
Abstract
Many experimental studies show that erythropoietin (EPO) has a neuroprotective action in the brain. EPO in acute and chronic neurological disorders, particularly in stroke, traumatic brain injury, Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, has neuroprotective effects. We previously reported the neuroprotective effect of NeuroEPO, a low sialic form of EPO, against oxidative stress induced by glutamate excitotoxicity. In this paper, we analyze the effect of NeuroEPO against apoptosis induced by glutamate excitotoxicity in primary neuronal cultures obtained from the forebrains of Wistar rat embryos after 17 days of gestation. Excitotoxicity was induced after nine days of in vitro culture by treatment with a culture medium containing 100μM glutamate for 15 min. To withdraw glutamate, a new medium containing 100 ng NeuroEPO/mL was added. Apoptosis was analyzed after 24 h. Images obtained by phase contrast microscopy show that neurons treated with glutamate exhibit cell body shrinkage, loss of dendrites that do not make contact with neighboring cells, and that NeuroEPO was able to preserve the morphological characteristics of the control. Immunocytochemistry images show that the culture is essentially pure in neurons; that glutamate causes cell mortality, and that this is partially avoided when the culture medium is supplemented with NeuroEPO. Activation of intrinsic apoptotic pathways was analyzed. The decreases in Bcl-2/Bax ratio, increase in the release of cytochrome c, and in the expression and activity of caspase-3 observed in cells treated with glutamate, were restored by NeuroEPO. The results from this study show that NeuroEPO protects cortical neurons from glutamate-induced apoptosis via upregulation of Bcl-2 and inhibit glutamate-induced activation of caspase-3.
INTRODUCTION
During the last 30 years, erythropoietin (EPO) has improved the quality of life of more than a million patients with anemia due to chronic renal failure [1]. EPO is a single polypeptide chain of 165 amino acids folded into four α helices that are linked by two disulphide bridges between cysteines 6 and 161, and between cysteines 29 and 33 [2] with a molecular mass of 30 kDa, depending on the carbohydrate content. The composition of EPO is about 60% protein and 40% carbohydrate, containing four glycosylated chains including three N-linked and one O-linked acidic oligosaccharide side chains. N-linked glycosylation sites occur at the positions 24, 38, and 83 of aspartyl residues, while that O-linked glycosylation site is at serine 126 [3]. Each N- and O-glycosylation site can accommodate up to four and two sialic acid residues, respectively [4, 5]. Initially known for its essential role in the regulation of erythropoiesis, many studies have shown that EPO also possesses various other biological functions, including antioxidant, anti-apoptotic, and tissue protective effects [6–9].
Neuroprotective effects of EPO in preclinical studies have been widely reported for a variety of neurodegenerative pathologies, including stroke, Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and traumatic brain injury [10–14]. All of them are neuropathologies that differ in the causes that originate them, but all share excitotoxity as one of the main mechanisms that lead to the death of neurons. These neuroprotective effects of EPO on cells of the nervous system and the fact that EPO is already widely used in clinical practice, especially for the treatment in patients with anemia associated with chronic kidney failure [7] makes EPO a highly attractive candidate drug for neuroprotection/neuroregeneration. However, although EPO is a promising molecule for neuroprotection, the use of EPO in the treatment of neurological diseases requires high doses and prolonged application, which can produce an increase in the hematocrit and viscosity of the blood and might lead to serious cardiovascular events such as infarction or stroke [15, 16]. Therefore, conventional EPO due to the cross-talk with hematopoietic activity may not be suitable for the treatment of neurodegenerative disorders [11]. To address this problem, one approach may be the use of non-hematopoietic EPO derivatives that produce tissue protection [17, 18]. In the last few years, non-hematopoietic EPO analogues, which include asialoerythropoietin (asialoEPO) [19], carbamylated EPO (CEPO) [17], and rHU-EPO with a low sialic acid content (NeuroEPO) [20], and that do not exhibit erythropoietic activity, have been tested in the treatment of neurodegenerative diseases.
