
Abstract
PRESENILIN 1 (PSEN1) and PRESENILIN 2 (PSEN2) genes are loci for mutations causing familial Alzheimer’s disease (fAD). However, the function of these genes and how they contribute to fAD pathogenesis has not been fully determined. This review provides a summary of the overlapping and independent functions of the PRESENILINS with a focus on the lesser studied PSEN2. As a core component of the γ-secretase complex, the PSEN2 protein is involved in many γ-secretase-related physiological activities, including innate immunity, Notch signaling, autophagy, and mitochondrial function. These physiological activities have all been associated with AD progression, indicating that PSEN2 plays a particular role in AD pathogenesis.
INTRODUCTION
PRESENILINs are essential components of the γ-secretase complex and both the PRESENILIN 1 (PSEN1) and PRESENILIN 2 (PSEN2) genes are loci for mutations causing familial Alzheimer’s disease (fAD) [1]. Interestingly, there have been nearly 200 potentially pathogenic mutations identified in PSEN1, but only 16 pathogenic mutations (Fig. 1 and Table 1) have been reported in PSEN2 to date [2, 3]. It appears that, although these two PRESENILIN proteins are highly homologous (they share 62% identity at the amino acid residue sequence level [4]), they can contribute to different biological processes [5, 6]. However, the complete function of the two PRESENILIN genes and how they contribute to fAD pathogenesis has not yet been determined. Due to the nearly 200 pathogenic mutations in PSEN1 this gene has been extensively covered in the literature, while PSEN2 has not received as much attention. This review provides a summary of the overlapping and independent functions of the PRESENILINs, with a focus on the physiological activities of PSEN2.
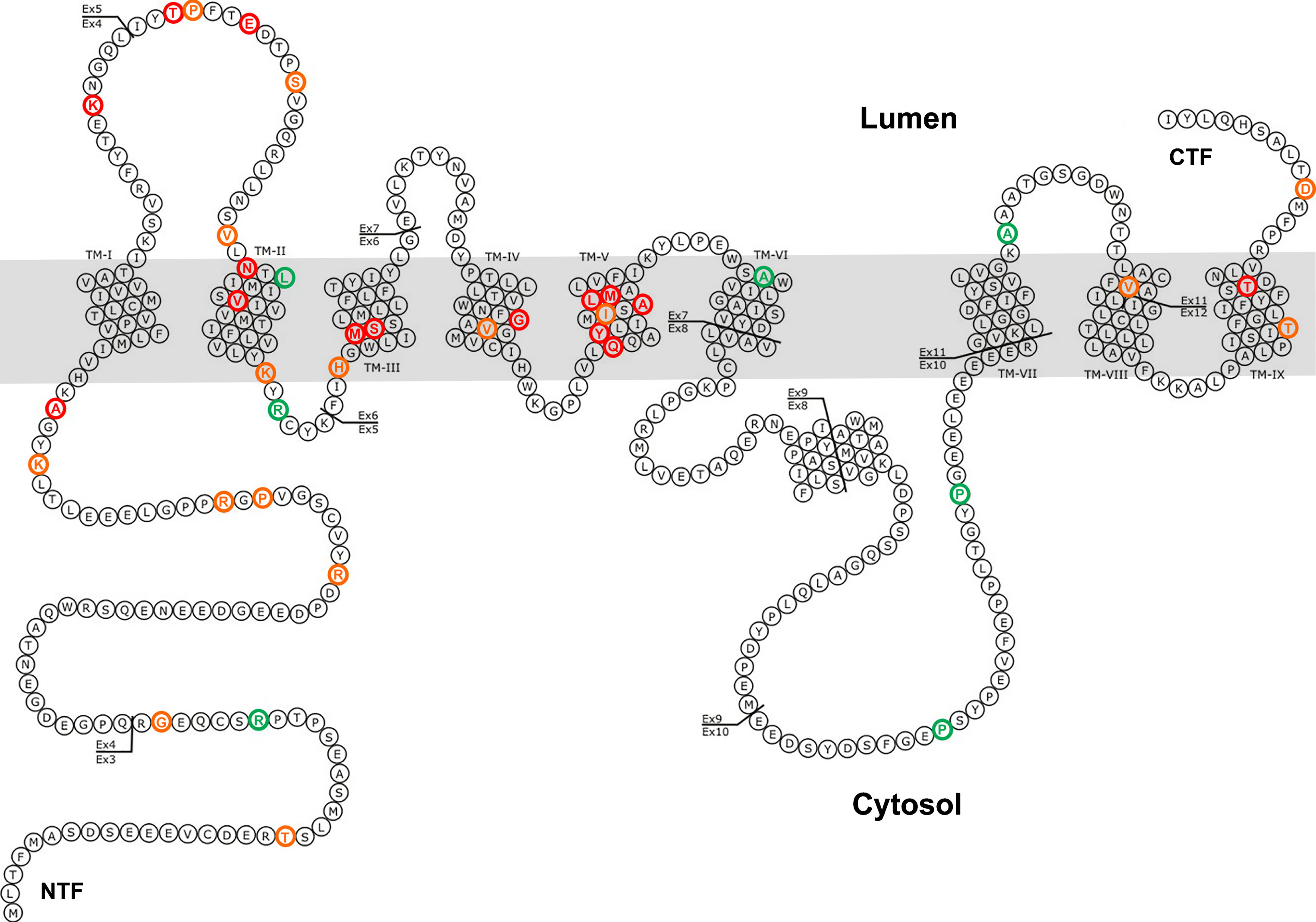
Mutations in PSEN2. Red: mutation is pathogenic; Orange: pathogenic nature is unclear or possible pathogenic; Green: mutation is not pathogenic. Adapted from http://www.molgen.vib-ua.be/ADMutations to include all mutations from Table 1 [3].
