
Abstract
Alzheimer’s disease (AD) afflicts more than 46.8 million people worldwide, with a newly diagnosed case every 3 seconds and no remission in the disease progression. The discovery of disease-modifying drugs is now on the summit of the neuropharmacological research priorities. The long-lasting derivatives of the insulinotropic incretin hormones—glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP)—have repeatedly been shown to cross the blood-brain barrier and counteract an array of deleterious effects across a range of experimental models of neuronal degeneration. Clinical trials for the efficacy of GLP-1 agonists in Alzheimer’s and Parkinson’s diseases have revealed beneficial effects of these anti-diabetic agents in halting neuronal degeneration progression. Herein, we examine whether the chronic treatment with the novel dual GLP-1/GIP receptor agonist DA-CH3 can restore the cognitive decline and AD-like cerebral pathology of the APPSWE/PS1ΔE9 mouse model at the age of 10 months old. We report that once-a-daily, eight-week intraperitoneal administration of 25 nmol/kg of the novel DA-CH3 dual-incretin analog rescues the spatial acquisition and memory impairments of this murine model that corresponds to the attenuation of the excessive plaque deposition, gliosis and synaptic damage in the APPSWE/PS1ΔE9 brain. The amelioration of the AD-related pathology reflects the resolution of the endoplasmic-reticulum stress and derailed autophagy that both lay downstream of the rectified Akt signaling. Collectively, our findings endorse the beneficial effects of the incretin-based therapeutic approaches for the neurotrophic support of the AD brain and for the first time associate the incretin-induced neuroprotection with the proteostasis machinery in vivo.
INTRODUCTION
Alzheimer’s disease (AD) afflicts more than 46.8 million people worldwide, with a newly diagnosed case every 3 seconds [1] and no remission in the disease progression [2–4]. By the mid-century, it will be a global epidemic with more than 130 million sufferers and will pose a public health crisis [1]. Therapeutic interventions able to delay or reverse the AD pathology and cognitive decline will spare patients and their families the debilitating emotional and financial burden of the disease. As such, the discovery of disease-modifying drugs is now on the summit of the neuropharmacological research priorities [2–4].
Endogenous neurotrophic factors—long-ago considered central for the neuronal differentiation, survival, and connectivity during development—can restore the morphology and function of degenerating neurons during adulthood when administered exogenously. Over the last two to three decades with numerous pre-clinical studies in rodent and primate models of the β-amyloidosis or cholinergic lesions, an abundant research history has acquired regarding the therapeutic potential of the neurotrophins in the AD treatment [5–7]. However, their efficacy and delivery to the brain remain significant disappointments at a clinical level [8, 9].
Derivatives of the insulinotropic incretin glucagon-like peptide 1 (GLP-1), currently approved for type II diabetes mellitus (T2DM) management, may offer an efficacious alternative for the neurotrophic support of the AD brain [10]. They cross the blood-brain barrier and preserve normal population and activity of cortical and hippocampal synapses, impede hippocampal atrophy while halting excessive Aβ deposition and gliosis to rescue spatial cognitive decline in the APPSWE/PS1ΔE9 mouse model for AD [11–14] and the senescence-accelerated mouse prone 8 (SAMP8) model for age-related sporadic AD [15]. In line with preclinical findings, the long-lasting GLP-1 derivative, Liraglutide, has attenuated the impairments in cerebral glucose consumption in AD patients, signifying improved neural energetics and activity in areas previously correlated with cognitive decline and disease progression [16] (also refer to [10] for a review). Accordingly, the stable analog of the glucose-dependent insulinotropic polypeptide (GIP), DAla2-GIP has alleviated neuroinflammatory responses, oxidative stress, and cerebral amyloid plaque load to attenuate the poor performance of the APPSWE/PS1ΔE9 mouse model of AD in the Morris water-maze and novel object recognition paradigms [17, 18]. These effects have been further correlated to enhanced synaptogenesis and restored synaptic plasticity events in the cortex and hippocampus of the middle-aged and aged APPSWE/PS1ΔE9 mice upon chronic DAla2-GIP treatment [18]. The induction of GLP-1 and GIP receptors confers trophic and survival signals through a downstream signaling cascade that converges into the extracellular signal-regulated kinases 1&2 (ERK1/2), the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt), and the signal transducer and activator of transcription 3 (STAT3) factor [10, 19–22]. Mechanistically, the incretin signaling modulates proteostatic responses, such as the unfolded protein response (UPR), the protein quality control machinery and autophagy, to elicit neuronal survival in vitro [21]. Recently novel GLP-1/GIP dual receptor agonists (DA) have deployed [10, 23] with an improved anti-hyperglycemic and insulinotropic efficacy compared to the GLP-1 mono-therapy, rendering them potent candidates for the T2DM treatment [23]. Notably, pilot pre-clinical work has unraveled the restorative effects of the dual incretins in the neuronal degeneration processes and functional decrements in a murine model of mild traumatic brain injury (DA–JC1) [24], the rat model of streptozotocin-induced insulin-desensitization in the brain and neurodegeneration (DA–JC4) [25], in neurotoxin-based rodent models of Parkinson’s-disease (DA–JC1, DA–JC4, DA–CH3, DA–CH5) [26–30], and in the APPSWE/PS1ΔE9 mouse model (DA–CH5) [31].
In the light of these encouraging findings, herein, we have examined whether the chronic DA–CH3 dual incretin treatment can restore the cognitive decline and Alzheimer-like cerebral pathology of the APPSWE/PS1ΔE9 mouse model at the age of 10 months old. We have additionally correlated the phenotype to the cellular responses for balancing protein synthesis and degradation. We report that once-a-daily intraperitoneal administration of 25 nmol/kg of this novel GLP-1/GIP dual receptor agonist rescues spatial acquisition and memory impairments of this murine model that corresponds to the attenuation of the excessive plaque deposition and gliosis and the preserved availability of vesicle and scaffolding proteins for synapse integrity and function in the APPSWE/PS1ΔE9 brain. The amelioration of the AD-related pathology reflects the resolution of the UPR and suppressed autophagy that both lay downstream of the rectified Akt signaling. Collectively, our findings endorse the beneficial effects of the incretin-based therapeutic approaches for precluding neuronal degeneration [10] and for the first time associate the incretin-induced neuroprotection with the proteostasis machinery in vivo.
MATERIALS AND METHODS
Materials
The primary monoclonal antibodies for the immunoglobulin heavy-chain-binding protein [BiP; C50B12, #3177], CAAT/enhancer-binding protein (C/EBP) homologous protein [Chop; L63F7, #2895], voltage-dependent anion channel [VDAC; D73D12, #4661], beclin-1 [D40C5, #3495], autophagy-related protein 3 [Atg3; #3415], Atg7 [#8558], phosphorylated glycogen synthase kinase 3β (GSK-3β) at the serine 9 (Ser9) residue [D85E12, #5558], phosphorylated Akt at the threonine 308 (Thr308) residue [D25E6, #13038], phosphorylated ERK1/2 at the Thr202 and tyrosine 204 (Tyr204) residues [D13.14.4E, #4370], post-synaptic density protein 95 [PSD95; D27E11, #3450] and for the β-actin protein [8H10D10, #3700] along with the primary polyclonal antibody for the microtubule-associated proteins 1A/1B light chain 3B [LC3B; #2775), the horseradish peroxidase (HRP)-linked secondary antibodies against the corresponding species IgG of the primary antibodies [#7074 & #7076], the PathScan® Sandwich ELISA lysis buffer [1X; #7018], and the Protease/Phosphatase Inhibitor Cocktail [100X; #5872] procured from Cell Signaling Technology (CST; New England Biolabs UK Ltd, Hertfordshire, UK). Polyclonal antibodies caspase 12 [CASP12; ab62484], synaptophysin [Sys; ab7837] and the glial fibrillary acidic protein [GFAP; ab7260] along with normal goat serum obtained from Abcam (Cambridgeshire, UK). Amyloid-β (Aβ) polyclonal antibody (71-5800) acquired from Thermo Fisher Scientific (Renfrewshire, UK). Ionized calcium-binding adapter molecule 1 (IBA-1) polyclonal antibody (016-20001) purchased by Wako Pure Chemicals GmbH (North Rhine-Westphalia, Germany).
The Quick Start Bradford protein assay kit procured by BIO-RAD Laboratories Ltd (Hertfordshire, UK). Amersham ECL Prime western blotting detection reagent kit was acquired from GE Healthcare Life Sciences (Buckinghamshire, UK). The iBlot® 2 Dry Blotting System, iBlot® 2 Transfer Stacks with integrated nitrocellulose transfer membranes, pre-cast polyacrylamide Bolt 4–12%gradient Bis-Tris Plus gels, and Restore PLUS Western Blot striping buffer purchased from Fisher Scientific UK Ltd (Leicestershire, UK). Bovine serum albumin (BSA), tris-buffered saline (TBS; pH 8.0) supplemented or not with 0.05%Tween® 20, phosphate buffered saline (PBS; pH 7.4), phosphate buffer (0.1M, pH 7.4), heparin sodium salt from porcine intestinal mucosa, sucrose, ethylene glycol (anhydrous, ≥99.8%), glycerol for molecular biology (≥99%), trisodium citrate, hydrogen peroxide solution (50%), and paraformaldehyde obtained from Sigma-Aldrich Corporation (Dorset, UK). VECTASTAIN Elite® ABC-HRP Kit (Peroxidase, Rabbit IgG) acquired from Vector Laboratories Ltd (Cambridgeshire, UK). Rest of the materials for western blotting and histology procured from Fisher Scientific UK Ltd (Leicestershire, UK), unless otherwise stated.
Animals
APPSWE/PS1ΔE9 male mice with a C57 black 6 (C57BL/6) background acquired from the Jackson Laboratory (Maine, US). Wild-type (WT) C57BL/6 female mice obtained from Charles River UK, Ltd (Kent, UK). Heterozygous APPSWE/PS1ΔE9 males were bred with WT females C57BL/6 in the Lancaster University animal unit. Their offspring were ear-punched and genotyped for the APP sequence (Forward “GAATTCCGACATGACTCAGG”, Reverse: “GTTCTGCTGCATCTTGGACA”) by polymerase chain reaction with sequence specific primers (PCR-SSP), as previously described [14]. Genotyping results were confirmed by the histological staining of Aβ plaques in mouse brains at the end of the study (please refer to “Brain tissue homogenization and protein extraction” below). Mice negative for the transgene in PCR-SSP served as WT controls while their littermates positive for the transgene served as the experimental groups in our study. Animals of both genders used in our study (Table 1) and single-housed in individually ventilated cages (IVCs) in a temperature-controlled colony room (21±2°C). Animals housed in an enriched environment with standard nesting material, NestPak® bedding material, chewing stick and non-toxic polycarbonate play tunnel. They were under a 12 h light/dark cycle, with the lights coming on at 07.00 AM. Food and water were provided ad libitum. Daily handling of the animals for two weeks preceded the initiation of the treatments. Treatments started when the mice were at the age of 10-month-old. Animals were administered Saline (0.9%Sodium Chloride) or DA–CH3 GLP-1/GIP Dual receptor Agonist [10] (Chinapeptides Co., Ltd, Shanghai, China) once-a-daily via the intra-peritoneal (i.p.) route at a dose of 10 ml/kg/day or 25 nmol/kg/day, respectively. The purity of the peptide analyzed with reverse-phase high performance liquid chromatography (HPLC) and characterized using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. The purity of the peptide was 96.86%. The peptide maintained in powdered, desiccated form at –80°C and reconstituted in Gibco® Water for Injection for Cell Culture (Fisher Scientific UK Ltd, Leicestershire, UK) to the appropriate dose at the beginning of the study. Daily dose was selected based on previous experiments that demonstrate the neuroprotective and restorative effects of the incretin mimetics in the aforementioned murine model [11–14, 32] and in accord with the Good Practice Guidelines for the administration of substances in laboratory animals [33–35]. Animals were monitored for their body weight weekly over the treatment period. Mice received the treatments for 8 weeks prior to the assessment of the animals’ exploratory behavior, anxiety, learning and memory. All procedures carried out during the light phase of the cycle, with at least an hour gap from the light/dark transition.