NeuroEPO is a recombinant human glycoprotein produced in Chinese hamster ovary (CHO) cells (not a commercial product; the patent is PCT/cu2006/000001 Patent 20050138) supplied by the Centre of Molecular Immunology (CIM, from its Spanish initials) (Havana, Cuba). It is characterized by its low sialic acid content, which means that NeuroEPO lacks erythropoietic activity while exhibiting neuroprotective properties [20]. NeuroEPO can rapidly reach the brain after intranasal delivery [21], and has actually proven to be effective in different biomodels of neurodegenerative diseases in preclinical studies. NeuroEPO has been shown to exert neuroprotective effects such as improving viability, neurological status, and cognitive functions in animal models of stroke [20]. Additionally, this compound is currently used in both transgenic and non-transgenic murine models of Alzheimer’s disease [22, 23]. With a view to achieving the use of NeuroEPO as a neuroprotector, it is first critical to understand the cellular pathways that may mediate neuronal injury and are subsequently susceptible to modulation by NeuroEPO. Thus, the aim of the present study is to investigate the neuroprotective effect of NeuroEPO against excitotoxicity–induced apoptosis in an in vitro model using cortical neuronal culture and the possible mechanisms for these effects.
Excitotoxicity appears as a common pathophysiological mechanism that plays an important role in the neurotoxicity of both acute and chronic neurological diseases [24–27]. Excitotoxicity is defined as cell death caused by excessive stimulation of neurons by excitatory amino acids neurotransmitters. The excessive excitation of a neuron via the transmitter glutamate leads to cell death through many distinct cascades, all of which originate at ionotropic glutamate receptors and all excitotoxic paradigms share the mechanism of at least some overstimulation of these receptors. Because glutamate is the major excitatory neurotransmitter in the mammalian central nervous system, excitotoxicity usually refers to neuron death when glutamate extracellular level increased above its threshold value causing overstimulation of glutamate receptors, mainly NMDA receptor [28, 29]. Excitotoxicity death requires the excessive influx of extracellular Ca2 + via receptor-operated channels or sensitive Ca2 + channels [26]. The excessive accumulation of Ca2 + into the cytosol initiates a series of biochemistry and molecular events that culminate in neuronal death [29–31].
It is well known that over-activation of NMDA glutamate receptors in response to glutamate activation is closely coupled to the generation of nitric oxide (NO) by activation of neuronal nitric oxide synthase (nNOS), an enzyme which is tethered to the NMDA receptor complex by the postsynaptic density protein-95 (PSD95) [31–33], and to Ca2 + intracellular overload [31]. The increase in cytoplasmic Ca2 + levels in response to glutamate receptor activation can induce homeostatic mechanisms from mitochondria to try to maintain the homeostatic levels of intracellular Ca2 + concentration trough the uptake of Ca2 + into mitochondria [29, 34] which, if excessive, can induce mitochondrial dysfunction [29, 36]. In this condition, the mitochondria increase the formation of reactive oxygen species (ROS) and activate pro-apoptotic proteins of the Bcl-2 family, triggering the apoptotic cascade that leads to the death of the neuron [37, 38].
There is abundant evidence that apoptosis induced by excitotoxicity plays a pivotal role in cell death in different neurodegenerative diseases of the central nervous system, including stroke, Alzheimer’s disease, amyotrophic lateral sclerosis, and Parkinson’s disease [39–42]. Glutamate excitotoxicity has been linked to oxidative stress and mitochondrial dysfunction both in acute as well chronic neurodegenerative disorders [43–45]. Modifications of this interconnection seem to be sufficient to induce the apoptotic pathway [46].
In a previous study, we found that NeuroEPO protects neurons from excitotoxic death and reduce the oxidative stress induced by glutamate excitotoxicity [47]. Oxidative stress together with mitochondrial dysfunction, are the main mechanism by which excitotoxicity causes cell death [44, 48]. In this study we focus in the possible effect of NeuroEPO on the apoptotic mechanisms induced by the mitochondrial intrinsic pathway.
MATERIAL AND METHODS
All experimental procedures were carried out following the guidelines of the Committee for the Care of Research Animals of the University of Barcelona, in accordance with the directive of the Council of the European Community (2010/63 y 86/609/EEC) on animal experimentation. The experimental protocol was approved by the local University Committee (CEEA-UB, Comitè Ètic d’Experimentació Animal de la Universitat de Barcelona) and by the Catalan Government (Generalitat de Catalunya, Departament de Territori I Sostenibilitat) with the approval number #9431.
Primary cortical cells culture
To study the effect of NeuroEPO on the morphological changes induced by glutamate excitotoxicity in neuronal culture, we used primary rat cortical cultures subjected to a treatment with cytosine arabinoside (AraC) to prevent glial proliferation. This treatment has the advantage, that eliminating glial cells, we can better observe the presence of apoptosis in neurons and to determine if NeuroEPO acts directly on neurons. Primary cortical cells were obtained from the cerebral hemisphere of embryos of Wistar rats (Harlan, Spain) after 17 days of gestation. The rats were killed with CO2 and the fetuses were decapitated. Dissociated cortical cells were plated in cell culture plates at a density of 105 cells/cm2 in 9.6 cm2 Corning Costar cell culture plates (Madison, WI, USA). Previous to its use, each well was coated with a 1 mL poly-L-ornithine solution (0.01%) for 1 h in the incubator at 37°C, the poly-L-ornithine solution was removed and washed three times with sterile water. Once the water was removed, the wells were dried in the sterile chamber with ultraviolet light.