fAD mutations in PSEN2
Data adapted from PSEN2 mutations recorded in the Alzheimer Disease & Frontotemporal Dementia Mutation Database (AD/FTD database); http://www.molgen.ua.ac.be/ADMutations/default.cfm [3] except those indicated by “*”. Mutations are based on PSEN2 transcript NM_000447.2. ExAC Browser; http://exac.broadinstitute.org/ and ClinVar; https://www.ncbi.nlm.nih.gov/clinvar databases were searched to identify allele frequency information (if available) for each mutation/variant. Clinical significance information was inferred from the AD/FTD database (database terms used: pathogenic, pathogenic nature unclear and not pathogenic) and ExAc Browser (database terms used: pathogenic, possible pathogenic and likely pathogenic), see references for further detailed clinical information for each mutation. AD, Alzheimer’s disease, N/A (and “?”), not available.
FAMILIAL ALZHEIMER’S DISEASE MUTATIONS IN PSEN2
PSEN2 was first identified as STM2, a candidate gene for the chromosome 1 AD locus, when a point mutation resulting in the substitution of an isoleucine for an asparagine (N141I) was found in a Volga German AD family in 1995 [7]. The fAD mutations in PSEN2 are notable for a number of reasons. Unlike fAD mutations in PSEN1, PSEN2 variants demonstrate incomplete penetrance [2, 9] with the mean [10] age at symptom onset of the fAD mutations in PSEN2 as a group being more than 10 years later than the mean for PSEN1 fAD mutations [10]. (The one exception is the PSEN2 N141I variant that shows nearly complete penetrance with few “escapees” beyond age 80). Variable expressivity and clinical overlap with late onset “sporadic” AD suggest it is possible that PSEN2 variants may be more common in the general AD population than previous early onset family studies have indicated.
All but one of the known fAD mutations in PSEN2 are similar to those in PSEN1 in that they follow the “fAD mutation reading frame preservation rule” [11]. The mutations permit production of transcripts encoding proteins that include C-terminal sequence. The apparent exception is the K115fs mutation that results from a two-nucleotide deletion near the downstream end of exon 4 [2]. This mutation is interesting in that it appears to mimic expression of a naturally truncated isoform of PSEN2 denoted PS2V ([12, 13], see later). However, in the presence of the two-nucleotide deletion of K115fs, the alternative splicing event that generates PS2V (by exclusion of exon 5 sequence) actually reinstates the original reading frame of the gene so that such transcripts may encode a mutant protein that includes the normal C-terminal sequence. Therefore, the K115fs mutation may be the exception that proves the “fAD mutation reading frame preservation rule”.
PSEN2 PROTEIN AND THE γ-SECRETASE COMPLEX
Presenilins are the catalytic core of the γ-secretase complex that functions to cleave a number of type I membrane proteins. The other three structural subunits of the γ-secretase complex are nicastrin (NCT), PSEN enhancer 2 (PSENEN or PEN-2), and either anterior pharynx 1 (APH-1) a or b [14]. NCT functions as a γ-secretase substrate receptor [15]. PSENEN is required for the stabilization of the heterodimer of PRESENILIN N- and C-terminal fragments within the γ-secretase complex [16]. APH-1 stabilizes newly synthesized PRESENILIN holoprotein, and is possibly able to downregulate the activity of uncleaved PRESENILIN holoprotein that antagonizes cell proliferation (or promotes cell death) [17].
The PSEN2 protein is a 50 to 55kDa protein consisting of nine transmembrane domains (TMDs), a cytosolic N-terminus, a lumenal C-terminus and a “cytosolic loop” between the sixth and seventh TMD [2]. The active site of the γ-secretase complex is thought to be formed by two aspartyl residues in adjacent TMDs, residues D263 and D366 of PSEN2 (residues D257 and D385 in PSEN1) [18, 19]. PSEN2 can be cleaved by γ-secretase itself, within the cytosolic loop, to generate a longer N-terminal fragment (NTF) and a shorter C-terminal fragment (CTF) [20, 21]. Two well-studied substrates of γ-secretase are the amyloid precursor protein (AβPP) and Notch. A cytosolic CTF is normally generated by the cleavage of these transmembrane proteins by γ-secretase. For Notch, this fragment is identified as Notch intracellular domain (NICD) [22]. The CTF produced by γ-secretase is then translocated to the nucleus to modulate gene expression [23].
A well-studied substrate of the γ-secretase complex is AβPP, a type I membrane protein that can be cleaved through either non-amyloidogenic or amyloidogenic pathways (Fig. 2). The non-amyloidogenic pathway is more prevalent in most cell types [24]. In this pathway, AβPP is first cleaved by α-secretase into a soluble N-terminal fragment (sAβPPα) and an 83 amino acid C-terminal fragment (named as α-CTF or C83). The C83 fragment is then cleaved by γ-secretase into an AβPP intracellular domain (AICD) and a p3 peptide. The amyloidogenic pathway is an important processing pathway in neurons. In this pathway, AβPP is first cleaved by β-secretase into sAβPPβ and β-CTF (also named C99). The C99 fragment is then cleaved by γ-secretase into AICD and Aβ [25]. Cleavage at three major sites in AβPP is mediated by γ-secretase (Fig. 2), namely cleavage at the ɛ-site to produce a 49 amino acid residue (aa) form of Aβ (Aβ49), ζ-site cleavage (Aβ46) and γ-site cleavage (Aβ42/40) [26, 27].