Animal population per genotype, assigned treatment, and gender, as used in the present study
Ethics statement
Experimental protocols and research purposes were approved and licensed by the Home Office, UK (PPL70/8250). Animal handling and experiments conducted in accordance with the “Animal (Scientific Procedures) Act of 1986”. Animal health state monitored daily throughout the study course. Neither treatment-induced harmful effects nor signs of pain noted. No mice prematurely euthanized. However, the hemizygous APPSWE/PS1ΔE9 mice on the congenic C57BL/6 background exhibit a quite high incidence of epileptic seizures that results in the premature death, with a mortality rate of 38%or higher [36–38]. Herein, we observed a mean mortality rate of 24%in the transgenic animal groups. All the casualties have reported to the corresponding ethics committee.
Open field test
Exploratory behavior and anxiety assessed in the open field test 8 weeks post initiation of the treatments, as previously described [39]. The open field test conducted in a grey-colored, circular aluminum arena (58 cm in diameter) surrounded by a continuous 31-cm-high wall. The open field apparatus was illuminated by a 60-W light bulb fixed to the ceiling, 2.1 m above the center of the arena. Prior to the commencement of the behavioral task, mice were acclimated to the experimental room for 20 min. Each mouse was placed into the center of the open field and allowed to explore freely the apparatus for 10 min, with the experimenter out of the animal’s sight. A Python-based video tracking/analyzer system interfaced with a monochrome digital camera (Point Grey, ClearView Imaging, Oxfordshire, UK) was used to automatically detect, record and process mouse movements in the arena. Each generated track represented the total distance traveled over the course session and analyzed for the time spent, velocity and path length in the center (inner zone with a radius of 10 cm) and periphery (outer zone with a radius of 10 cm) of the open field (Supplementary Figure 1). Rearing (when the mouse raises its forepaws, stands on its hind paws and extends its head upwards) and grooming (when the mouse licked/scratched its fur, washed its face, and/or licked its genitalia) events recorded manually during each session. The experimenter for scoring mouse behavior was blind to the animal group and treatment design. Upon task completion, the mouse was gently removed from the open field and returned back to its home cage. Any fecal boil deposits were manually counted and removed, and the apparatus was thoroughly cleaned with 70%ethyl alcohol to eliminate residual olfactory cues.
Morris water-maze task
The Morris water maze was employed to evaluate spatial learning and reference memory [40] 8 weeks post initiation of the treatments. It conducted in a circular white plastic pool with a diameter of 150 cm and a depth of 60 cm. The pool was filled with water at 22±1°C to preclude animal hypothermia and at that depth in which the escape platform (10 cm in diameter; made of clear acrylic glass) was submerged 1 cm below the water surface. The water opacified with a non-toxic white dye that rendered the escape platform invisible. The maze apparatus illuminated by four 60-W light bulbs fixed on the floor of the pool perimeter. Extra-maze visual cues positioned in the sidewalls of the apparatus and triangulated the center of the pool.
Two principal axes of the maze were designated, with each axis bisecting the apparatus perpendicular to one another and creating an imaginary “+”. The ends of each axis demarcate four cardinal points along the circumference of the apparatus –North (N), South (S), East (E), and West (W) that did not refer to true magnetic compass directions. Instead, they relied on the experimenter’s position, with S being the experimenter’s position, N being opposite to S point, and E and W being to the experimenter’s right and left, respectively [41]. These points served as starting positions for the mice in apparatus. Having further divided the maze in that way created four equal quadrants for platform positioning and animal tracking analysis. Swimming activity of each mouse monitored with a monochrome digital camera (The Imaging Source Europe GmbH, Bremen, Germany), mounted overhead (2.1 m above the center of the apparatus) and relayed video signals to a Python-based, water-maze tracking/analyzer system software.
The Morris water-maze task initiated a day post the completion of the open field task and lasted 17 days. It consisted of three phases, the visible-platform training, the spatial acquisition and the spatial reversal phase, as described below. On each day of testing, all mice were acclimated to the experimental room for 20 min prior to the commencement of the task. At the end of each trial, over the testing period, mice were gently removed from the pool, returned to their home cages, and placed under a 60-W infrared heat lamp for 1-2 min. Mice were visually inspected to ensure thorough dryness. All testing was conducted at roughly the same time each day in order to minimize variability in performance due to time of day.
Visible-platform training
Mice were initially trained to locate the escape platform cued with a flag within 60 s. If an animal could not locate the visible platform over the allotted time, it was gently guided onto platform and allowed to re-orient to the distal visual cues for additional 30 s. Visible-platform training aimed to detect any potent sensorimotor and motivational/emotional defects which could significantly impact the upcoming spatial acquisition phase [42]. The path length and the amount of time required to locate the platform along with the swim velocity and thigmotactic behavior were recorded. Thigmotactic behavior was evaluated by the swim time along the perimeter of the pool. The latter describes a peripheral zone with a radius of 10 cm in the tracking/analyzer system software. Mice assigned to the visible-platform training for four consecutive days. Over the training course, animals run in total 9 trials, with one trial on the first day, two trials on the second day, three trials on third and fourth day. Pairs of trials were separated by an inter-trial interval of approximately 30 min. The position of the cued, visible platform and the starting position of each mouse were randomly alternated to avoid animal habituation to a specific area of the pool.
Spatial acquisition
Mice were next trained to locate an invisible escape platform fixed on a target position in the northwest quadrant and 30 cm away from the wall of the apparatus. Animals were allowed to search for the invisible escape platform for 60 s and gently guided onto the platform if they could not locate it in the allotted time. Animals run 3 trials per day with an inter-trial interval of approximately 30 min. The animal starting position was randomized among the trials per day. Acquisition phase continues for nine additional days for a total of 30 training trials. Swim time and the path traveled to the platform along with the swim velocity and thigmotactic behavior were recorded during each trial. The daily mean swim time (hereinafter referred to as the escape latency) and path length to locate the platform were monitored to evaluate spatial learning [41]. A retention probe trial was held on the 11th day to assess spatial reference memory. The escape platform removed and the swimming pattern of each mouse recorded in a single trial of 60 s. The percentage of the swim time spent in the quadrant that formerly contained the platform along with the average distance of each mouse from the center of platform were considered as indexes of memory retention [40, 43].
Spatial reversal
A day after the retention probe trial, the escape platform was placed back to the maze though re-located to the opposite, southeast quadrant and fixed 30 cm away from the wall of the apparatus. Mice were subjected to a set of 3 trials per day for five additional days for a total of 15 trials. Spatial reversal training was conducted similarly to the spatial acquisition training. The spatial reversal phase was aimed to study animals’ cognitive flexibility to extinguish their initial learning of the platform’s position and acquire a direct path to the new goal position [41]. As in the acquisition phase, at the end of the reversal learning, a reversal probe trial conducted 24 h later. The percentage of the swim time spent in the quadrant that formerly contained the platform along with the average distance of each mouse from the center of platform similarly reflected memory retention [40, 43].
During each session, animal activity recorded with a monochrome digital camera (Point Grey, ClearView Imaging, Oxfordshire, UK) mounted on the ceiling, 2.1 m above the center of the open field arena. The camera was interfaced with a Python-based video tracking/analyzer system that allowed us the automatic detection of the exploration time allotted in each object during the acquisition and retention trials. A mouse was considered to explore an object when its nose was directed towards the object at a distance ≤2 cm [44]. Data was processed for the calculation of the Recognition Index that describes the percent ratio of the amount of time spent in exploring the familiar or novel object over the total time spent in object exploration during the retention trial course [44]. Recognition index for the novel object was then compared to the corresponding index for the familiar object within each animal group. Novelty preference signified task acquisition and recognition memory retrieval [44]. The velocity and total path length were also recorded and compared among the groups to determine whether lack of motivation or emotional defects interfered with the object recognition task acquisition and memory testing.
Brain tissue collection
Following the behavioral/cognitive assessment, animals were transcardially perfused under deep urethane anesthesia (i.p. injected) with sterile-filtered PBS, supplemented with 20 IU/ml of heparin. Brains were removed from the skull and sagittally dissected into the two hemispheres. Collected right hemispheres were processed for immune-histochemical studies. Left hemispheres were snap frozen in liquid nitrogen and stored at –80°C until processed further for homogenization and protein extraction.
Histology
Upon collection, the right hemispheres were immersion-fixed in 4%paraformaldehyde for 48 h at 4°C. They were subsequently cryoprotected into 30%sucrose in PBS solution at 4°C, until sunk to the bottom. Brain tissue was embedded on the specimen stage using Tissue-Tek® O.C.T. compound, snap frozen with Ice-It Freezing Spray and sectioned in the coronal plane at 40μm on a Leica cryostat (Leica Microsystems Ltd, Buckinghamshire, UK). Sections were sampled in accordance to the stereological rules [45], with the first one retained randomly and every 12th section afterward that rendered 7–12 coronal sections throughout the neocortex and limbic system per animal for quantitative analysis. Sections were preserved in 30%ethylene glycol and 25%glycerol in 0.1 M phosphate buffer (pH 7.4) at –20°C until processed for immune-histochemical assessment of Aβ plaques and neuroinflammation. We evaluated neuroinflammation by monitoring the immune-reactivity of IBA-1 that is involved in membrane ruffling and phagocytosis of activated microglia [46, 47] and combined with the immune-reactivity of GFAP. The latter describes a hallmark intermediate filament protein of astrocytes with a profound role in of reactive astrogliosis and glial scar formation [48, 49]. We performed avidin-biotin-peroxidase complex (ABC) staining method for detecting and amplifying each target antigen signal.
Upon thawing, sections were washed three times in 1X TBS for 10 min each and subsequently incubated with pre-warmed 0.05 M trisodium citrate (pH 9.0) at 90°C for 30 min to enhance antigen recognition. When cooled down to room temperature, the sections were washed three times in 1X TBS for 10 min each and incubated with 0.6%hydrogen peroxide for 30 min with gentle agitation at room temperature to quench endogenous peroxidase activity. Following three washes in 1X TBS for 10 min each, the sections were treated with 0.3%Thermo Scientific Triton-X 100 for 10 min with gentle agitation at room temperature to promote cell permeabilization. Non-specific antibody binding prevented by blocking the sections in 5%normal goat serum in TBS for 30 min with gentle agitation at room temperature. The sections were then incubated with the primary antibody against GFAP, IBA-1, or Aβ overnight at 4°C. All primary antibodies were generated in rabbit and diluted in 2%normal goat serum in 1X TBS with 0.3%Thermo Scientific Triton-X 100 at 1:500, 1:2000, and 1:250 ratio, respectively. Following the primary antibody incubation, sections were washed three times in 1X TBS for 10 min each and incubated with a biotinylated goat secondary antibody to rabbit IgG (1:360) for 90 min at 4°C with gentle agitation. Three washes in 1X TBS followed to remove the unbound antibody and tissue incubated with a pre-formed ABC reagent (VECTASTAIN® Elite® ABC-HRP Kit) for 90 min at 4°C with gentle agitation. The ABC reagent was prepared as per manufacturer’s instructions 30 min before use that allowed the biotinylated horseradish peroxidase to irreversibly link to the multiple biotin-binding sites available in each tetravalent avidin molecule. Biotinylated peroxidase enzyme and avidin were mixed at the manufacturer’s recommended ratio that favored the formation of lattice enzyme-avidin complexes in the solution though preventing avidin saturation. The remaining biotin-binding sites on the avidin molecule could bind to the biotinylated antibody that had been already bound to the tissue. As a result, a greater concentration of peroxidase localized at the antigen site, increasing sensitivity [50]. Following the ABC incubation, sections were washed three times in 1X TBS for 10 min each and flooded with Vector® DAB (3,3-diaminobenzidine) peroxidase substrate working solution, freshly prepared as per manufacturer’s instructions. Color development ceased after 7-8 min by washing the sections three times in tap water for 10 min each. Sections were mounted on silanized slides and cover-slipped using VectaMount AQ aqueous mounting medium.
