBS), showing a clear impairment of LTP cortical-like plasticity and a relative sparing of LTD mechanisms, reflecting the AD murine models of altered hippocampal plasticity [15–17]. There is also evidence that spike-timing dependent plasticity (STDP) is compromised in AD patients, as revealed by paired associative stimulation (PAS) protocols coupling an electrical peripheral nerve stimulation with a central TMS pulse over the primary motor cortex (M1) [18, 19]. However, a fundamental aspect of AD pathophysiology is based on the dysfunction of long-range cortical networks. It has to be noted that the memory loss typical of AD is linked to early degenerative changes involving not only the hippocampus but also the associative cerebral cortices; indeed, since the early stages of AD posterior cortical regions, including the posterior parietal cortex (PPC) are affected by prominent atrophy and neuropathological abnormalities [20]. In this view, AD can be considered as the result of a disconnection between different neuronal systems [21] suggesting a dysfunction of large-scale networks underlying memory processes. In this regard, PPC exerts a key role in retrieval of episodic memory and especially in supporting attentional process to memory retrieval [22]. Therefore, abnormalities in neural networks originating form PPC may be considered responsible for many of the cognitive function deficit observed in AD such as working memory, episodic memory, and attention/executive function impairments [23].
Recently, we developed a new TMS protocol investigating STDP within the connections between the PPC and M1 in healthy conditions [24]. The direction of the after-effects reflected the order of the two stimuli: PPC-to-M1 decreased MEPs suggesting LTD-like plasticity, whereas M1-to-PPC elicited MEP facilitation suggesting LTP-like plasticity [24]. Here we aimed to investigate these mechanisms of cortico-cortical STDP in AD patients.
MATERIAL AND METHODS
Fifteen consecutive patients were recruited at the memory clinic of the University Hospital Tor Vergata, admitted for complaining symptoms. After the first visit to our Centre, all patients underwent for diagnostic purposes a complete clinical investigation in a period not superior to 60 days, including medical history, neurological examination, Mini-Mental State Examination (MMSE), a complete blood screening, neuropsychological assessment, magnetic resonance or CT imaging, ApoE sampling, and lumbar puncture for CSF analysis [25] (Table 1). Patients fulfilled the clinical criteria of dementia as defined by the DSM-IV and typical Alzheimer’s disease according to the criteria of the International Working Group (IWG) for New Research Criteria for the Diagnosis of AD [26]. Neurophysiological examinations were performed at the Santa Lucia Foundation within 30 days from CSF sampling. Patients included in this study did not received drugs that could have modulated cerebral cortex excitability such as acetylcholinesterase inhibitors [27], L-DOPA [28] or dopamine agonists [29], antidepressants or any other neuroactive drugs (i.e., benzodiazepines, anti-epileptic drugs or neuroleptics) in the 90 days prior to TMS evaluation. Exclusion criteria were the following: patients with isolated cognitive deficits, patients with clinically manifest acute stroke in the last 6 months showing a Hachinsky scale score >4, and a radiological evidence of ischemic lesions, Aβ1 - 42 CSF values >600 pg/mL. Ten age- and sex-matched healthy subjects (HS) were recruited as control group. All participants or their legal guardian provided written informed consent after receiving an extensive description of the study. The study was performed according to the Declaration of Helsinki. The ethics committee of the Santa Lucia Foundation IRCSS approved this protocol.
Demographic and clinical characteristic of AD patients and Healthy Subjects
aFisher’s exact test. n, numbers; y, years; m, months; CSF, cerebrospinal fluid; CDR, Clinical Dementia Rating; ADL, Activities of Daily Living; IADL, Instrumental Activities of Daily Living; MMSE, Mini–Mental State Examination; SD, standard deviation.
TMS
We used bifocal TMS to repeatedly activate the connections between the PPC and M1 of the left-dominant hemisphere [30, 31]. Left PPC TMS preceded or followed the M1 stimulation by 5 ms, respectively PAS +5 and PAS – 5. We then tracked the time course of the aftereffects of PAS protocol by measuring motor-evoked potentials (MEPs) amplitude as an index of M1 excitability. Notably, we assumed that the inputs from PPC to M1 may discharge the same neurons that are activated by the M1 TMS itself [30].