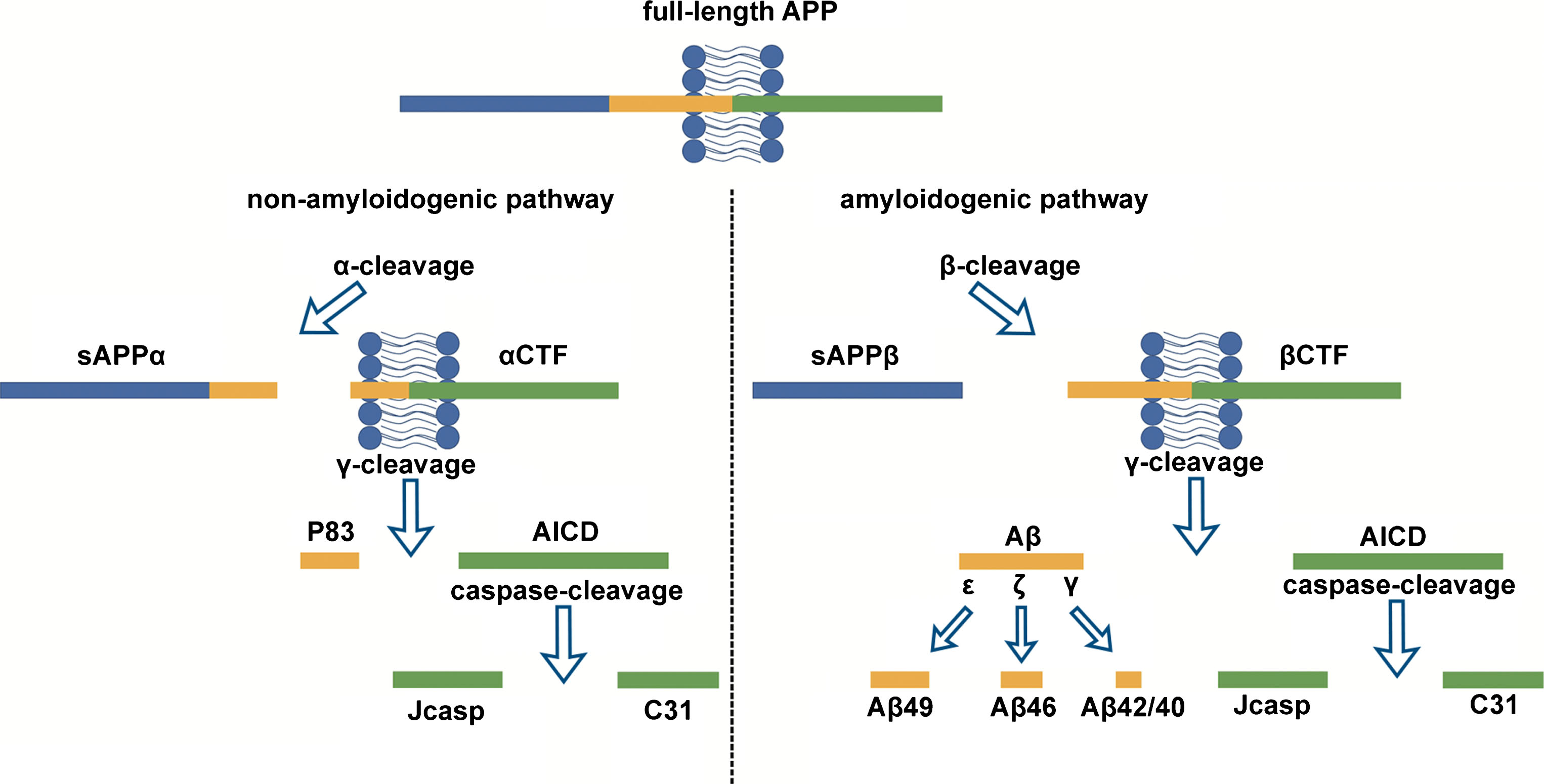
Processing of AβPP through either the non-amyloidogenic or amyloidogenic pathways.
Aβ40 (the 40aa form of Aβ) is the predominant Aβ species produced, while Aβ42 is a minor product of AβPP cleavage. Aβ40 and Aβ42 are produced from two different “production lines” of sequential cleavage by the γ-secretase enzyme, Aβ49 >Aβ46 >Aβ43 >Aβ40 and Aβ48 >Aβ45 >Aβ42 >Aβ38. In these production lines, long Aβs are shortened by consecutive carboxypeptidase-like γ-cleavages, and the hydrophobicity of Aβs can be decreased during this process, so that these Aβs are more easily moved into the extracellular environment [28]. Aβ42 has a much stronger tendency to aggregate than Aβ40 [29], as the biophysical and biochemical properties of Aβ vary strongly with its length [30, 31]. Normally, Aβ42 only constitutes about 10% of total secreted Aβ [32]. However, since longer Aβ peptides promote aggregation and neurotoxicity, an increase in the relative amount of Aβ42 versus Aβ40, (caused either by an increase in Aβ42 production or by a decrease in Aβ40 levels), has been proposed to be involved in the pathogenesis of AD [33]. Some of the fAD mutations in PSEN2 (and PSEN1) have been reported to cause increased ratios of Aβ42/40 [27, 34–36].