Nikon Eclipse E600 polarizing microscope equipped with Zeiss AxioCam ICc1 was used for image acquisition. Three micrographs at random points over the cortex along with 2-3 micrographs over the hippocampus were captured at 10X magnification per section per slide per animal group for quantification. Image J was used to quantify the percentage of stained area on each micrograph. The captured micrographs were converted in 8-bit black and white images and automatically segmented into stained and unstained surface areas by the “MultiThresholder” Image J plug-in, employing maximum entropy thresholding [51]. Image acquisition and processing were performed in a blinded fashion.
Brain tissue homogenization and protein extraction
Left hemispheres were homogenized by mechanical shearing with a Wheaton Dounce tissue grinder in pre-cooled 1X PBS supplemented with protease/phosphatase inhibitor cocktail (1X) on ice. For every 100 mg of tissue, we added 500μl of the homogenizing buffer to acquire a homogenate of 20%(w/v). Following a freeze/thaw cycle, the homogenate was mixed at 1:1 ratio with pre-cooled 1X PathScan® Sandwich ELISA cell lysis buffer supplemented with protease/phosphatase inhibitor cocktail (1X), sonicated for 5 min (30 s ON; 30 s OFF –5 cycles) in an ultrasonic water bath (Ultrawave, South Glamorgan, UK) and gently agitated in a roller shaker (Stuart, Staffordshire, UK) for an hour at 4°C to complete the cell lysis. Protein was extracted by centrifugation at 16000× g for 15 min at 4°C. The Quick start Bradford protein assay conducted to estimate the protein concentration of the samples, as previously described [19]. Protein of mouse-brain, whole-cell lysate (20μg) subsequently processed for western blotting in accord with the previously published protocol by Panagaki et al. [21] (please, also refer to the Supplementary Methods for the protocol description). The primary antibodies used in the present study are summarized in Supplementary Table 1.
Statistics
All the results were expressed as the mean± standard error (SEM) of 11–15 animals for the behavioral studies or 6–8 animals for the histological and biochemical studies per group (Table 1). Differences among the means were considered significant, if p≤0.05. Upon the occurrence of overall significant differences, post hoc analysis ensued. The assumption of data normality examined with the Kolmogorov-Smirnov test. The animal-weight data and Morris water-maze acquisition endpoints analyzed with repeated measures two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-tests at each time point. The data from the Morris water-maze probe trials and open-field sessions processed with one-way ANOVA and a posteriori Bonferroni’s multiple comparison t-tests. Histological data processed with the Kruskal-Wallis test and a posteriori Dunn’s multiple comparison tests or the Mann-Whitney U test, depending on the number of groups compared. Biochemical findings analyzed with one-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-tests. Statistical calculations were performed in GraphPad Prism 5 (GraphPad Software Inc., San Diego, USA) for Mac OS X software.
Data availability statement
The present manuscript and its supplementary material accommodate all the data supporting our herein reported findings.
RESULTS
The DA–CH3 dual incretin treatment has a neutral effect on the body weight neutrality of the APP SWE /PS1 ΔE9 mice
Incretin-based therapies have been previously shown to convey regulatory effects in satiety and appetite via the central nervous system, resulting in a reduced food intake and weight loss in patients and animal models of diabetes and obesity [23, 53]. In this light, we monitored the body weight of the animals over the 8-week treatment administration period. Two-way ANOVA analysis has demonstrated no significant effect both of the animal genotype [F (1,25) = 1.06, p = 0.312] and assigned treatment [F (1,21) = 0.13, p = 0.724], as illustrated in Fig. 1.
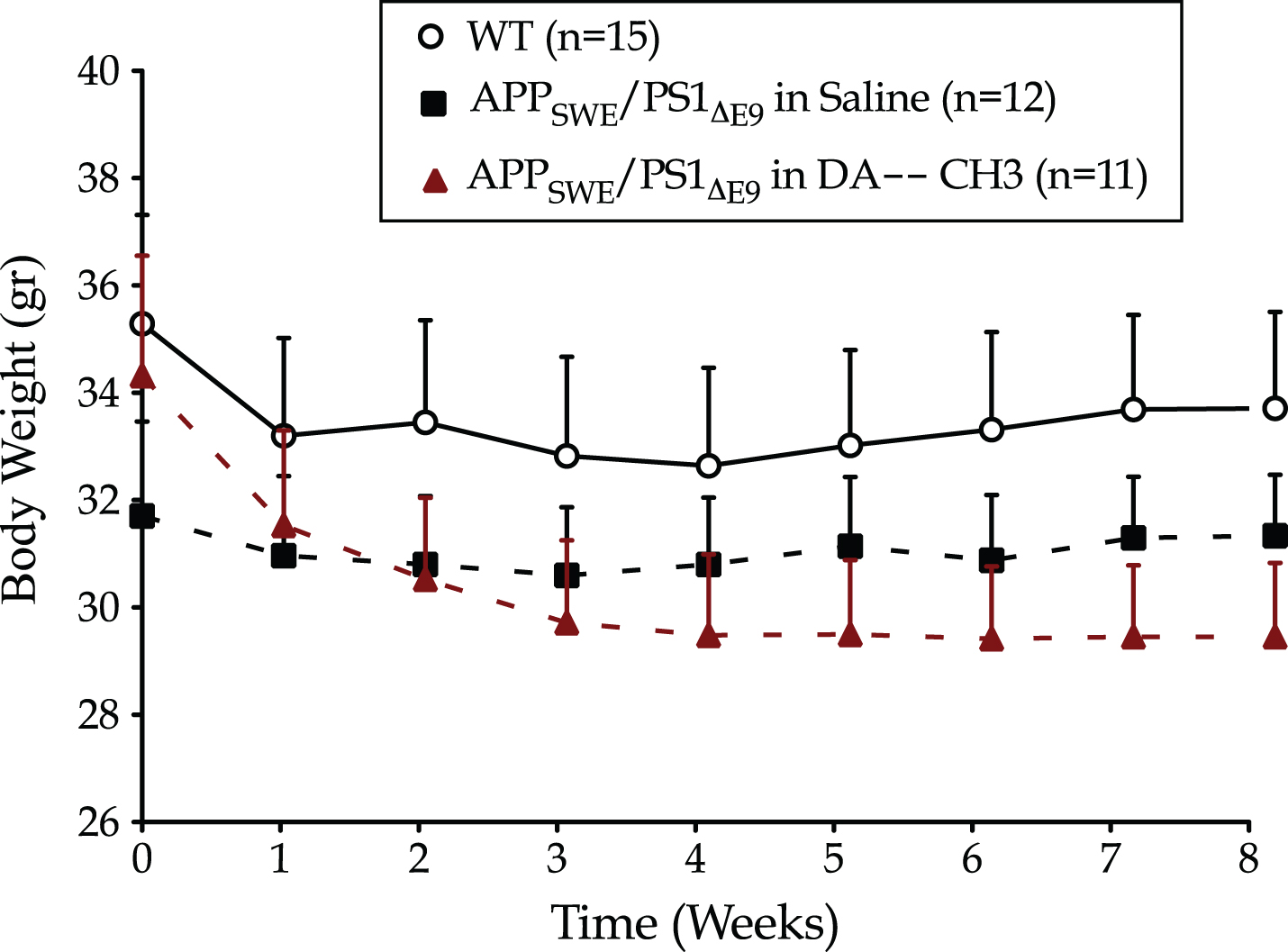
The novel DA–CH3 dual incretin does not impact the body weight of the APPSWE/PS1ΔE9 mouse model of Alzheimer’s disease. Each point represents the mean±SEM of wild type (WT) or APPSWE/PS1ΔE9 mice treated with 10 ml/kg/day of saline or 25 nmol/kg/day of DA–CH3 GLP-1/GIP Dual receptor Agonist. Data analyzed with repeated-measures two-way ANOVA.
The DA–CH3 dual incretin ameliorates the aberrant exploratory locomotion accompanying the APP SWE /PS1 ΔE9
Eight weeks after the initiation of saline and dual incretin treatments, animals were subjected to the open field test. In the experimental data presented herein (Fig. 2), APPSWE/PS1ΔE9 mice display an increased ambulatory activity in the arena with a total path of 5834±594.9 cm, as compared to the length of the path (4400±329.1 cm) traveled by their wild-type littermates during a 10-min session (post hoc; t = 2.497, p≤0.05). To further evaluate locomotion motivated by exploration, we monitored the vertical activity (rearing incidents) and the frequency of grooming activity of each animal over the open field session. One-way ANOVA analysis has revealed no significant differences in rearing incidents [F (2,35) = 1.240, p = 0.302] and grooming sessions [F (2,35) = 0.801, p = 0.457] among the animal groups.

The novel DA–CH3 dual incretin seems to ameliorate the aberrant exploratory locomotion of the APPSWE/PS1ΔE9 mouse model of Alzheimer’s disease. The open field test was conducted 8 weeks after the initiation of saline and DA-CH3 GLP-1/GIP Dual receptor Agonist treatments. A Python-based video tracking/analyzer system software interfaced with a monochrome digital camera was used to automatically detect, record and process mouse movements over a 10-min session, as shown in the representative tracks (a). Data analyzed with one-way ANOVA, as summarized in the table (b).
We next assessed the activity in the center of the open field arena as a measure of anxiogenic behavior in mice [54]. One-way ANOVA analysis overall demonstrates differences in the activity in the center of the open field arena among groups (Fig. 2b) that correspond to the significantly shorter path traveled by the APPSWE/PS1ΔE9 mice (post hoc; t = 2.479, p≤0.05). The DA–CH3 dual incretin treatment does not alter the emotionality of the animals. Post hoc analysis demonstrates no significant differences in the total path (t = 2.15, p > > 0.05) and percent ratio of the path length in the center (t = 1.79, p > > 0.05) between the wild type and dual incretin-treated APPSWE/PS1ΔE9 mice. Instead, it may ameliorate the impaired exploratory locomotion accompanying the APPSWE/PS1ΔE9 genotype though no significant differences noted in the aforementioned parameters between the saline- and DA–CH3-treated APPSWE/PS1ΔE9 mice.
The DA–CH3 dual incretin reverses the spatial learning and memory impairments of the APP SWE /PS1 ΔE9 mouse model
A significant effect of the animal genotype in the escape latency [F (1,25) = 17, p = 0.0004] and length of the swimming path [F (1,25) = 22.44, p < 0.0001] was present over the Morris water-maze acquisition phase. As illustrated in Fig. 3a and b, APPSWE/PS1ΔE9 mice have required longer sessions and swum longer paths to locate the escape platform, relative to wild-type counterparts. Post hoc analysis at each time point reveals that the APPSWE/PS1ΔE9 mice display prominent spatial acquisition impairments at the 2nd (escape latency: p≤0.05; swimming path: p≤0.01), 4th (p≤0.05), 9th (p≤0.001), and 10th (p≤0.01) days of the course. The spatial acquisition impairments reported herein do not stem from defects in locomotor coordination during swimming, visual acuity, and/or in motivation. APPSWE/PS1ΔE9 and wild-type mice feature similar swim patterns in the Morris water-maze visible-platform training (Supplementary Figure 2), with an insignificant effect of the genotype over time in the repeated measures two-way ANOVA analysis of the swim time [F (3,75) = 0.20, p = 0.90], path length to locate the cued escape platform [F (3,75) = 0.19, p = 0.90], swim velocity [F (3,75) = 1.83, p = 0.148], percentage of thigmotaxis [F (3,75) = 0.40, p = 0.755]. Similarly, the APPSWE/PS1ΔE9 and wild-type mice display comparable swim velocity and thigmotactic behavior over the 10-day course of the Morris water-maze spatial acquisition, as shown in Supplementary Figure 3.