The cells were cultured for 9 days in growth medium for Neurobasal ® neurons containing Penicillin / Streptomycin, enriched with GlutaMAX ® and B27 ® (Antioxidants) and supplemented with 1/5 of Neurobasal Medium plus GlutaMAX ® and B27 ® previously conditioned in astrocyte cultures, in culture incubator at 37°C in a humidified atmosphere containing 5% CO2/95% air (pH 7.2). To halt the proliferation of non-neuronal cells, 3 days after plating, half the culture medium was replaced with medium containing AraC (final concentration: 10μM). On day 5 after plating, half of the medium was replaced with Neurobasal ® medium containing 2 mM glutamine and 2% B27 (AO+), and the neurons were analyzed at day 9.
Glutamate exposure to induce excitotoxicity and neuroprotective effect of NeuroEPO
In a previous study, in the same conditions as those used here, we tested the effect of different concentrations of glutamate on cell viability 24 h after a 15 min exposure to glutamate, as well as the effect of different concentrations of NeuroEPO after treatment with glutamate [47]. From these results, we chose to use as concentration and exposure time to induce excitotoxicity, glutamate 100μM for15 min, and for protection 100 ng/mL of NeuroEPO.
On the 9 day, the medium was removed, neurons were washed once with pre-warmed (37°C) basic saline solution (BSS) containing (in mM) 137 NaCl, 3.5 KCL, 0.4 KH2PO4, and 0.33 Na2HPO4.7H2O followed by conditioned Neurobasal Medium containing 2 mM glutamine and 2% B27 (AO+), excitotoxicity was induced by adding glutamate (100μM final concentration) and cells were further incubated for 15 min at 37°C. After this incubation, the medium containing glutamate was removed, and after two washes with BSS, replaced by the Neurobasal Medium containing 2 mM glutamine and 2% B27 (AO+), and the cells were further incubated for 24 h at 37°C. The neuroprotective effect of NeuroEPO was tested by adding 100 ng/mL of NeuroEPO to the culture after the glutamate was removed, as is summarized in Fig. 1. In some experiments the treatment time and concentration with glutamate was modified, being indicated on each case in the figure legend. After 24 h, the medium was removed, and the cells washed with PBS, and immediately placed into 500μl of homogenization buffer (0.3 M sucrose in 0.1 M phosphate-buffered saline (PBS) with complete protease inhibitor cocktail (Catalog No. P2714-BTL, Sigma-Aldrich) per 10 ml, and separated from the plate by cell lifter (Sigma Catalog No. SIAL0008). The cells were stored at –20°C until further analysis.
Schematic representation of the in vitro experiments with neuron cortical cultured cells. After 9 days of culture, the cells were incubated in: medium basal (Control); medium containing a final concentration of NeuroEPO 100 ng/mL (NeuroEPO); medium containing a final concentration of 100μM glutamate for 15 min, after which the medium is replaced by basal medium (Glutamate); medium containing a final concentration of 100μM glutamate for 15 min, after which the medium is replaced by medium containing a final concentration of NeuroEPO 100 ng/ml (Glutamate + NeuroEPO). The cells were always cultured at 37°C in an atmosphere of 5% CO2. The cells were treated for 24 h. The results correspond to 3 independent experiments performed on different dates. Each data is the result of three experiments with 3 wells for each group in each experiment (control, glutamate and glutamate plus NeuroEPO), as indicated in figure, and the measurement in triplicate of samples of each well.