The contribution of PSEN1 and PSEN2 to γ-secretase activity has been studied and quantified in various systems. The loss of PSEN1 is thought to reduce the activity of γ-secretase complexes [37, 38], and the loss of both PSEN1 and PSEN2 is thought to eliminate the γ-secretase activity of such complexes completely [39, 40]. Although these two PRESENILINs are thought to complement each other functionally, they also have distinct individual functions. Psen1 is thought to contribute more to γ-secretase-associated AβPP and Notch cleavage than Psen2 since knockout of Psen1 in mice resulted in an embryonic lethal phenotype and a significant decrease in Aβ levels [41, 42], while knockout of Psen2 resulted in no overt embryonic phenotype or change of Aβ levels [43]. In mouse blastocyst-derived (BD) cells that were transiently transfected with the C100 fragment of APP (C99 with an additional translation start codon), the relative cellular activity of Psen1-associated-γ-secretase complexes (in Psen1 +/–Psen2–/– BD cells) was found to be ∼38-fold greater than that of Psen2-associated-γ-secretase complexes (in Psen1–/–Psen2 +/+ BD cells), indicating that Psen1 may be more active than Psen2 toward AβPP’s C100 fragment [5]. The activities of Psen1-associated γ-secretase complexes and Psen2-associated γ-secretase complexes could be discriminated based on their susceptibility to potent γ-secretase inhibitors, therefore these two classes of γ-secretase complexes probably have different active sites with different substrate preferences. This study also revealed that only ∼14% of the Psen1 in wild-type BD cells was engaged in active γ-secretase complexes, suggesting that the remaining Psen1 serves other biological functions [5]. Expression of human PSEN1 in Psen1–/–Psen2–/– mouse fibroblasts, could generate more Aβ (resulting from γ-site cleavage) than expressing human PSEN2, while the levels of the physiologically active AβPP intracellular domain (AICD) product (resulting from ɛ-site cleavage) provided by these two PRESENILINs were the same [44]. In a yeast reconstitution system, it has been found that PSEN1-associated-γ-secretase complexes can generate ∼24-fold more total Aβ than PSEN2-associated-γ-secretase complexes (measured by secreted Aβ per γ-secretase complex). However, the amount of PSEN1 in the γ-secretase complexes was ∼28 times higher than that of PSEN2, suggesting that PSEN1 does not have higher activity than PSEN2 in Aβ production [45].
Interestingly, one hypothesis suggests that, since PSEN2 generates less Aβ than PSEN1, mutations in PSEN2 that cause AD must result in a more severe impact on γ-secretase than those in PSEN1, so that the effect on Aβ levels can be strong enough to produce the disease in the presence of normal PSEN1 alleles [46]. Functional analyses of putative PSEN2 fAD mutations also supported this hypothesis, as these mutations cause dramatic changes in the Aβ42/40 ratio [46] and this ratio is proposed to be related to development of AD [47, 48]. Furthermore, these analyses revealed that most of the PSEN2 mutations also show parallel decreases in the generation of CTFγ, a CTF generated from the cleavage of AβPP by γ-secretase, as well as decreased Notch intracellular domain (NICD) production. Some very early-onset PSEN1 fAD mutations also show similar effects on CTFγ and NICD production [46].
THE ROLE OF PSEN2 IN INNATE IMMUNITY VIA γ-SECRETASE ACTIVITY
Dysfunction of the immune system has been considered a major factor in AD [49]. As one of the important immune cell types, microglia are essential for surveillance, and rapidly respond to changes in the central nervous system (CNS) [50]. However, in some neurodegenerative diseases, microglia may also cause neuronal injury through upregulating production of inflammatory cytokines, neurotoxins, excitotoxins, and reactive oxygen species [51, 52]. In AD, microglia are able to bind to Aβ through cell-surface receptors and drive Aβ fibrils into the endolysosomal pathway [53]. Thus, microglia are key components for Aβ clearance.
Psen2 may play a specific role in CNS innate immunity not performed by Psen1. In a mouse microglial cell line, Psen1 and Psen2 show compensatory regulation (i.e., knockdown of one leads to upregulation of the other); however, only Psen2 knockdown results in markedly decreased γ-secretase activity that leads to an exaggerated proinflammatory cytokine release from microglia [51]. miR146, a negative regulator of monocyte pro-inflammatory response, is constitutively down-regulated in microglia isolated from Psen2 knockout mice [54]. While reduced responsiveness to lipopolysaccharide (LPS) and reduced pro-inflammatory cytokine production together with decreased nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and mitogen-activated protein kinase (MAPK) activity has been reported in Psen2- (but not Psen1-) knockout mice [55]. These studies suggest that Psen2 may be the predominant γ-secretase in microglia and that deficiency of only Psen2 function (not Psen1) is associated with exaggerated microglial pro-inflammatory responses.
PSEN2 AND NEURAL DEVELOPMENT
In mice, the absence of both Psen1 and Psen2 genes simultaneously results in embryonic lethality, indicating that Presenilins are essential for embryonic development [41, 56]. The embryos of Psen1 and Psen2 double knockout mice display a loss of neural progenitor cells (NPCs) and disrupted neuronal migration at embryonic day 11 [57]. The Notch signaling pathway, which controls cellular fate choices throughout neurodevelopment [58], was also found to be blocked in double knockout mouse embryos, indicating that Presenilins are essential for neural development [57, 59]. Premature differentiation of NPCs and inhibition of the Notch signaling pathway (as well as skeletal defects [42]) were also observed in Psen1 knockout mice and resulted in perinatal lethality [41, 60]. In comparison, Psen2 knockout mice are viable and fertile [43] and do not show any effect on the physiologically important process of apoptosis during embryonic development [61]. This indicates that, in mice, only Psen1 is essential for neural development, in contrast to Psen2 [43].