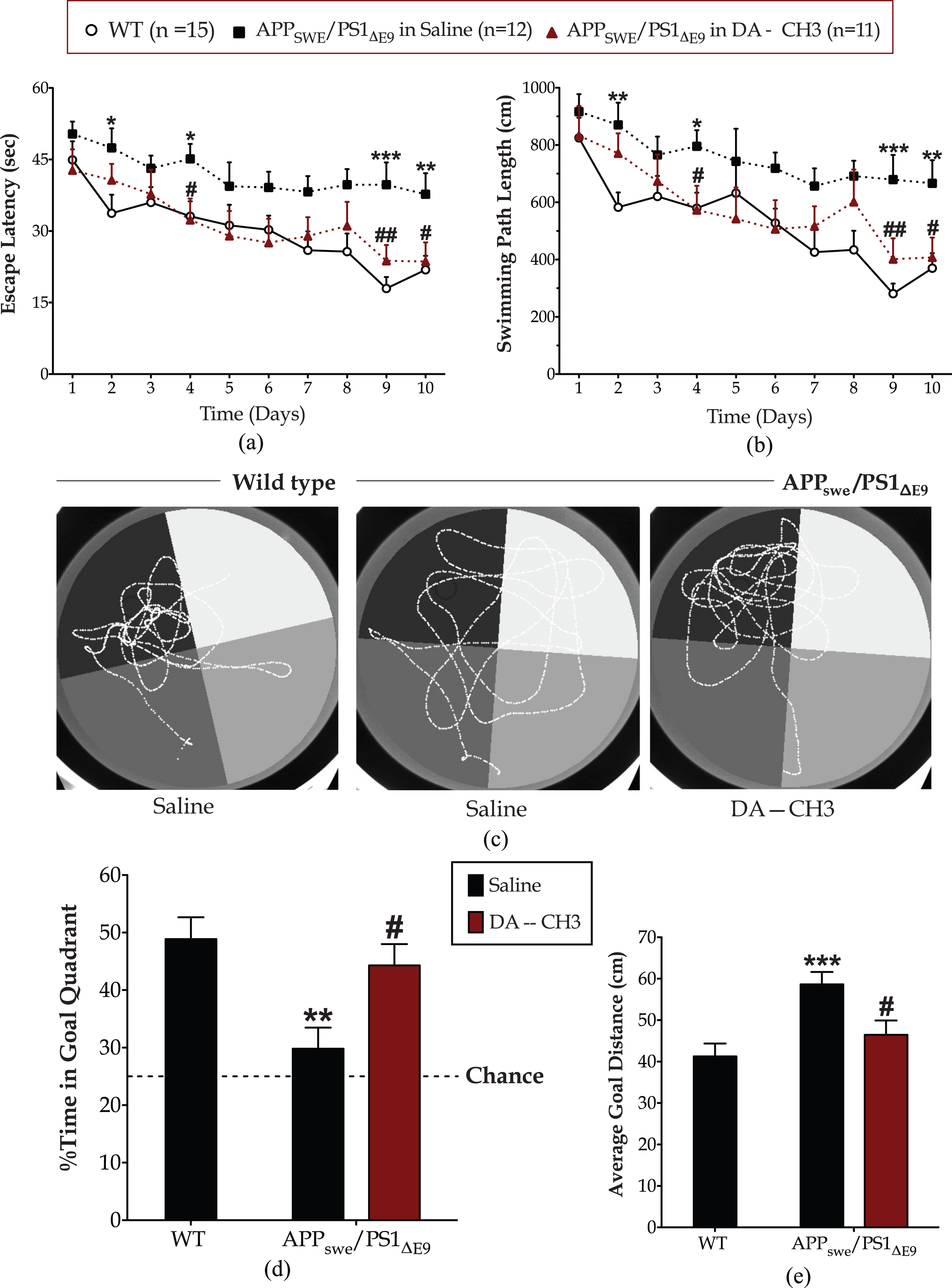
The novel DA–CH3 dual incretin restores the spatial acquisition and reference memory impairments of the APPSWE/PS1ΔE9 mouse model of Alzheimer’s disease. Nine weeks after the initiation of saline or DA-CH3 dual incretin treatments, animals were trained to locate an invisible escape platform fixed on a target position in the northwest quadrant of the Morris water-maze apparatus. Animals were given 3 trials per day with an inter-trial interval of approximately 30 min for 10 consecutive days. The escape latency (a) along with the daily mean of the length of the swimming path (b) were monitored to evaluate spatial learning. A retention 60-s probe trial conducted on the 11th day during which the swimming pattern of each mouse tracked in the absence of the escape platform, as illustrated in the representative tracks in panel (c). The percentage of the swim time spent in the goal quadrant that formerly contained the platform (d) along with the average distance of each mouse from the center of platform (e) considered indexes of memory retention. Acquisition data analyzed with repeated measures two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-tests at each time point. Probe trial data was analyzed by one-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-tests (*p≤0.05, **p≤0.01 & ***p≤0.001 compared to wild type (WT); # p≤0.05 & # # p≤0.01 compared to the saline-treated APPSWE/PS1ΔE9 group).
Spatial acquisition impairments have accordingly impacted the performance of APPSWE/PS1ΔE9 mice in the Morris water-maze probe trial (Fig. 3c). One-way ANOVA has demonstrated overall significant differences in the time spent in the quadrant that formerly contained the escape platform [goal quadrant; F (2,35) = 7.09, p = 0.003] and the average goal distance [F (2,35) = 8.04, p = 0.001] among the groups. Wild-type mice have devoted half of the allotted time in the goal quadrant, acquiring relative positions during the probe trial with an average distance of 41.26±3.092 cm from the center of the platform. Contrarily, the APPSWE/PS1ΔE9 mice have spent less than a third of the allotted time in the goal quadrant, having scored slightly above the chance level with an average distance from the center of the platform of 58.65±2.975 cm. As such, post hoc analysis demonstrates significant differences in the percentage of time spent in the goal quadrant (p≤0.01) (Fig. 3d) and average goal distance (p≤0.001) (Fig. 3d) between the two groups that signifies defective reference-memory consolidation and recollection of the APPSWE/PS1ΔE9 mice.
Intriguingly, the DA–CH3 dual incretin treatment reverses the spatial learning and reference memory defects of the APPSWE/PS1ΔE9 mouse model in Morris water maze, as illustrated in Fig. 3. During the spatial acquisition phase, the DA–CH3 dual incretin-treated APPSWE/PS1ΔE9 mice have required less time (Fig. 3a) and swum shorter paths (Fig. 3b) to locate the invisible escape platform as time progresses. Repeated measures two-way ANOVA analysis demonstrates a significant effect of the treatment in the escape latency [F (1,21) = 11.80, p = 0.0025] and the length of the swimming path [F (1,21) = 7.77, p = 0.011] during the task acquisition phase. Post hoc analysis reveals that the restorative effects of the GLP-1/GIP dual receptor agonist DA–CH3 in spatial learning are prominent on the 4th (p≤0.05), 9th (p≤0.01), and 10th (p≤0.05) days of the training course. The dual incretin-treated APPSWE/PS1ΔE9 mice have accordingly displayed significantly improved recall of the escape platform position (p≤0.05) in the probe trial (Fig. 3d). They have spent approximately half of the allotted time in the goal quadrant (Fig. 3c), with an average distance from the center of the platform of 46.46±3.465 cm (Fig. 3e), and therefore scored similar to the wild-type littermates in the probe-trial endpoints.
Furthermore, repeated measures two-way ANOVA analysis demonstrates significant effects of the genotype over time in the escape latency [F (4,100) = 4.51, p = 0.002] and length of the swimming path [F (4,100) = 3.83, p = 0.006] during the reversal spatial acquisition of the Morris water-maze task. As illustrated in Fig. 4a, the APPSWE/PS1ΔE9 mice have required longer sessions to locate the invisible escape platform, with their learning impairments becoming significantly prominent at the 3rd (p≤0.05), 4th (p≤0.05), and 5th (p≤0.01) days of the training course. Accordingly, the APPSWE/PS1ΔE9 mice acquired longer paths to the escape platform, particularly at the last day of the training course (post hoc; p≤0.01 versus wild-type) (Fig. 4b). In the retention probe trial, the APPSWE/PS1ΔE9 mice spent approximately 24%of the allotted time to the goal quadrant, performing at the chance level (Fig. 4c, d) with an average distance from the center of the platform of 63.7±3.25 cm (Fig. 4e). In contrast, the wild types devoted half of the allotted in the quadrant that formerly contained the escape platform (Fig. 4c, d) with an average goal distance of 47.6±2.129 cm (Fig. 4e). As such, post hoc analysis demonstrates significant differences in the percentage of time allotted in the goal quadrant (p≤0.001) and average goal distance (p≤0.001) between the two groups.