Immunocytochemistry
Cells grown on coverslips pre-treated with poly-L-ornithine (9 days) were washed in PBS, fixed with 4% paraformaldehyde in PBS (40 min at 4°C) and again washed three times with PBS. For permeabilization, and blocking of nonspecific proteins, three new washes were made in blocking solution: 0.1% Triton X-100 in PBS and 10% horse serum for 30 min. Then, cells were incubated with the primary antibodies: monoclonal anti-MAP-2 (NB600-1372, Novusbio Biotechnology) at a dilution 1:200 (neuronal protein) and the polyclonal anti-GFAP (NB300-141, Novusbio Biotechnology) at a dilution 1:1000 (glial protein) in horse serum at 1% PBS at 4°C overnight. After 3 washes with 0.1% Triton-X PBS, the coverslips were incubated with secondary anti-mouse and anti-rabbit antibodies with the coupled fluorochromes (Mouse Conjugate DyLight 650 dilution 1:1000 in PBS and Rabbit Conjugate DyLight 488 dilution 1:1000 in PBS, Bethyl Laboratories Inc. TX, USA), for 60 min. Finally, the coverslips were dried and mounted in slide with Mowiol. The cells were visualized with Leica confocal microscope (TCS-NT Leica Microsystems AG, Wetzlar, Germany) and after being digitized, the images were processed with Image-J 2.0. software.
The nuclear chromatin was assessed with DAPI (4’6-diamidino-2-phenylindole dihydrochloride, Sigma Aldrich Inc. USA). Cells grown on poly-L-ornithine-coated coverslips were washed in PBS, fixed with 4% paraformaldehyde in PBS (40 min at 4°C) and again washed three times with PBS, followed by permeabilization with 0.1% Triton X-100 in PBS and blocking of nonspecific proteins with 10% horse serum for 30 min and washed three times with PBS followed by incubation in a DAPI labeling solution (1:200 in PBS from a DAPI stock solution of 400μg DAPI/ml) for 30 min at room temperature and in the dark, and subsequently washed again with PBS. The coverslips were mounted on the glass slides with Mowiol. DAPI is a molecule with the ability to intercalate between base pairs Adenine - Thymidine DNA [49] and with light of appropriate wavelength (λ= 460 nm), emits a bright blue fluorescence directly proportional to the amount of DNA present. Fluorescence microscopy was used to perform differential counting of cells, identifying them by the morphology and fluorescence intensity of the nucleus. The quantification of apoptotic and necrotic nuclei was performed by counting the total cells in five fields/slide, three slide per treatment from n = 3 independent experiments, using a Leica confocal microscope (TCS-NT), (Leica Microsystems AG, Wetzlar, Germany), under a 460 nm filter, and after being digitized, the images were processed with Image-J 2.0 software [49]. The nuclei of healthy cells can be differentiated morphologically from the nuclei of necrotic and apotoptic cells according to the size of the nucleus, the intensity of the staining and the condensation of DNA.
Western blot analysis
Samples of cells were lysed in RIPA buffer (Catalog No. R 0278 Sigma-Aldrich). Cells were thawed and homogenized for 3 min using a pellet pestle (catalog No. 2359947, Sigma-Aldrich), then centrifuged for 10 min at 10.000×g, 4°C in a centrifuge 5415 R (Eppendorf). The supernatant was collected and protein concentration was determined by the Bradford method (Bio-Rad protein assay kit) and using a Tecan spectrophotometer (Quad4 Monochromators brochure - Infinite® 200 (Tecan® Group, Ltd) at 595 nm. Equal amounts of protein (15μg) were fractionated by electrophoresis for 1.5 h at 125 V on SDS-polyacrylamide gel, and electrophoretically transferred to a polyvinylidene difluoride PVDF membrane (Millipore, Bedford, MA, USA). The membranes were blocked with 5% defatted milk in TBS-Tween (TBS-T) (50 mM Tris, pH 7.6, 150 mM NaCl, 0.1% Tween-20) and incubated overnight at 4°C with the corresponding primary antibodies: in the 12% gels (β-actin 1:200, Bcl-2 1:500 and Bax 1:500) and 15% gels (cytochrome c 1:80 and caspases-3 1:500) (Santa Cruz, Biotechnology, CA, USA). β-actin was used as a load control of the amount of sample protein. The membranes were then incubated with their respective secondary antibodies anti-rabbit IgG (1:8000/12000) or anti-mouse IgG (1:7000) (Cell Technology Signaling, MA, USA) in TBST for 1.5 h at room temperature, in gentle agitation. The bands were then visualized by chemiluminescence substrate using an ECL (Cell Signaling) immunoblot kit. The bands were scanned and subjected to densitometric analysis that was carried out using software Image-J 1.42. All data were expressed as the relation to the optical density (OD) values of the corresponding controls for statistical analyzes.