We used a bifocal stimulation technique based on two high-power Magstim 200 machines. The magnetic stimuli had a nearly monophasic pulse configuration with a rise time of 100μs, decaying back to zero over 0.8 ms. To measure the MEPs, electromyographic (EMG) traces were recorded from right first dorsal interosseous (FDI) muscles using 9-mm-diameter, Ag–AgCl surface cup electrodes. The active electrode was placed over the belly muscle, whereas the reference electrode was located over the metacarpophalangeal joint of the index finger. Responses were amplified using a Digitimer D360 amplifier through filters set at 20 Hz and 2 kHz with a sampling rate of 5 kHz and then recorded by a computer using SIGNAL software (Cambridge Electronic Devices). For M1 TMS, the coil was positioned over the hand motor area of left M1, defined as the point at which stimulation evoked the largest MEPs from the contralateral FDI muscle. The stimulator for M1 was connected to a small custom-made figure-of-eight-shaped coil (50 mm external diameter). The intensity of M1 TMS was adjusted to evoke an MEP of 1 mV peak to peak in the relaxed contralateral FDI muscles. The coil was positioned to induce a posterior–anterior (PA) directed current, being held posterolaterally at an angle of 45° to the midline.
To best activate the ipsilateral PPC–M1 connection, the conditioning stimulus (CS) was applied over the left PPC at an intensity of 90% of the ipsilateral resting motor threshold (RMT). RMT was defined as the lowest intensity that evoked five small responses (50μV) in the contralateral FDI muscle, in a series of 10 stimuli when the subject kept the FDI muscles relaxed in both hands [32]. The RMT was assessed at the beginning of each experimental session for each stimulating coil (PPC coil and M1 coil) separately. The magnetic pulse over PPC was applied using a second small custom-made figure-of-eightshaped coil (50 mm external diameter). We used a neuronavigation system (Softaxic; E.M.S.) to precisely position the coil over the PPC sites, using individual T1-weighted magnetic resonance imaging volumes as anatomical reference; this technique has been described previously in detail [33–35].
In the cortico-cortical PAS (ccPAS) protocol, 100 pairs of stimuli were continuously delivered at a rate of 0.2 Hz for ∼8.3 min. The PPC stimulus preceded (PPC–M1 PAS +5 ms) or followed (PPC–M1 PAS–5 ms) the M1 TMS by 5 ms. Each subject underwent the two PAS conditions (PPC–M1 PAS +5 ms and PPC–M1 PAS – 5 ms) in two different sessions performed at least 1 week apart.
In each session, 20 MEPs were collected and averaged at baseline and at T0′, T10′, and T20′ minutes after PAS. For measuring MEPs, the coil was positioned over the M1 cortical site with the same PA orientation as for the PAS using a standard 7 cm figure-of-eight coil connected with a Magstim 200 stimulator.
Data analyses
Data were analyzed using SPSS for Windows version 11.0 on the mean MEPs amplitude in each condition. A three-way repeated measures ANOVAs was performed on MEP amplitude expressed as percentage of change in comparison to baseline for each PAS PROTOCOL (ccPAS +5 ms and ccPAS – 5 ms) with TIME (baseline and T0, T10 and T20 mins after ccPAS protocols) and GROUP (AD and HS) as between subjects factor.
The Greenhouse–Geisser correction was used for nonspherical data. When a significant main effect was reached, paired t tests with Bonferroni’s correction were used to characterize the different effects of the specific ISIs. Mauchley’s test examined for sphericity. For all statistical analyses, a p value <0.05 was considered to be significant.
Pearson’s r correlation coefficients were used to explore any influence of age, education, disease duration, MMSE, CSF Aβ1 - 42, tTau, and pTau181 levels on the individual amount of change in MEPs.