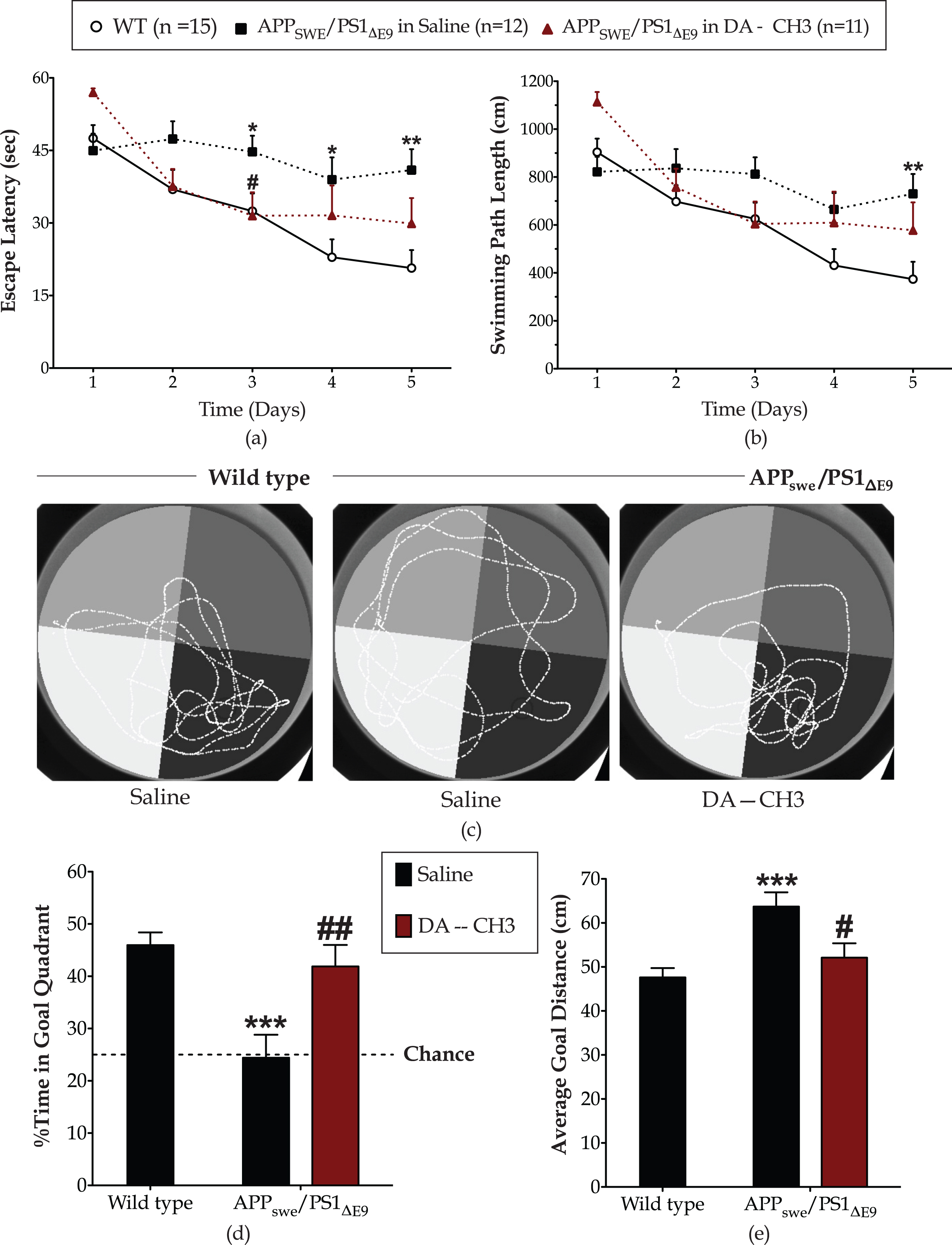
The novel DA–CH3 dual incretin promotes cognitive flexibility of the APPSWE/PS1ΔE9 mouse model of Alzheimer’s disease. Mice were next assigned to the spatial reversal phase of Morris water-maze task during which an invisible escape platform was fixed on a target position in the southeast quadrant. The escape latency (a) along with the daily mean of the length of the swimming path (b) were monitored to evaluate spatial learning. Twenty-four hours post the last day of the training, a reversal probe trial of 60 s conducted during which the swimming pattern of each mouse tracked in the absence of the escape platform, as illustrated in the representative tracks in panel (c). The percentage of the swim time spent in the goal quadrant that formerly contained the platform (d) along with the average distance of each mouse from the center of platform (e) considered indexes of memory retention. Each graph point and bar represents the mean of the corresponding parameter for the wild-type (WT) or APPSWE/PS1ΔE9 mice treated with 10 ml/kg/day of saline and APPSWE/PS1ΔE9 mice treated with 25 nmol/kg/day of the DA–CH3 GLP-1/GIP dual receptor agonist. Acquisition data analyzed with repeated measures two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-tests at each time point. Reversal probe trial endpoints processed with one-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-tests (*p≤0.05, **p≤0.01 & ***p≤0.001 compared to WT; # p≤0.05 & # # p≤0.01 compared to the saline-treated APPSWE/PS1ΔE9 group).
The dual incretin treatment promotes cognitive flexibility of the APPSWE/PS1ΔE9 mouse model, with a significant effect of the treatment over time in the escape latency [F (4,84) = 4.98, p = 0.0012] and the length of the swimming path [F (4,84) = 3.64, p = 0.009]. Although post hoc analysis indicates a prominent restorative effect of the treatment solely in the escape latency at 3rd (p≤0.05) day of the training course (Fig. 4a), the DA–CH3 dual incretin-treated APPSWE/PS1ΔE9 have successfully acquired a strategy to the new goal and prominently recalled of the escape platform position in the reversal probe trial (Fig. 4c). They have spent approximately half of the allotted time in the goal quadrant (Fig. 4d) with an average distance from the center of the platform of 52.11±3.27 cm (Fig. 4e). As such they have scored similarly to the wild-type littermates in the trial endpoints.
The novel DA–CH3 dual incretin ameliorates AD-related pathognomonic features in the APP SWE /PS1 ΔE9 mouse brain
We next immunolabelled coronal brain sections from the APPSWE/PS1ΔE9 mice for Aβ –the AD pathognomonic signature [55] –and evaluated the cerebral amyloid plaque load. As illustrated in Fig. 5a, the APPSWE/PS1ΔE9 brain features an excessive Aβ plaque deposition, which notably lessens after the unimolecular GLP-1 and GIP co-administration. Mann-Whitney U test demonstrates significant differences between the treatment groups, with the DA–CH3 dual incretin treatment nearly halving the pathologic plaque deposition in the APPSWE/PS1ΔE9 cortex [U = 728.0, p < 0.0001; two-tailed] and hippocampus [U = 260.0, p < 0.0001; two-tailed] (Fig. 5b).
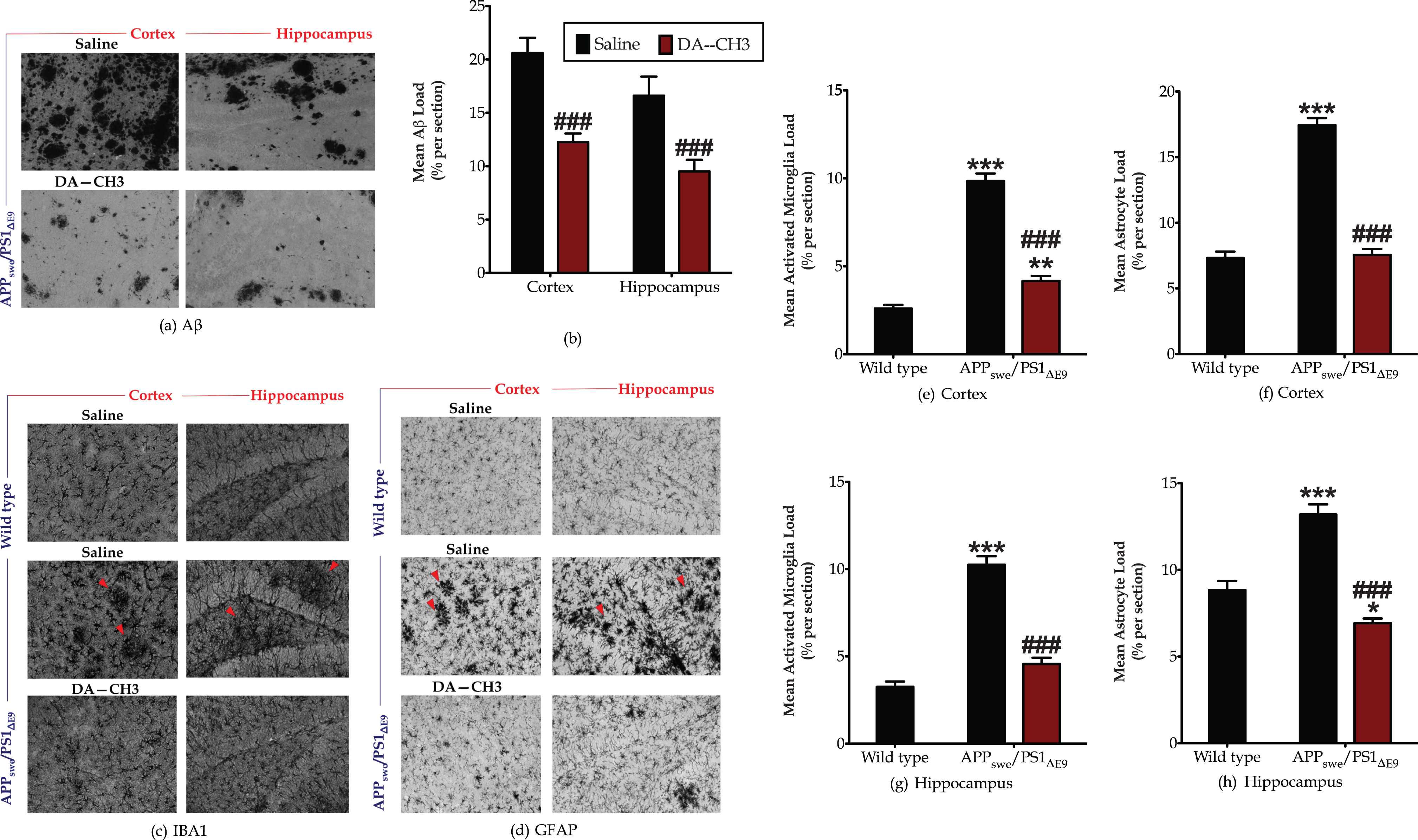
The novel DA–CH3 dual incretin treatment ameliorates the excessive β-amyloid deposition and aberrant neuroinflammation in the APPSWE/PS1ΔE9 cortex and hippocampus. Three micrographs over the cortex along with 2–3 micrographs over the hippocampus captured per section and converted in 8-bit black and white images, as shown in the representative panels (a), (c), and (d). Image J was used to segment each micrograph into stained and unstained surface areas and automatically measure the Aβ (b), microglia (e and g) and astrocyte (f and h) immune-positive load. Each bar represents the mean±SEM of the percentage of stained area per section from wild-type (n = 8) or APPSWE/PS1ΔE9 mice (n = 6) treated with 10ml/kg/day of saline and APPSWE/PS1ΔE9 mice treated with 25 nmol/kg/day of the DA–CH3 dual GLP-1/GIP receptor agonist (n = 6). Differences in Aβ plaque deposition between the saline- and DA-treated APPSWE/PS1ΔE9 mice determined with the Mann-Whitney two-tailed test. Rest histological data analyzed with the Kruskal-Wallis test, followed by post hoc Dunn’s multiple comparison tests (*p≤0.05, **p≤0.01 & ***p≤0.001 compared to wild type; # # # p≤0.001 compared to the saline-treated APPSWE/PS1ΔE9 group).
In addition to the Aβ pathology, microglia from the APPSWE/PS1ΔE9 cortex and hippocampus display an amoeboid morphology with decreased branching and enlarged soma that all signal for microglia activation [56, 57]. Quantitative analysis reveals a more than three-fold increase in the microglia load in the APPSWE/PS1ΔE9 cortex (Fig. 5e) and hippocampus (Fig. 5g), which significantly differs from the corresponding load in the retrospective brain areas from the wild-type mice (post hoc; p≤0.001). Accordingly, the astrocytes in the APPSWE/PS1ΔE9 cortex and hippocampus assume an immune-activated phenotype with the processes emerging from their soma tending to be thicker and present in bundles, when compared to the astrocytes from wild-type mice that feature a stellate morphology with emerging processes at regular intervals (Fig. 5d). Kruskal-Wallis test demonstrates significant differences in the cortical [H (3) = 79.99, p < 0.0001] and hippocampal [H (3) = 52.05, p < 0.0001] astrocyte load. The APPSWE/PS1ΔE9 genotype nearly doubles the percentage of GFAP stained area in the cortex and hippocampus (Fig. 5f, h), rendering it significantly different from the control in both examined brain areas (p≤0.001). The DA–CH3 dual incretin decreases the amoeboid reactive microglia load in the cortex and hippocampus of the APPSWE/PS1ΔE9 mouse model (Fig. 5c) that corresponds to an approximately 50%drop in the percentage of IBA-1 stained area [post hoc; p≤0.001] in the aforementioned brain areas (Fig. 5e, g). It additionally culminates in a restoration of the cortical (Fig. 5f) and hippocampal (Fig. 5h) loads of the astrocytes that mainly feature a normal stellate morphology (Fig. 5d).