Assay caspase-3 activity
Cell samples were homogenized in 50 mM Tris-150 mM NaCl buffer, pH 7.2, and sonicated. After centrifugation at 15,000 g for 15 min at 4°C, supernatants were used for caspase-3 activity assay according to manufacturer’s instructions (CASP3C kit, Sigma, Saint Louis, MO, USA). The caspase 3 colorimetric assay is based on the hydrolysis of the peptide substrate acetyl-Asp-Glu-ValAsp p-nitroanilide (Ac-DEVD-pNA) by caspase 3, resulting in the release of the p-nitroaniline (pNA)moiety. p-NA has a high absorbance at 405 nm (ɛ mM = 10.5).
Statistical analyses
Statistical analysis was performed using the Statistical Package for Social Sciences (SPSS), version 21.0. Completion of the Kolmogorov-Smirnov normality test showed normal distribution of the mean values evaluated. Therefore, we used the parametric one-way ANOVA test with post hoc analysis using the Bonferroni test to perform comparison hypothesis testing between Control, Glutamate, NeuroEPO and Glutamate + NeuroEPO groups. Statistical differences were considered significant when the P-value was determined to be <0.05. Results were expressed as the mean±S.E.M. of 3 independent experiments n = 9
RESULTS
Protective effects of NeuroEPO against glutamate excitotoxicity: Cytological analysis
The observation using phase-contrast microscopy showed that untreated cortical neurons maintained with Neurobasal Medium, retained normal morphology, which is characterized by many large cells with thick and abundant cellular extensions (dendrites) that make contact with the neighboring cells (Fig. 2A). Exposure to glutamate showed an evident morphological deterioration. Neurons exhibited cell body shrinkage, formation of cell surface “blebs”, and a dramatic loss of dendritic processes in the cell culture were found. Small cells, retracted with thin processes that do not make contact with neighboring cells could be observed (Fig. 2B). Cortical neurons exposed to glutamate followed by 24 h in Neurobasal medium containing 100 ng/mL of NeuroEPO showed a tendency to retain the morphology seen in the control: large cells had thick processes and preserved contact with neighboring cells (Fig. 2C).