RESULTS
The three-way ANOVA did not show effect for the GROUP main factor (F(1,23) = 0.81902, p = 0.37) and for the TIME within subject factors (F(3.69) = 2.3971, p = 0.075). There was a significant effect for the PROTOCOL main factor (F(1,23) = 5.4229, p = 0.03); the PROTOCOL X GROUP (F(1,23) = 12.8, p = 0.00159) and TIME X PROTOCOL (F(3,69) = 2.288, p = 0.025) interactions were significant, while the TIME X GROUP (F(3.69) = 0.63045 p = 0.156) interaction was not significant. The triple interaction PROTOCOL X GROUP X TIME was significant (F(3,69) = 3.816, p = 0.013). Post hoc analyses with Bonferroni correction showed that in HS for PPC–M1 ccPAS +5 ms there was an inhibition of MEP amplitude compared with baseline at T10′ (p = 0.03) (Fig. 1B) while for ccPAS – 5 ms, there was a facilitation of MEP amplitude compared with baseline at T20′ min (p = 0.02) (Fig. 1A). Instead, when considering ccPAS protocols after-effects in AD patients we did not observe the same MEP modifications. Therefore, ccPAS protocol induced in HS, but not in AD group, the expected bidirectional cortico-cortical associative plasticity in the PPC–M1 network: when PPC preceded M1 stimulation (ccPAS +5 ms), there was a long-lasting decrease of the excitability of M1, indicating an LTD-like effect; conversely, when PPC followed M1 stimulation (ccPAS – 5 ms), there was a long-lasting increase of the excitability of M1, indicating an LTP-like effect.
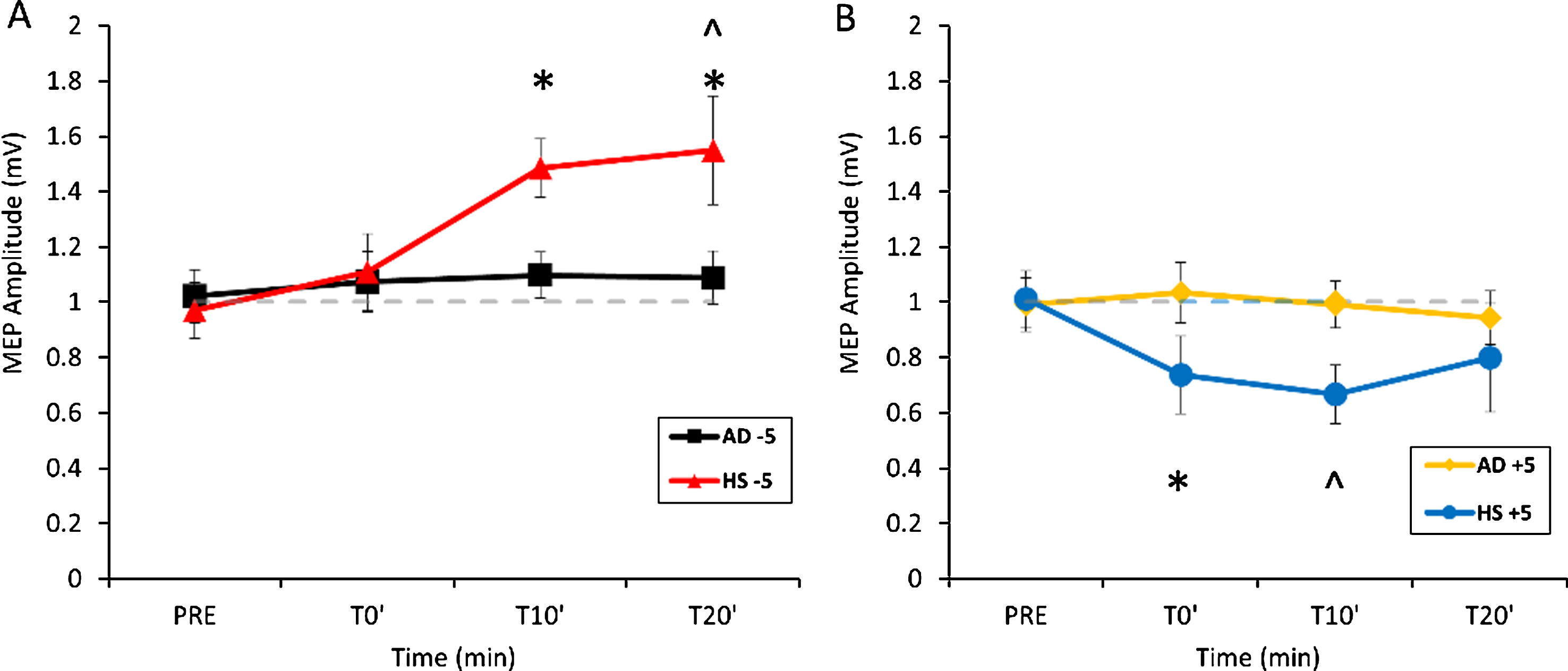
Aftereffects of PPC-M1 ccPAS – 5 ms (A) and +5 ms (B) in AD and HS. Error bars indicate SEM. *p < 0.05 compared to AD; ∧p < 0.05 compared to baseline.