The novel DA–CH3 dual incretin preserves synapse integrity in the APP SWE /PS1 ΔE9 mouse brain
Synapses are the earliest sites of pathology, with their loss being considered the best clinical correlate of dementia [55, 58]. Herein, we report overall significant differences in the cerebral expression levels of the presynaptic vesicle protein –synaptophysin [F (2,26) = 9.175, p = 0.001] and the scaffolding protein for the assembly and function of the post-synaptic density –PSD95 [F (2,17) = 6.78, p = 0.007], as per one-way ANOVA analysis. The APPSWE/PS1ΔE9 genotype provokes a 25%drop in the synaptophysin and PSD95 protein levels, as compared to wild-type littermates and illustrated in Fig. 6. The DA–CH3 dual-incretin analog restores the suppressed synaptophysin [post hoc; p≤0.001] and PSD95 [post hoc; p≤0.01] protein expressions back to control levels (Fig. 6) and thus preserves synapse integrity in the APPSWE/PS1ΔE9 mouse brain.
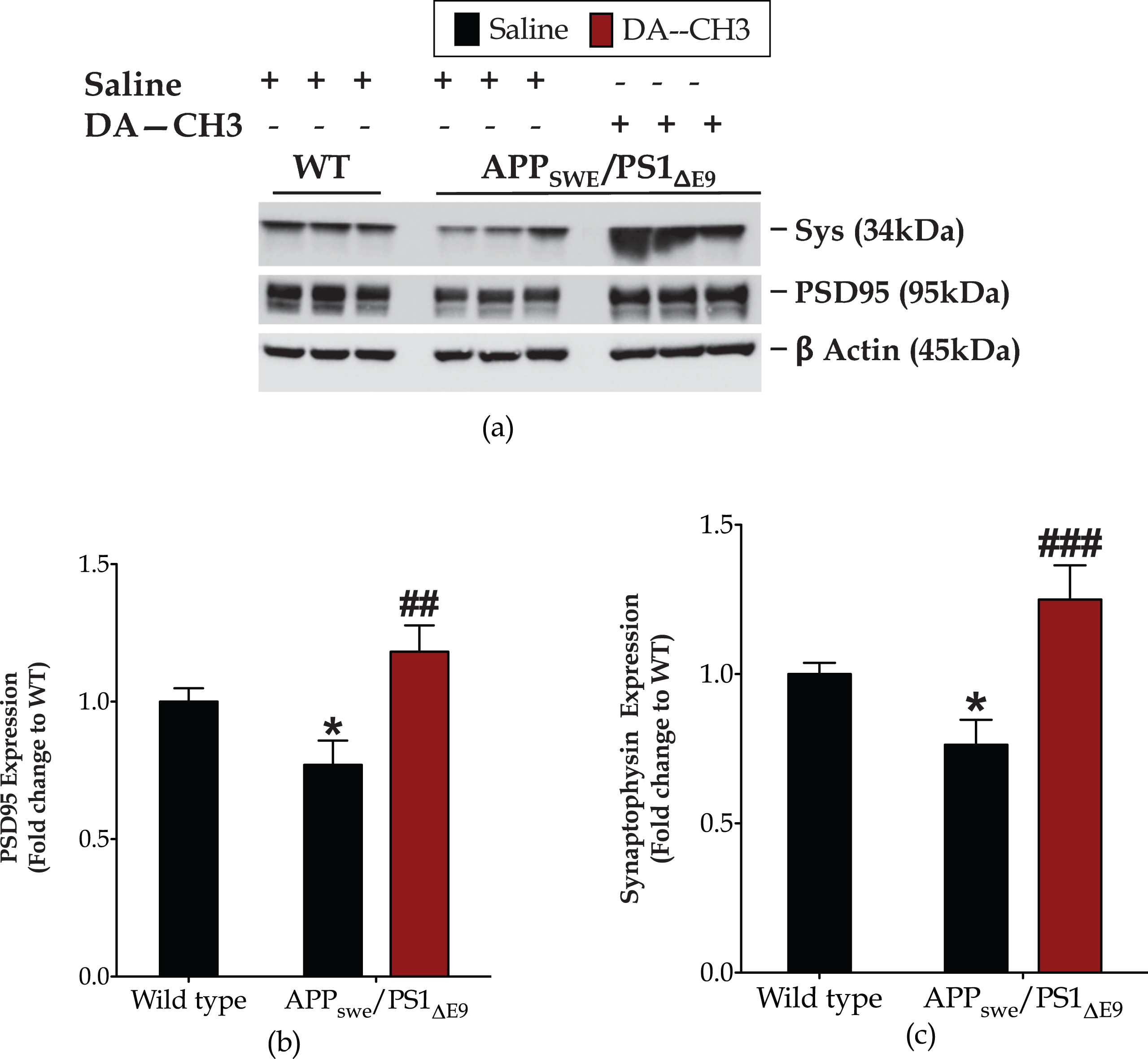
The novel DA–CH3 dual incretin promotes synapse homeostasis in the APPSWE/PS1ΔE9 mouse brain. Protein of mouse brain whole cell lysate (20μg) assayed for the expression of the 95-kDa post-synaptic density protein (PSD95, a and b) and synaptophysin (Sys, a and c) with western blotting. β-Actin used as loading control to all western blot analyses. Each bar represents the mean±SEM of the normalized-to-wild-type protein expression from wild type (WT; n = 8) or APPSWE/PS1ΔE9 mice (n = 6) treated with 10ml/kg/day of saline and APPSWE/PS1ΔE9 mice treated with 25 nmol/kg/day of the DA3–CH GLP-1/GIP dual receptor agonist (n = 6). Data analyzed with one-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-tests (*p≤0.05 compared to WT; # # p≤0.01 & # # # p≤0.001 compared to the saline-treated APPSWE/PS1ΔE9 group).
The novel DA–CH3 dual incretin alleviates endoplasmic reticulum (ER) stress and autophagy impairments in the APP SWE /PS1 ΔE9 mouse brain
One-way ANOVA analysis demonstrates overall significant differences in the molecular chaperone BiP [F (2,17) = 11.89, p = 0.0006], the transcription factor Chop [F (2,17) = 3.8, p = 0.0421] and the ER-resident CASP12 [F (2,17) = 6.56, p = 0.008]. The APPSWE/PS1ΔE9 mouse brain features a nearly one-fold increase in the protein levels of BiP (Fig. 7a) that indicates ER stress [59]. A significant upregulation in protein expression of the pro-apoptotic UPR mediators Chop (p≤0.05) (Fig. 7b) and CASP12 (p≤0.01) (Fig. 7b) is additionally prominent in the APPSWE/PS1ΔE9 mouse brain.
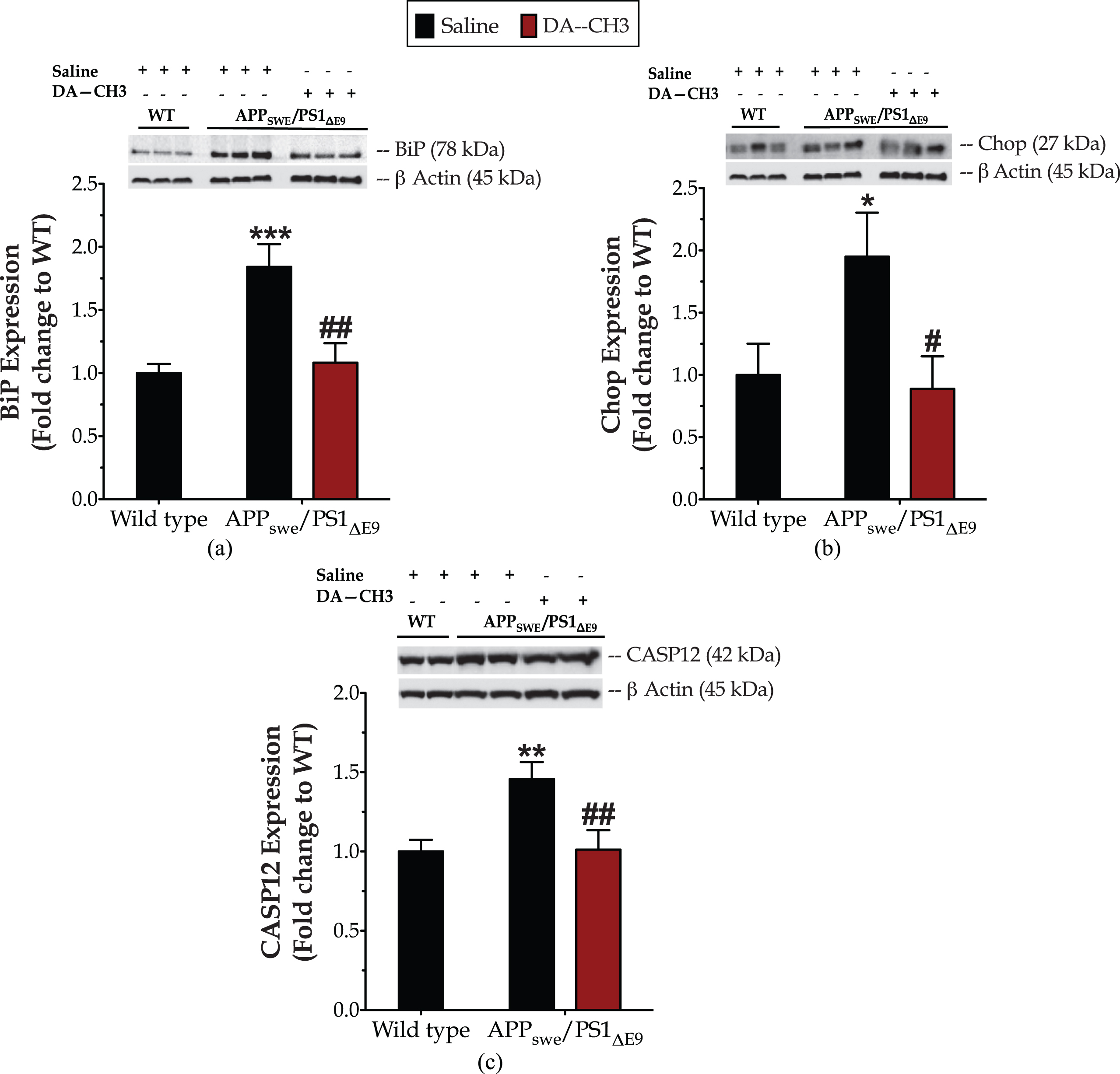
The novel DA–CH3 dual incretin resolves ER stress in the APPSWE/PS1ΔE9 mouse brain. Protein of mouse brain whole cell lysate (20μg) assayed for the expression of the ER-resident molecular chaperone BiP (Binding immunoglobulin protein, a), the transcription factor CCAAT-enhancer-binding protein homologous protein (Chop, b) and the ER-resident caspase 12 (CASP12, c) with western blotting. β-Actin used as loading control to all western blot analyses. Each bar represents the mean±SEM of the normalized-to-wild-type protein expression from wild type (WT; n = 8) or APPSWE/PS1ΔE9 mice (n = 6) treated with 10ml/kg/day of saline and APPSWE/PS1ΔE9 mice treated with 25 nmol/kg/day of the DA–CH3 GLP-1/GIP dual receptor agonist (n = 6). Data analyzed with one-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-tests (*p≤0.05, **p≤0.01 & ***p≤0.001 compared to WT; # p≤0.05 & # # p≤0.01 compared to the saline-treated APPSWE/PS1ΔE9 group).
The presence of the ER stress in the APPSWE/PS1ΔE9 mouse brain correlates to an approximate 25%drop in the expression of the autophagy-related proteins beclin-1, Atg3, Atg7, and LC3 when compared to the controls (Fig. 8, Supplementary Table 2). Intriguingly, the unimolecular GLP-1 and GIP co-administration normalizes the aberrant BiP, Chop and CASP12 expression levels (Fig. 7) that signals for resolution of the ER stress response in the APPSWE/PS1ΔE9 mouse brain. It additionally rescues the suppressed expression of beclin-1 (p≤0.05) (Fig. 8a), Atg3 (p≤0.001) (Fig. 8b), Atg7 (p≤0.01) (Fig. 8c), and LC3 (p≤0.01) (Fig. 8d) proteins. The latter signifies the restoration of the machinery that regulates the catabolic elimination and recycling of cytoplasmic components, preserves structures and functioning of sub-cellular organelles (e.g., mitochondria and ER), and determines cell adaptation in response to stress [60, 61].