Morphological analysis of glutamate-mediated neurotoxicity in cortical neuronal cultures, after 9 days of culture. A) Photomicrographs of cortical neurons maintained with Neurobasal Medium, showed the presence of large cells with thick and thin processes that make contact with the neighboring cells. B) Photomicrographs of cortical neurons exposed to 100μM glutamate for 15 min, followed by 24 h in Neurobasal Medium. Small cell with thin processes fragmented that do not make contact with neighboring cells, and with cell body shrunk, can be observed. C) Photomicrographs of cortical neurons exposed to 100μM glutamate for 15 min followed by 24 h in Neurobasal Medium containing 100 ng/mL of NeuroEPO. Cells have thick and thin processes and preserve contact with neighboring cells. Scale bar, 100μm.
By using neuronal (MAP-2) and astrocytic (GFAP) specific immunostaining, we showed that inhibition of glial proliferation by AraC leads to neuron-enriched cultures containing very low glial cells (Fig. 3A). Treatment with glutamate showed a decrease in the number of neurons confirming that the excitotoxicity causes cell death (Fig. 3B) and that this is partially avoided when NeuroEPO is added to the culture medium just after exposure to glutamate (Fig. 3C).
Characterization of cortical cells cultures. Cultures of 9 days were stained with two antibodies, anti-MAP2 (red) and anti-GFAP (green). Cells not exposed to glutamate (A), cells exposed to glutamate 100μM for 15 min (B) and cells exposed to Glutamate 100μM for 15 min followed treatment with NeuroEPO 100 ng/ml (C). Staining for the neuronal marker, MAP2, showed the presence of neurons. The glial marker, GFAP, reveals that only a very small number of glia were present in the culture. Scale bar, 100μm.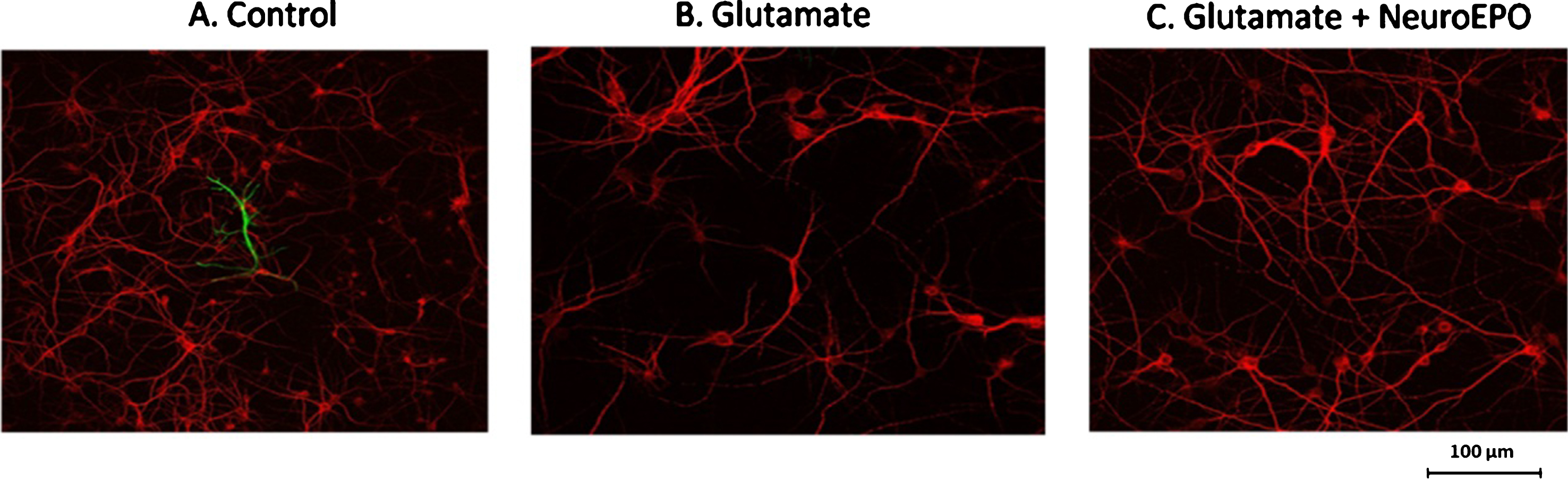
Glutamate excitotoxicity can cause neuronal death both by necrosis and by apoptosis, depending on the intensity and duration of the excitotoxic insult. Death due to necrosis is associated with intense stimulation and is an early death that makes difficult to achieve effective therapy. Using MAP-2 we showed that, compared with control (Fig. 4A, 1), glutamate caused neuronal cell killing (Fig. 4A, 2). Treatment with NeuroEPO added after glutamate, protected the cells from this glutamate-induced neuronal death (Fig. 4A, 3). Using DAPI we found that, compared to the control (Fig. 4A, 4), glutamate increased the number of pyknotic nuclei in which a reduction in its size, DNA condensation and a greater intensity of staining was observed (Fig. 4A, 5). NeuroEPO reduced the number of nuclei with small size and greater intensity of glutamate-induced staining (Fig. 4A 6). Table 1 shows the number of photomicrographs of nuclei per group and the numerical data resulting from the count of each photomicrographs. The data with the mean±S.E.M. are shown in Fig. 4C.