THE ROLE OF γ-SECRETASE IN TUMORIGENESIS
An inverse association between cancer and AD has been observed in a population-based cohort study, which revealed that patients with prevalent cancer had a 43% lower risk of developing AD, while those with AD had a 69% lower risk of having cancer [62]. Unsurprisingly, there are several lines of evidence linking Presenilins and γ-secretase activity to tumorigenesis.
Presenilins are thought to have an anti-oncogenic function since the overexpression of both Presenilin genes promotes cellular apoptosis [63, 64], and the knockout of either Presenilin gene results in higher rates of tumorigenesis [65–67]. The AICD produced through γ-secretase cleavage of AβPP was found to be able to regulate epidermal growth factor receptor (EGFR) transcription (which is upregulated in a wide variety of tumors) via binding to the EGFR promoter to provide a tumor suppressive effect [68]. Overexpression of both wild type and mutant PSEN2 (N141I) in cultured neural cells (PC12 cells) was found to induce apoptosis in the absence of an apoptotic insult [69, 70], while no spontaneous apoptosis was observed in PC12 cells with overexpression of either wild type or fAD mutant PSEN1 (either the L286V or M146V mutation) [71]. Furthermore, the anti-apoptotic oncogene bcl-2 was found to be downregulated during PSEN2-mediated apoptosis in primary cultured neurons [72]. A recent study by Park et al. demonstrated decreased expression of Peroxiredoxin 6 (PRDX6) (a tumor-promoting protein), in AD patients with mutation of PSEN2 [73]. Further analysis of a mutant PSEN2 (N141I) mouse model verified that reduced PRDX6 activity occurred due to an increased γ-secretase activity and this resulted in a reduced incidence of spontaneous and carcinogen-induced lung tumor development [73].
DOES PSEN2 HAVE A ROLE IN AUTOPHAGY?
Autophagic/lysosomal dysfunction is thought to be extensively involved in the neurodegenerative process in AD since the endosomal-lysosomal system is a prominent site of AβPP processing, Aβ uptake, and Aβ production [74]. The transport of autophagic vacuoles (AVs) and their maturation to lysosomes may be impaired in AD brains [75], and significant accumulations of AVs have also been detected in AD brains [76].
Lee et al. [77] showed that deletion of the Psen1 gene in murine blastocysts caused complete loss of macroautophagy without affecting non-lysosomal forms of proteolysis. This was due to the failure of the V0a1 subunit of v-ATPase to become N-glycosylated in the endoplasmic reticulum (ER), resulting in a selective impairment of autolysosome acidification and cathepsin activation [77]. While there is data showing involvement of PSEN2 in autophagy (see below) its role in lysosomal acidification has not yet been tested.
In human fibroblasts, fAD mutations in PSEN1 resulted in even more severe impairment of lysosomal/autophagic functions compared to the affects seen in PSEN1 knockout cells [77]. This effect was independent of γ-secretase activity since chemical inhibition or the absence of Nicastrin (an essential component of the γ-secretase complex) did not affect autophagy [77]. However, the roles of PRESENILINs in lysosomal/autophagic functions are still controversial. Another study by Neely et al. [78] revealed that, although the deletion of either Psen1 or Psen2 in mouse embryonic fibroblasts caused impaired autophagic function, no deficits in lysosomal acidification were found [78]. In that study, it was also found that deletion of both Presenilins in mouse fibroblasts (which resulted in loss of γ-secretase activity) led to impaired autophagic function. As previously demonstrated by Lee et al., the 2011 Neely et al. study also showed that γ-secretase inhibitors did not adversely impact autophagy [77], indicating that Presenilins are involved in the autophagic function in a γ-secretase-independent manner [78]. It was also revealed that, although both Presenilins are involved in regulating autophagy, one cannot compensate for the loss of the other [78], suggesting separate independent functions for the Presenilins in autophagy. Additionally, in PSEN1–/– hippocampal neurons, a greatly decreased lysosomal calcium release that altered ion channels in the endoplasmic reticulum has been reported [79]. Similar alterations in calcium homeostasis and reduced autophagic function were also observed in Presenilin-double knockout mouse embryonic fibroblasts, which supports that Presenilins are necessary for calcium homeostasis [80].
In a major paper in 2016, Sannerud et al. [81] demonstrated that an evolutionarily conserved, unique acidic-dileucine sorting motif near the N-terminal of PSEN2 interacts with the trans-Golgi network (TGN)/endosomal adaptor complex protein (AP) complex AP-1 in a manner modulated by phosphorylation. This directs PSEN2 protein specifically into the late endosome/lysosome system compared to PSEN1’s broader subcellular distribution. Interestingly, these researchers also saw that a number of fAD mutations in PSEN1 caused the distribution of PSEN1 protein to resemble more closely that of PSEN2. Melanophores are organelles containing the pigment melanin that are related to lysosomes (reviewed by [82]). Sannerud et al. saw that knockdown of PSEN2 expression in a human melanocyte cell line greatly affected γ-secretase cleavage of Tyrosinase-related protein 1 (TYRP1) and silver, mouse, homolog of (SILV) (also known as PMEL, PREMELANOSOME PROTEIN) that are both involved in melanosome biogenesis. The specific importance of PSEN2 for melanosome biogenesis is supported by our discovery that loss of psen2 gene function in zebrafish causes loss of surface melanotic pigmentation in adult fish [83].