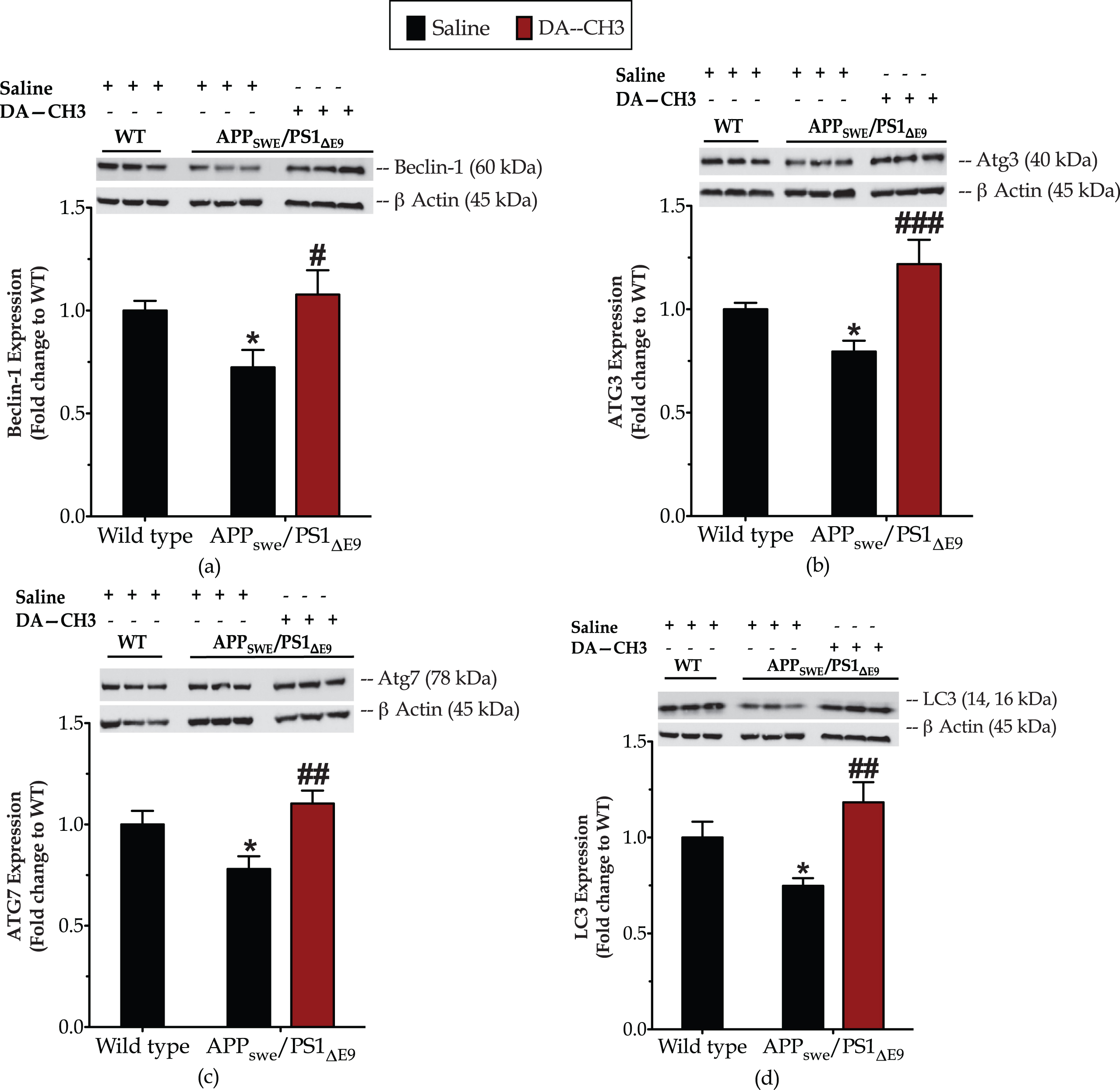
The novel DA–CH3 dual incretin restores the suppressed expression of ‘core’ autophagy-related (Atg) proteins in the APPSWE/PS1ΔE9 mouse brain. Protein of mouse brain whole cell lysate (20μg) assayed for the expression of beclin (a), Atg3 (b), Atg7 (c), and LC3 (d) with western blotting. β-Actin used as loading control to all western blot analyses. Each bar represents the mean±SEM of the normalized-to-wild-type protein expression from wild type (WT; n = 8) or APPSWE/PS1ΔE9 mice (n = 6) treated with 10ml/kg/day of saline and APPSWE/PS1ΔE9 mice treated with 25 nmol/kg/day of the DA–CH3 GLP-1/GIP dual receptor agonist (DA; n = 6). Data analyzed with one-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-tests (*p≤0.05 compared to WT; # p≤0.05, # # p≤0.01 & # # # p≤0.001 compared to the saline-treated APPSWE/PS1ΔE9 group).
The novel DA–CH3 dual incretin restores the derailed Akt signaling in the APP SWE /PS1 ΔE9 mouse brain
One-way ANOVA analysis demonstrates overall significant differences in Akt phosphorylation at Thr308 [F (2,26) = 9.14, p = 0.001] (Fig. 9a) but not in ERK1/2 phosphorylation at Thr202 and Tyr204 residues [F (2,24) = 0.88, p = 0.43] (Fig. 9c). Post hoc analysis points out a significant down-regulation of the Akt activation (p≤0.001) in the APPSWE/PS1ΔE9 mouse brain that further correlates to a 30%drop in the inhibitory phosphorylation of GSK3β at the Ser9 residue (Fig. 9b). The DA–CH3 dual-incretin analog normalizes the derailed cerebral Akt and GSK3β activity, as illustrated in Fig. 9.
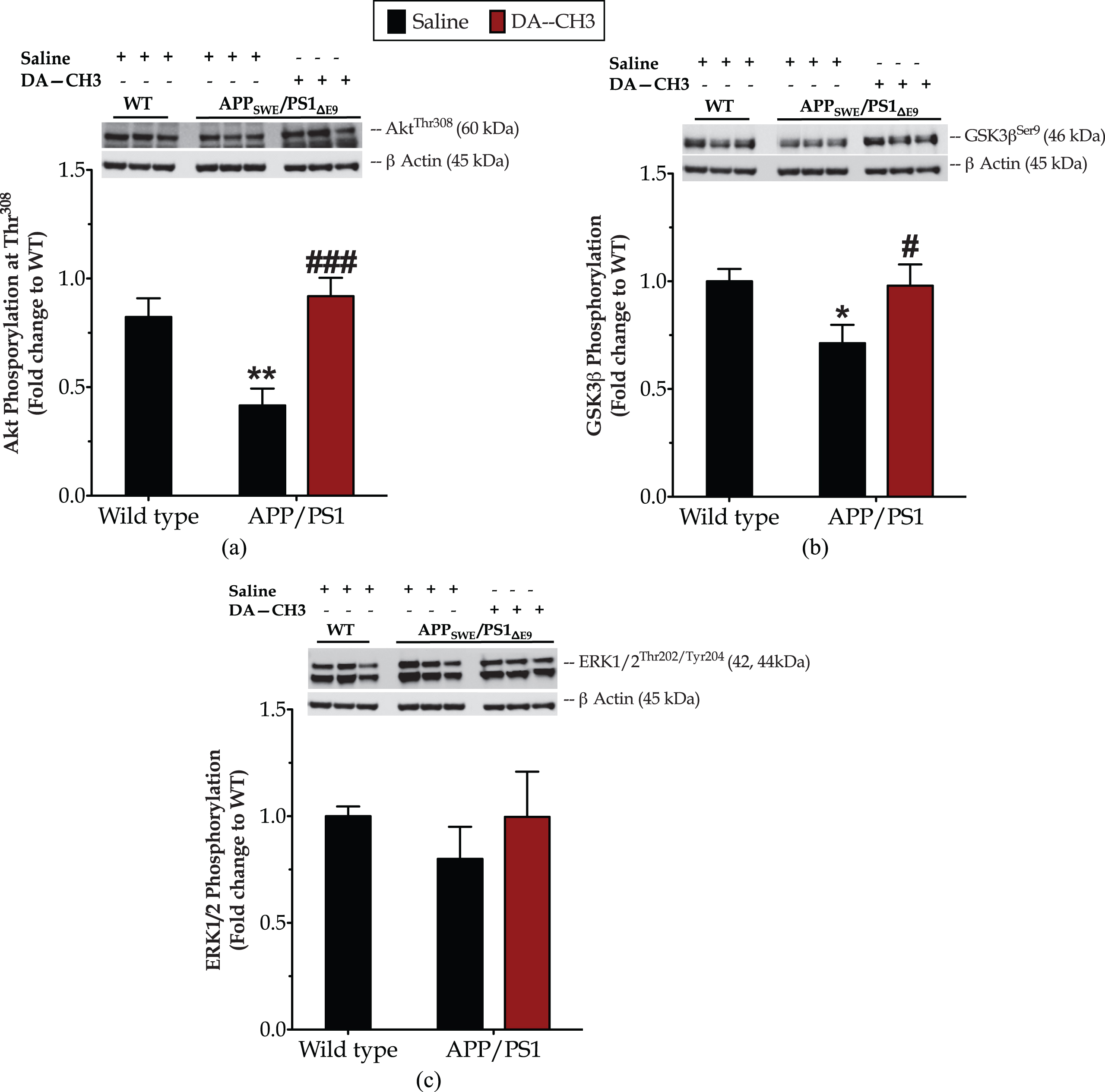
The novel dual incretin rectifies the impaired Akt signaling in the APPSWE/PS1ΔE9 mouse brain. Protein of mouse brain whole cell lysate (20μg) assayed for the expression of the phosphorylated Akt at the threonine 308 residue (Thr308) (a), extracellular signal-regulated kinase 1 & 2 (ERK1/2) at the threonine 202 and tyrosine 204 residues (Thr202/Tyr 204) (c), and glycogen-synthase kinase 3β (GSK3β) at the serine 9 residue (Ser9) (b) with western blotting. β-Actin used as loading control to all western blot analyses. Each bar represents the mean±SEM of the normalized-to-wild-type protein expression from wild type (WT; n = 8) or APPSWE/PS1ΔE9 mice (n = 6) treated with 10ml/kg/day of saline and APPSWE/PS1ΔE9 mice treated with 25 nmol/kg/day of the GLP-1/GIP Dual receptor Agonist (DA; n = 6). Data analyzed with one-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-tests (*p≤0.05 & **p≤0.01 compared to WT; # p≤0.05 & # # # p≤0.001 compared to the saline-treated APPSWE/PS1ΔE9 group).
DISCUSSION
Herein, we demonstrate the neuroprotective and restorative effects of the novel DA–CH3 dual-incretin analog in the APPSWE/PS1ΔE9 mouse model of AD along with the potential underlying mechanism for the first time. Ten-month-old APPSWE/PS1ΔE9 mice assigned to the DA–CH3 or saline treatments for 8 weeks before the initiation of the experiments. The co-stimulation of the GLP-1 and GIP receptors resolves the ER stress and autophagy impairments to attenuate cognitive decline and Alzheimer-like pathology in the middle-aged APPSWE/PS1ΔE9 mice.