Staining of neurons and nuclei in cortical cells exposed to glutamate followed treatment with NeuroEPO. A) Cultures were exposed to glutamate 100μM for 5 min and subsequently reincubated in their original culture medium for 24 h. The cultures were then fixed, permeabilized, stained with the anti-MAP2 neuronal antigen (images 1–3) and cells nuclei labeled with DAPI (images 4–6), and examined by confocal microscopy. (1 and 4) Control at 24 h; (2 and 5) 100μM glutamate removed after 5 min and replaced by basal medium at 24 h; (3 and 6) 100μM glutamate for 5 min and replaced with basal medium containing 100 ng/ml of NeuroEPO. Scale bar 100μm. B) Glutamate induce death of cortical cells. Data obtained from (A, 1–3), after digitation and processed with Image-J 2.0 software. Data were standardized relative to untreated (100% cell viability). Glutamate exposure caused a significant decrease of viability while that NeuroEPO attenuate the loss of viability induced by glutamate treated with glutamate and glutamate followed NeuroEPO. Data are the mean±SEM values. Each data is the result of 3 wells for each group (n = 9) in each experiment (control, glutamate and glutamate plus NeuroEPO) from 3 independent experiments *p≤0.005. C) Neuronal death induced by glutamate and effect mitigated by NeuroEPO in primary culture of cortical cells. The histograms show the relation, expressed as of healthy nuclei and damaged nuclei in each group, obtained from the numerical data of Table 1. Neuronal death induced by glutamate is mitigates by NeuroEPO. The table shows the data obtained by counting the number of nuclei from 5 microphotographs per group (control, glutamate and glutamates and NeuroEPO) selected randomly from 9 microphotographs obtained from 3 slice per group and 3 microphotographs per slice, after digitation and processed with Image-J 2.0 software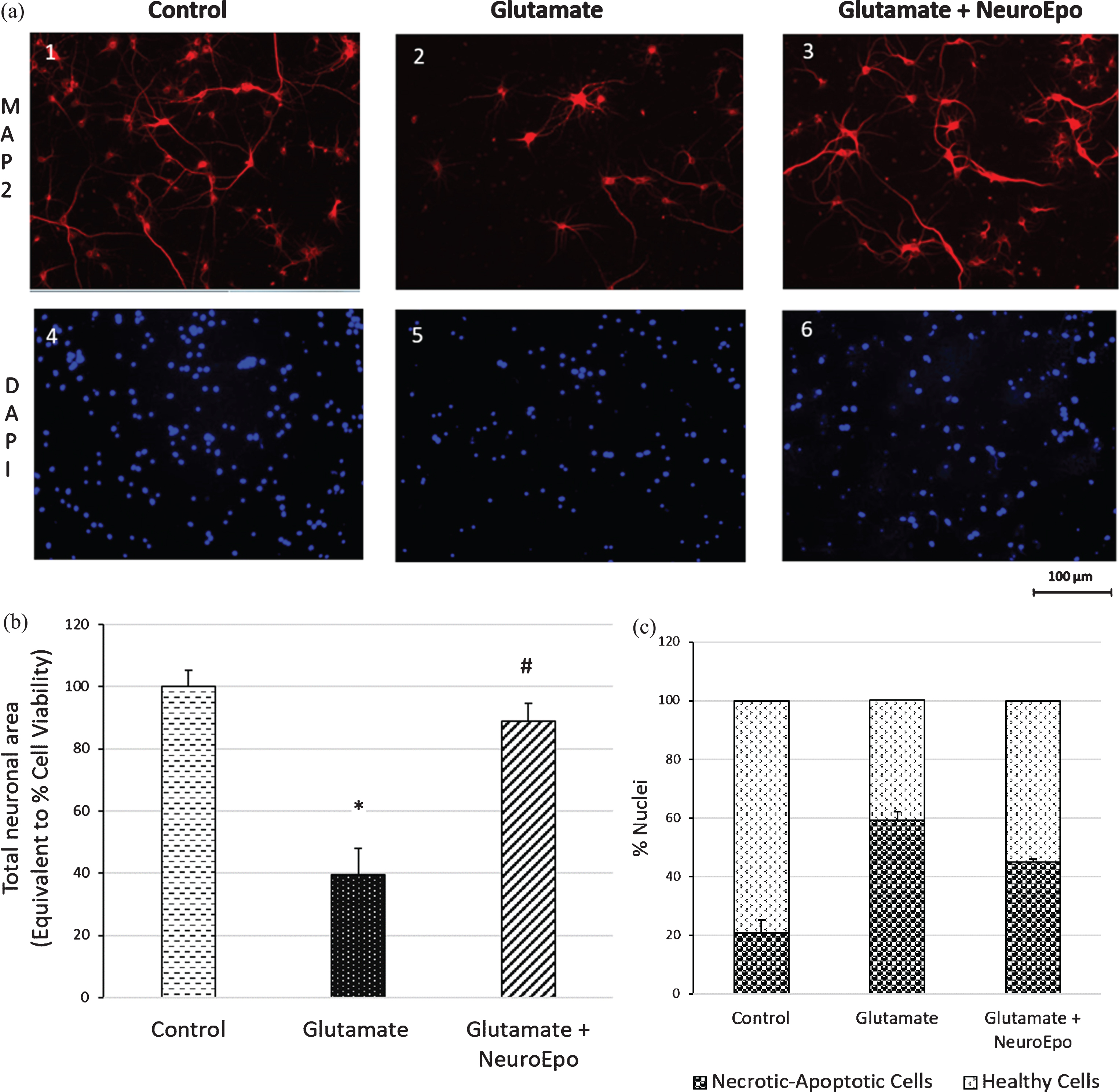
Protective effect of NeuroEPO against glutamate excitotoxicity: Biochemical analysis
Along with oxidative stress, the activation of the mitochondrial apoptotic pathway is another mechanism that plays an important role in neuronal death induced by glutamate excitoxicity. The permeabilization of the outer mitochondrial membrane is considered as “the point of no return”, which irreversibly leads to cell death [50]. An indirect way of determining alterations in the outer mitochondrial membrane is achieved by assessing the levels of proteins involved in the mitochondrial membrane.