PSEN2 AND CALCIUM HOMEOSTASIS
Calcium homeostasis has been found to play an important role in AD progression, and calcium dysregulation may contribute to increased Aβ production [84], enhanced vulnerability of neurons to apoptosis [85], and numerous processes underlying aging-related changes in the brain [86]. Moreover, changes in the concentration of calcium ions not only affect membrane channels but also diverse intracellular calcium-regulating structures and systems, such as mitochondria, and enzymes such as calcium-dependent ATPases [87].
Independent of their role in γ-secretase activity, PRESENILINs also appear to function as passive Ca2 + leak channels in the ER [88]. Mutations in both PSEN1 and PSEN2 have been found to affect intracellular calcium homeostasis [89, 90]. Overexpression of fAD PSEN1 mutations in cultured neural cells resulted in increased cytoplasmic calcium and induced calcium release from the ER when stimulated by agonists (such as carbachol and bradykinin) [91]. It was reported that such elevations of calcium can induce oxidative stress [92, 93], which mediates staurosporine-induced mitochondrial dysfunction and apoptosis [94]. Mice with a knock-in of the fAD PSEN1 mutation, M146V, show elevation of cytoplasmic calcium levels in synaptosomes, but this was not seen in M146L knockin mice (possessing a different point mutation at the same amino acid position) that express lower and closer-to-physiological levels of mutant PSEN1 [95]. Mitochondrial dysfunction and caspase activation were also observed in synaptosomes of M146V knockin mice [95, 96].
The level of calcium in the ER is also dependent on plasma membrane store-operated calcium channels (SOCCs) during capacitative calcium entry (CCE) (also known as store-operated calcium entry) which is triggered by depletion of intracellular calcium stores) [97]. In PSEN1M146V knockin mouse fibroblasts, elevated ER calcium levels and deficits in CCE (but functional SOCCs) were detected [98]. Additionally, in CHO cells that stably overexpress wild type AβPP, both overexpression of the PSEN1 mutation, D257A, and co-expression of D257A and the PSEN2 mutation, D366A (mutations that decrease γ-secretase activity) resulted in reduced intracellular calcium stores and enhanced CCE compared to overexpression of wild type PSEN1. However, expression of another dominant negative isoform of PSEN1, ΔTM1-2 (which also abrogates γ-secretase activity), led to reduced intracellular calcium stores but with a deficit of CCE [99]. Thus, modulation of CCE appears to be independent of the function of PRESENILINs in γ-secretase activity [99].
The knockdown of the gene PSENEN in Hela cells results in inhibition of proteolytic processing of PRESENILINs [100], thus, increasing the holoprotein form of these proteins. The holoprotein form of Psen1 in Aph-1abc–/– mouse embryonic fibroblasts (where γ-secretase activity is completely absent and Psen1 is present as a holoprotein) was found to increase greatly the calcium leak rate, strongly suggesting that the holoprotein forms of the Presenilins are the functional forms in calcium leakage [88].
Knockdown of PSEN2 activity in Hela cells was found to reduce dramatically the ER calcium leak rate and to produce a large increase in ER calcium load [100]. The PSEN2 fAD mutation, N141I, was found to induce ER calcium leak and reduce the ER calcium pool [88, 101]. However, in SH-SY5Y neuroblastoma cell lines, PSEN2, but not PSEN1, modulates the shuttling of Ca2 + between the ER and mitochondria since mitochondrial Ca2 + dynamics were reduced by PSEN2 down-regulation and enhanced by the expression of the PSEN2 mutant forms, N141I, D366A and T122R [102]. This specific function for PSEN2 in Calcium transport reinforces that the PRESENILINs have separate independent functions.
PRESENILINS ARE IMPORTANT FOR MITOCHONDRIAL FUNCTION
Mitochondria are essential for cellular energy supply and for other cellular processes such as apoptosis, reactive oxygen species production and calcium homeostasis [103]. The limited glycolytic capacity of neurons means that their energy supply is highly dependent on aerobic oxidative phosphorylation (OXPHOS) that occurs in mitochondria [104]. The “mitochondrial cascade hypothesis” postulates that mitochondrial dysfunction is a primary event in both sporadic and familial forms of AD [105, 106]. Although mutations in both PSEN1 and PSEN2 have been shown to sensitize cells to apoptosis through impaired mitochondrial function, there is evidence suggesting that PSEN2-associated γ-secretase activity may play a specific role required for proper mitochondrial function [63]. Overexpression of the PSEN2 fAD mutation, N141I, has been shown to induce apoptosis in PC12 cells [70] and to increase γ-secretase activity in mouse brain mitochondria [63]. This induced the production of Aβ42 peptides and contributed to mitochondrial dysfunction and AD pathology [63]. In mouse embryonic fibroblasts (MEFs), it has been found that knockout of Psen2 caused a significantly lower basal respiratory rate compared to knockout of Psen1 or compared to wild type cells, and the percentage of fully functional mitochondria in Psen2 knockout cells (and Presenilin double knockout cells) was much lower than that in wild type cells or cells lacking only Psen1 [107]. A separate MEF study showed that a deficiency of Psen2 led to a reduction in the subunits responsible for mitochondrial OXPHOS, an altered morphology of the mitochondrial cristae and an increase in glycolytic flux. This indicated that absence of Psen2 causes an impairment in respiratory capacity but also an increase in glycolytic flux to support energy needs [108].