The APPSWE/PS1ΔE9 mouse model over-expresses the Swedish mutation of the amyloid precursor protein (APP) together with the presenilin-1 (PS1) exon 9 deletion that both drive the generation of the highly fibrillogenic Aβ42 over Aβ40. The latter progressively culminates into the parenchymal amyloid deposition [62, 63] surrounded by glial cytopathology in brain areas serving memory and cognition [63–65], as evident in our histological assessment, as well. As such, these mice manifest spatial acquisition and reference memory impairments in the Morris water-maze task at the earliest test age of 7 months, which persist over the rest of the life of this murine model [66, 67]. Consistently, we have observed that the 12-month-old APPSWE/PS1ΔE9 mice display decrements in their Morris water-maze performance, with longer latency and path lengths to the escape platform over the spatial acquisition phase, as well as with shorter percentages of time in the goal quadrant and greater average distance from the center of the platform over the retention probe trial too. These deficits reflect a general cognitive decline of these mice, rather than defects in sensorimotor coordination and/or increased emotionality. Indeed, the APPSWE/PS1ΔE9 mice have acquired the visual platform training similarly to their control littermates while featuring normal swim velocity and thigmotactic behavior over the Morris water-maze spatial acquisition phases. This general cognitive decline seems to further restrain animals’ executive function, as seen in AD patients [67]. Herein, we show that the middle-aged APPSWE/PS1ΔE9 mice have failed to develop a new spatial strategy to locate the escape platform over the Morris water-maze reversal spatial acquisition phase that signifies cognitive inflexibility and thus defects in higher cognitive processes.
Intriguingly, the chronic DA–CH3 dual-incretin treatment restores learning and memory impairments of the APPSWE/PS1ΔE9 mouse model during the Morris water-maze spatial acquisition phase. It additionally promotes cognitive flexibility in the spatial reversal phase of the Morris water-maze task. The GLP-1/GIP dual receptor agonist-treated APPSWE/PS1ΔE9 mice have been able to extinguish from their initial training, developed a new spatial strategy to locate the escape platform—evident by the gradual decrease in the total duration of sessions and the swimming path length over the training period—and scored similarly to their wild-type littermates at the reversal probe-trial endpoints.
Histologically, the co-stimulation of GLP-1 and GIP receptors results in a prominent decrease of Aβ and reactive microglia loads in the cortex and hippocampus of the APPSWE/PS1ΔE9 mouse model. These histological results further demonstrate a restoration of the excessive astrogliosis in the aforementioned brain areas that serve learning and memory, and to the normalization of synaptophysin and PSD95 protein expressions. It has been repeatedly shown that Aβ accumulation in the brain of mice and non-human primates potentiates the aberrant reactivity of microglia and astrocytes [63–65, 69] that, in turn, stimulates pro-inflammatory signaling [56, 70]. The latter, when persists, hampers Aβ clearance, exacerbates Aβ and tau pathologies, and compromise synapse homeostasis, which ultimately altogether prompt cognitive defects [56, 72]. Therefore, our findings collectively suggest that the DA–CH3 dual-incretin analog precludes the excessive cerebral amyloid disposition and neuroinflammatory processes to rescue cognitive decline of this murine model. In accord with our observations, Cao et al. have recently reported that the DA–CH5 dual-incretin analog decreases hippocampal senile plaques and aberrant tau phosphorylation while normalizing late-phase long-term potentiation within hippocampal synapses to restore working-memory and long-term spatial memory decrements of the middle-aged APPSWE/PS1ΔE9 mice [31]. Similarly, previous studies have demonstrated that novel dual agonists of the GLP-1 and GIP receptors alleviate the increased reactivity of glial cells and disorder-related pathognomonic features to restore cognitive or behavioral impairments of a mouse model of mild traumatic brain injury [24], a rat model of streptozotocin-induced insulin desensitization and neurodegeneration in the brain [25] and neurotoxin-based rodent models of Parkinson’s disease [26–29].
Intracellularly, the DA–CH3 dual incretin modulates effectors and components for proteostatic responses to attenuate the AD-like pathology of the APPSWE/PS1ΔE9 mouse model. The UPR activation has been repeatedly detected in the postmortem brain samples from affected patients [73–78] and animal models of AD [79–83]. Increased BiP expression and phosphorylation of the UPR effectors (IRE1α, PERK, and eIF2α) occur in morphologically intact neurons at the early disease stages [73–75], preceding the overt amyloid deposition in AD brain [73, 84]. Meanwhile, the upregulation of pro-apoptotic UPR components, including the Chop expression and CASP12 proteolysis occurs at the late disease stages (Braak stage VI), signifying the occurrence of persistent ER stress in the AD brain [76, 78]. Similarly, the APPSWE/PS1ΔE9 mouse brain features elevated BiP and Chop expressions along with the enhanced activity of the ER-resident CASP12 [80], as prominently seen in our findings, too. CASP12 provokes a downstream caspase cascade to initiate programmed cell death (apoptosis) [85, 86] that may play a physiological role in eliminating cells unresponsive to the UPR [87]. However, in the AD, the apoptosis initiation is more likely to drive neuronal degeneration, in which the synapse is the initiation point [87–89]. Indeed, Aβ potentiates an ER stress-dependent synaptosomal mitochondrial dysfunction and pre-synaptic vesicle depletion. These are accompanied by a decrease in the protein expression of synaptophysin and cytoskeletal components and mediated by local caspase activity [90]. Subsequent findings have revealed that Aβ requires CASP12 to induce the pre-synaptic toxicity in the triple transgenic mouse model for AD (3×Tg-AD) [91].
Furthermore, Chop downstream inhibits the expression of the anti-apoptotic Bcl-2 protein and/or engages the p53 upregulated modulator of apoptosis (PUMA) protein to signal to the mitochondrial apoptotic machinery and thus promote neuronal and astrocytic damage [92, 93]. Herein, we show a 30%increase in the protein expression of the mitochondrial voltage-dependent anion channel (VDAC) (Supplementary Figure 5) that interacts with Bcl-2 family members to regulate mitochondrial permeability and the subsequent release of pro-apoptotic factors [94]. Although the aforementioned enhanced expression has not reached the statistical significance level [APPSWE/PS1ΔE9 versus WT, p = 0.056], it may indicate an early mitochondrial dysfunction [95] in the middle-aged APPSWE/PS1ΔE9 mouse brain. In addition to the core apoptosis machinery, Chop can limit autophagy through the transcriptional regulation of autophagy-related components for phagophore elongation and maturation into the autophagosome, including the Atg7 gene [96, 97]. Atg7 deficiency potentiates a spontaneous accumulation of protein aggregates, neuronal degeneration, and loss in mice [98].
Notably, the GLP-1/GIP dual receptor agonist DA–CH3 restores the excessive BiP and Chop expressions and aberrant CASP12 activity back to control levels that signal for resolution of UPR in the APPSWE/PS1ΔE9 mouse brain. The DA–CH3 dual incretin further normalizes the suppressed expression of the ‘core’ Atg proteins beclin-1, Atg3, Atg7, and LC3 that pivotally regulate the formation and maturation of the autophagosome [60]. In postmortem brain samples from affected patients and animal models of AD, autophagic vacuoles accumulate in swollen dystrophic neurites that may reflect defective maturation and retrograde transport of the autophagosome [99, 100]. A substantial suppression of beclin-1 protein levels occurs in the midfrontal cortex of the human AD brains that parallels the disease progression [101]. Mechanistically, beclin-1 determines the formation of the phagophore (that initiates autophagic process) [60] and regulates APP processing and turnover [102]. As such, the genetic reduction of beclin-1 compromises neuronal autophagy and exacerbates the intra-neuronal Aβ accumulation and extracellular Aβ deposition in the brain of the transgenic APPsweInd mouse model (J20) for AD [101]. Beclin-1 deficiency additionally suppresses the expression of synaptophysin, the dendrite-specific microtubule-associated protein 2 (MAP2) and the calcium-binding protein, calbindin to provoke the degeneration of neuronal synapses [101] while eliciting phagocytic deficits in microglia to impair Aβ clearance in vivo. Taken together, our findings suggest that the co-stimulation of GLP-1 and GIP receptors promotes the homeostasis of UPR and autophagy machinery—of which sustained deregulation favors a vicious circle of defective proteostasis and persistent ER stress in the brain—and thereby rescues neuronal and glial dysfunction in the APPSWE/PS1ΔE9 mouse model.
Mechanistically, the DA–CH3 dual-incretin treatment rectifies the suppressed Akt signaling to resolve ER stress and defective proteostasis in the APPSWE/PS1ΔE9 mouse brain. Akt (also known as protein kinase B –PKB) is a serine/threonine kinase member of the AGC protein kinase family with a profound function in growth-factor signaling for neuronal survival [104] and synaptic delivery of PSD95 [105]. It is also a central effector of the neuroprotective signaling of the incretin mimetics [19–21, 31]. In response to the PI3K stimulation, Akt is recruited to the cell membrane where it can undergo phosphorylation at the Thr308 site that critically determines the activation of the kinase [104]. Activated Akt can downstream phosphorylate and inhibit GSK3β kinase [104] that pivotally drives neurodegenerative processes in AD [106] and neuronal apoptosis following ER stress [21, 107–110]. Accumulating evidence from diverse neuronal cell lines, primary neuronal cultures, and ER insults have demonstrated that the UPR abrogates the inhibitory phosphorylation of GSK3β at Ser9 [21, 107–110] to favor CHOP expression and the dominance of pro-death signals during unmitigated ER stress [21, 108]. In the AD brain, GSK3β immunoreactive granules occur in neurons with activated PERK signaling [75, 111]. The latter exploits the autosomal-lysosomal pathway to selectively remove the inactive (phosphorylated at Ser9) kinase form and thus engage the GSK3β activity [111]. Exacerbated GSK3β activity in turn impairs the Morris water-maze performance and provokes excessive astrogliosis and aberrant microglia reactivity in vivo [106, 112]. Restoring normal levels of GSK3β activity reverses spatial learning and reference memory deficits and precludes reactive gliosis [106, 112], as prominent herein too.
Akt may also phosphorylate and inhibit PERK kinase [113], providing an additional mechanistic link on how DA–CH3 confers its restorative effects on the APPSWE/PS1ΔE9 mouse model. Deregulated PERK signaling parallels the temporal and spatial pattern of pathologic protein accumulation and aggregation in AD brain. There, it triggers general translation repression and downregulation of the plasticity-related cyclic AMP response-element-binding protein (CREB), through its downstream targets eIF2α and ATF4, respectively, to provoke PSD95 deficiency, synapse damage, and cognitive decline [88]. PERK over-activation can further elevate the expression of the β-site amyloid precursor protein cleaving enzyme 1 (BACE-1) to accelerate amyloidogenesis [88, 115]. Although we have not assessed the expression of phosphorylated PERK levels, transcriptional and translational regulation of Chop expression primarily lies downstream of the PERK arm [116].
In conclusion, our study demonstrates the neuroprotective and restorative effects of the novel DA–CH3 dual-incretin receptor agonist in the APPSWE/PS1ΔE9 mouse model of AD. It further unravels that the co-stimulation of GLP-1 and GIP receptors modulates the ER stress response and autophagy machinery to elicit homeostasis of the synapse and glial cells and reverse the AD-related pathology and cognitive decline in vivo. Importantly, the unimolecular co-agonism of GLP-1 and GIP receptors does not correlate with body-weight alterations. That is in close accordance with previous reports that the systemic and sustained administration of incretin-based treatments does not impact body-weight regulation, food intake, and glycemic levels in non-obese and non-diabetic animals [15, 117]. Our findings overall endorse the pleiotropic effects of the incretin signaling for neuronal survival and functioning, with a possible clinical relevance for the design of AD-modifying therapeutic strategies [10].