Moreover, Bonferroni correction showed some direct differences in PPC-M1 STDP between AD and HS. In fact, for ccPAS – 5 ms protocol AD patients differed from HS at T10′ (p = 0.034) and T20′ (p < 0.01) (Fig. 1A), while for ccPAS +5 ms protocol, a significant difference was observed at T0′ between AD and HS (p = 0.037) (Fig. 1B). Taken together these data confirm that LTP mechanisms, investigated by exploring STDP within the PPC-M1 network, are deeply altered in AD patients compared to HS, while LTD mechanisms, even if different between the two groups, seem to be less affected (Fig. 2).

Violin plot of the individual mean change of MEP amplitude (mean of MEP amplitudes recorded at T0′, T10′, and T20′ minutes after each ccPAS protocol respect to baseline).
These differences were confirmed when directly comparing PPC-M1 STDP between AD and HS with a two-way ANOVA. Indeed, for ccPAS – 5 ms protocol, the repeated measure ANOVA performed on the mean MEP amplitude percentage change showed a GROUP effect (F(1, 23) = 6.6990, p = 0.01644) and TIME X GROUP interaction (F(2, 46) = 3.3305, p = 0.04458). For ccPAS +5 ms protocol, the ANOVA showed an effect for GROUP F(1, 23) = 6.1155, p = 0.02122, but not for the TIME (F(2, 46) = 0.37879, p = 0.68680) and for GROUP X TIME interaction (F(2, 46) = 0.19742, p = 0.82154).
We also employed Pearson r correlation coefficient in univariate correlations in order to explore any relationship between the individual amount of change induced by both protocols of ccPAS in each AD patients and the different clinical and demographic factors. Correlation analyses performed in the AD sample did not find any significant association between the individual amount of change in MEPs (induced by both ccPAS protocols) and clinical and demographic factors: age (ccPAS +5: r = 0.13; p = 0.45; ccPAS – 5: r = 0.17; p = 0.41), education (ccPAS +5: r = 0.24; p = 0.34; ccPAS – 5: r = 0.21; p = 0.39), disease duration (ccPAS +5: r = 0.22; p = 0.40; ccPAS – 5: r = 0.12; p = 0.51), MMSE (ccPAS +5: r = 0.09; p = 0.81; ccPAS – 5: r = –0.16; p = 0.27),) and CSF Aβ1 - 42 (ccPAS +5: r = –0.26; p = 0.32; ccPAS – 5: r = –0.09; p = 0.71), tTau (ccPAS +5: r = 0.29; p = 0.21; ccPAS – 5: r = 0.08; p = 0.62), and pTau181 levels (ccPAS +5: r = 0.09; p = 0.83; ccPAS – 5: r = –0.10; p = 0.51).
DISCUSSION
Our findings indicate that the mechanisms of ccSTDP are altered in AD patients. Indeed, while in HS ccPAS protocols (both ccPAS +5 ms and ccPAS – 5 ms) were able to induce consistent aftereffects ascribable to STDP, in AD patients the same protocols did not evoke any signification modification of M1 activity. These findings are consistent with previous studies investigating mechanisms of cortical plasticity in AD patients and age matched HS, either with TBS technique [15–17] and with canonical PAS protocol coupling an electrical peripheral nerve stimulation with a M1 TMS pulse [18, 19]. The novelty of the present study is that we investigated cortical plasticity within the PPC-M1 connection, so directly testing the strength of a specific neural network in AD. Interestingly, ccPAS protocols showed clear differences among HS and AD patients, shedding light on the putative mechanisms of cortical impairment and cognitive decline.