PSEN2 IS ENRICHED IN MITOCHONDRIA-ASSOCIATED ER MEMBRANES
Mitochondria-associated ER membranes (MAM) are a sub-compartment of the ER juxtaposed to mitochondria [109]. This compartment is involved in various cellular activities, such as calcium homeostasis [109, 110], cholesterol metabolism [111], and apoptosis [112]. These functions of MAM are also related to those dysregulated in AD [113]. Upregulation of MAM-associated proteins in both AD brain and an AβPP mouse model, increased ER-mitochondrial coupling in human fibroblasts from both fAD and sporadic AD patients [114]. The enrichment of PSEN1 and PSEN2 in the MAM [115] strongly supports a role for MAM in AD pathology.
A study by Pera et al. [116] focused on the accumulation of the AβPP C-terminal fragment, C99, in the MAM (this fragment becomes a substrate for the γ-secretase complex in the amyloidogenic pathway of AβPP cleavage). In cells from AD patients, and in the embryonic cortical neurons of a PSEN1 fAD mutation (M146V) knockin mouse, the level of unprocessed C99 was increased in MAM, which then induced sphingolipid turnover and ultimately resulted in the dysfunction of mitochondria [116].
In mice, Psen2 has been shown to bind to Mitofusin 2 (Mfn2), one of the major regulators of ER-mitochondria [117] and to induce ER-mitochondrial coupling [118]. Although Psen2 is not necessary for the antagonistic effect of Mfn2 on MAM formation (Mfn2 can antagonize ER-mitochondrial coupling in the absence of Psen2), the presence of Psen2 modulates the effect of Mfn2. Surprisingly, an fAD mutant form of PSEN2 (N141I) was shown to be more effective than the wild-type form in regulating ER-mitochondrial coupling, as its binding to Mfn2 is favored [118]. Although PSEN1 has been suggested to play a similar role in regulating ER-mitochondrial coupling to PSEN2 [119], it showed no interaction with Mfn2 (nor with its homologue Mfn1) [118].
PSEN2 AND ISCHEMIC OXIDATIVE STRESS
Brain ischemia has long been considered one of the possible causes of AD [120]. Dysregulated AβPP, the PRESENILINs, β-secretase and Apolipoproteins (the APOE ɛ4 allele is considered to be the major genetic risk locus for late onset, sporadic AD [121]) have been observed in experimental models of incomplete brain ischemia [122]. In rats, after 10 min of global brain ischemia, the expression of Psen1 showed a modest trend of downregulation from days 2 to 7 post-ischemia, with an opposite trend at day 30. The expression of Psen2 showed significant upregulation on day 2 post-ischemia and was only modestly elevated on day 7 followed by a slight downregulation on day 30, indicating that the two Presenilins may have different roles during brain ischemia in rats [123]. This brain ischemia model supported that PSEN2 plays a role in the modulation of apoptosis [72], since neurons in ischemic injured areas of the brain began to die from days 2 to 7 post-ischemia [123]. Although the PRESENILINs appear to complement each other’s functions, PSEN1 seems to provide a basic constitutive function, while PSEN2 appears to operate as an emergency aid under ischemic oxidative stress [124].
PS2V is a truncated isoform of the PSEN2 protein that shows enhanced expression in AD brains [12]. PS2V is produced by alternative transcript splicing that excludes exon 5 sequence from PSEN2 mRNA (Fig. 3) [12]. It was demonstrated in human neuroblastoma SK-N-SH cells, that the expression of PS2V is induced by hypoxia, but not by many other stresses [125]. This was also supported by observations in the guinea pig [126] and zebrafish [13]. However, the PS2V isoform appears to be absent in mice and rats [13]. In zebrafish, the PS2V-equivalent isoform, PS1IV, is produced when hypoxia induces alternative splicing of transcripts of the psen1 gene rather than transcripts of psen2. Although PS1IV is far smaller than PS2V, it is functionally similar to PS2V in that it is able to stimulate γ-secretase activity and suppress the unfolded protein response [13]. Further study of PS1IV revealed that its absence under hypoxia-like conditions changes the expression of genes involved in inflammation such as the gene il1b, which is equivalent to IL1B in humans, that is recognized as an AD risk locus [127, 128].
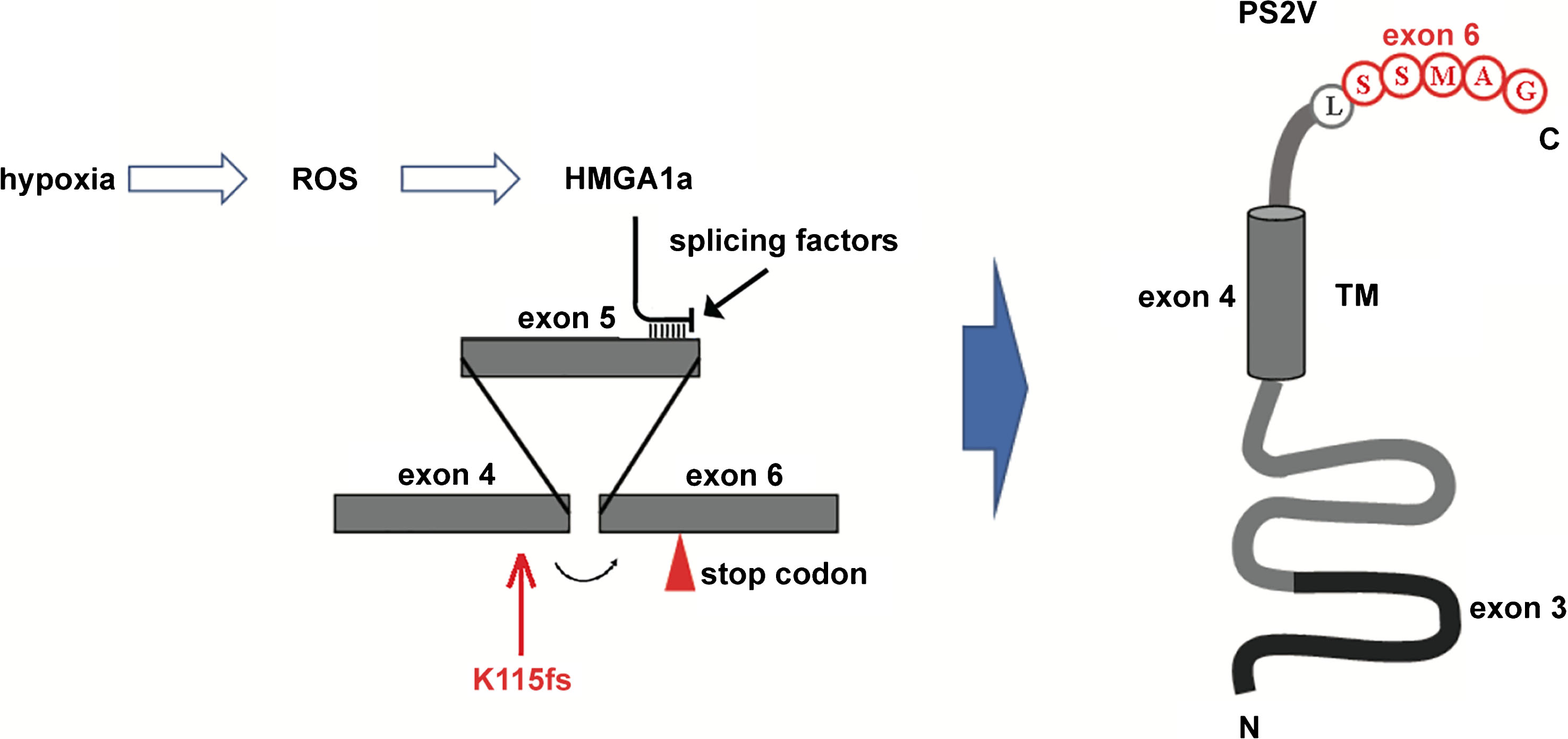
The generation of PS2V. Under hypoxia, reactive oxygen species production by mitochondria increases and induces the expression of HMGA1a that binds to PSEN2 transcripts at the 3’ end of exon 5. This excludes binding by splicing factors and causes exon 4 to be spliced to exon 6. This causes a frameshift that results in early termination of the coding sequence, translation of which produces PS2V [13]. The position of the 2-ncuelotide deletion K115fs mutation is indicated.
DIFFERENTIAL EXPRESSION OF THE PRESENILINS WITH AGE AND SEX
AD is more prevalent in women than men [129]. AD histopathology [130] and AD-related cognitive decline [131] reported to be greater in women than in men. Although the mechanism of this gender difference in AD risk is still unclear, sex steroid hormones are thought to be a contributing factor. Indeed, sex steroid hormones are known to be involved in neural development [132].
The expression of PRESENILINs has been found to change with age and sex. Thakur and Gosh have published a number of studies investigating Presenilin expression in the cerebral cortex of mice [133–135]. In aged mice, Psen1 is downregulated and Psen2 is upregulated in both sexes. However, in female mice, the expression level of Psen1 is relatively higher and the expression level of Psen2 is relatively lower compared to males of the same age [133]. Follow-up studies found that the expression of Psen1 is downregulated by gonadal hormones (17β-estradiol and testosterone) in female and male adult mice and upregulated in aged male mice only [134]. After gonadectomy in female and male adults there was a decrease in Psen2 expression but supplementation with gonadal steroids could upregulate the expression of Psen2 in both sexes [135]. A separate study found that there was no difference in the levels of PSEN1 (and APP) proteins in hypogonadal female mice, while male hypogonadal mice had increased levels of these proteins and this was confined to the hippocampal region [136]. Collectively, these results suggest a sex-dependent regulation of brain Presenilin expression during aging and that the regulation of the Presenilins by gonadal steroids may influence Presenilin associated brain functions.
CONCLUSION
PSEN2 is one of the genes implicated in fAD pathogenesis, but our understanding of how it contributes to AD progression is still limited. As one of the essential components of the γ-secretase complex, PSEN2 protein has been found to be involved in γ-secretase-related physiological activities that are associated with AD progression such as innate immunity, Notch signaling, tumorigenesis and autophagy. PSEN2 also plays a specific role(s) in mitochondrial function due to its regulation of calcium homeostasis. Research using animal models has found that expression of Psen2 changes with age and sex, and, significantly, is altered under ischemic oxidative stress which is likely a major contributor to AD pathogenesis. However, this may be only the tip of the iceberg of PSEN2’s functions, and more research must be done to fully identify and understand PSEN2’s roles in normal cell biology and AD.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0656r1).