In associative synaptic plasticity, simultaneous or rapid sequential activation of two connected neurons leads to a change in the strength of synapses between them. Continuous structural refinement of cortical synapses is essential in the formation of LTP, a dynamic enhancement of synaptic efficacy generally considered as a cellular correlate of learning and memory [12]. In this framework, computational models of brain functioning introduced the concept of functional connectivity, which is the influence that one neuronal system exerts over another [36]. Therefore, in this contest of long-ranged cortical networks the integrity of synaptic machinery results essential for a functional communication. In AD, cortical synaptic function becomes compromised prior to the physical disintegration of synapses [37], and synapse loss is disproportionate to the extent of neuronal demise [38]. Notably, these events occur early in different cortical areas, such as temporal, parietal and frontal lobes. In the last years, it has been advanced a new model for AD pathophysiology, namely “disconnection interpretation” which suggests that AD pathology would derive from the disturbance of the brain’s effective connectivity suggesting abnormal interactions between neuronal systems [21]. This intriguing theory has been supported by several neuroimaging data showing in AD patients a dysfunction of cortico-cortical connections, ascribed to the loss of afferent and efferent connections between different cortical areas [23, 40]. These connectivity abnormalities lead to the shrinkage of neural networks accountable for high order cognitive capacities such as working memory, episodic memory, and attention/executive functioning [41, 42], thus explaining the impairment of these cognitive systems since the early phase of AD. In this context the data from the current study, showing a weakened neural communication among two interconnected areas, suggest an impairment of a cortical network involved in many high cognitive processes, paralleling neuroimaging data obtained from AD patients. The neurophysiological reflection of connectivity is LTP (measured with either TBS and ccPAS or canonical PAS protocol) and our data reinforce the concept of LTP impairment as a key neurophysiological phenomenon in early stages also in AD patients.
Indeed, our data are in line with animal models of AD pathology. In previous studies on transgenic murine models of Aβ-induced AD pathology electrophysiological recordings failed to manifest an increase in STDP compared to their age matched controls in both Layer 2/3 [43] and Layer 5 of neocortical pyramidal cells [44] even at younger ages, suggesting that impairment of cortical plasticity machinery is an early event in the cascade of physiopathological processes leading to neurodegeneration and finally to dementia. Noteworthy, also in mice models of tau pathology both morphological [45] and functional [46] abnormalities have been found on cortical layer V of pyramidal neurons. Interestingly, significant electrophysiological changes are present in the early stage and persist in the advanced stage and consist in depolarized resting membrane potential, an increased depolarizing sag potential and increased action potential firing rates— all indicative of hyperexcitability [47, 48]. Recent studies demonstrated that changes in glutamatergic transmission occur prior to spine loss, disrespectfully of the underlying pathology: indeed, the reduced surface expression of postsynaptic glutamate receptors (both NMDA and AMPA receptors) is associated with impaired basal synaptic transmission and LTP in the hippocampus in several animal models [46, 50]. In this view, the weakening of glutamatergic inputs, together with the scarcity of excitatory synaptic contacts in neocortical pyramidal cell networks [51] further magnifies the pathogenic burden of Ab and tau accumulation, and its capacity to significantly hinder information processing, detectable with electrophysiological tools both in animal models and in AD patients [46].
Furthermore, recent evidence shows that neocortex is richly innervated by basal forebrain cholinergic neurons in a layer-dependent manner, accordingly to the expression of nAChRs, resulting in a layer-specific control of synaptic plasticity by endogenous ACh [52]. Since activation of nAChRs located on presynaptic terminals can increase glutamate release from synapses [53] and presynaptic nAChRs containing α7 subunits elicits LTP of glutamatergic synapse in different brain regions [54, 55], it seems likely that the cholinergic signaling depauperization typical of AD, could contribute to the alteration of the cellular mechanisms underlying long term modification of synaptic efficacy.
A limitation of the current study is that we did not perform a paired-pulse TMS protocol to study functional interplay between PPC to ipsilateral motor cortex [30]. However in a previous paper from our group we already showed that parieto-frontal functional connectivity is altered in AD patients [56]; in light of these results, and in line with neuroimaging studies supporting a functional disconnection between different cortical areas [23, 40], we cannot rule out that the impairment of STDP found in our sample of AD patients by repeatedly stimulating two cortical areas highly interconnected could be driven by an altered functional connection between the two areas.
Conclusions
The present study represents a further step in investigating in AD patients the mechanisms of cortical plasticity impairment that, as assessed by several animal models, increasingly accounts for a key and early role in the pathophysiological cascade of events leading to neurodegeneration and finally to dementia. We demonstrate that in AD patients the time-locked activation of human cortico-cortical connections is not able to form STDP, reflecting an impairment of a multi-factor plasticity process that depends jointly on firing rate, spike timing, dendritic depolarization and synaptic cooperativity [57]. Since, STDP, based on computational and in vitro models, is thought to mediate learning and memory both in the hippocampal and the neocortex [58] as a cognitive and global cognitive map, disruption of spiking recurrent neural networks could take account for the memory deficits typical of AD patients.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0503r1).