One of the main accepted mechanisms of permeabilization of the mitochondrial membrane is the formation of pores by Bax and Bak proteins, which are found in the cytosol and in the outer mitochondrial membrane, respectively. When these two proteins are activated by an apoptotic stimulus, in our case by glutamate excitotoxicity, Bax is translocated from the cytosol to the mitochondria and forms a pore in the outer membrane, either alone or together with Bak, which allows the release of cytochrome c. This action in physiological situations is inhibited by anti-apoptotic proteins, mainly by Bcl-2, which is also found in mitochondria [51]. Intrinsic apoptotic pathways were assessed using Bcl-2/Bax expression. The results of our investigation show that the exposure of neurons to glutamate significantly decreased (*p≤0.05) the expression of Bcl-2 compared to the control (0.60±0.07 versus 1.09±0.08). The treatment with NeuroEPO to the culture of neurons pre-treated with glutamate significantly (#p≤0.05) reduces the decrease of Bcl-2 (1.23±0.11) (Fig. 5A). Treatment with glutamate induced a significant increase (*p≤0.05) in Bax expression compared to the expression of untreated neurons (control) (1.48±0.15 versus 1.08±0.05) (Fig. 5B). Subsequent treatment with NeuroEPO significantly reduced (#p≤0.01) the increase in Bax expression induced by glutamate alone (Fig. 5B). Compared with the control, a significant (*p≤0.05) decrease in Bcl-2/Bax ratio was induced by glutamate excitotoxicity (0.60±0.22 versus 1.08±0.27), which was normalized with the NeuroEPO treatment (1.22±0.37 versus 1.08±0.27) (Fig. 5C). Treatment with alone NeuroEPO increased significantly the Bcl-2/Bax ratio compared with untreated cells.
Expressions of anti-apoptotic (Bcl-2) and apoptotic (Bax) proteins induced by glutamate excitotoxicity in cortical neurons of rat. Cortical neurons were treated with 100μM glutamate for 5 min without or with NeuroEPO (100 ng/mL) for 24 h and with alone NeuroEPO. Western blots with homogenates of cortical neurons control, treated with glutamate, treated with glutamate and NeuroEPO and treated with NeuroEPO alone. Data are means±SEM values of 3 different samples/group from 3 independent experiments (n = 9). A) Treatment with glutamate decreased the Bcl-2 expression compared with control cells (*p < 0.05); treatment with glutamate and NeuroEPO reduced significantly (#p < 0.05) the decrease in Bcl-2 caused by glutamate. NeuroEPO alone induced a significant increase of the expression of Bcl-2 compared with control. B) Treatment with glutamate increased the Bax expression compared with control cells (*p < 0.05); treatment with glutamate and NeuroEPO reduced significantly (#p < 0.05) the effect caused by glutamate. NeuroEPO alone had no significant effect compared with control. C) Effect of glutamate excitotoxicity on the Bcl-2/Bax ratio. Treatment with glutamate reduced significantly the Bcl-2/Bax compared with untreated control group (p < 0.001); subsequent treatment with NeuroEPO reduces the effect of glutamate maintaining the Bcl-2/Bax ratio like to the level of control cells. Treatment with only NeuroEPO significantly increased the Bcl-2/Bax ratio compared to untreated cells.
The consequence of the alteration of the permeability of the outer mitochondrial membrane is the release of the cytochrome c protein to the cytosol where it activates the apoptosis cascade signaling. The results showed that the treatment with glutamate induced a significant increase (*p≤0.05) of the levels of cytochrome c in the cytosol compared with the levels observed in the control culture (1.54±0.20 versus 0.95±0.19) (Fig. 6). Subsequent treatment with NeuroEPO for 24 h caused a significant reduction in cytochrome c levels in the cytosol compared to the levels observed in cells exposed only to glutamate (Fig. 6).
Release of cytochrome c into the cytoplasm of cortical neurons. Cortical neurons were treated with 100μM glutamate for 5 min, followed without or with NeuroEPO (100 ng/mL) for 24 h. The levels of the cytochrome c were evaluated by analysis of data obtained from the Western blots. Glutamate induced significant increases of cytochrome c, while treatment with NeuroEPO prevented the increases of cytochrome c induced by glutamate. Data are means±SEM values of 3 different samples/group from 3 independent experiments (n = 9). *p < 0.05 compared with control, and #p < 0.001 compared with glutamate.
Finally, the activity and expression of caspase-3 was analyzed. A significant increase in caspase-3 activity is considered as the main effector of the pathway leading to the apoptotic death of the neuron. The assessment of caspase-3 expression showed a significant (*p≤0.001) increase in the expression of caspase-3 in cells exposed to glutamate compared to untreated cells (control) (2.21±0.20 versus 0.99±0.07). The presence of NeuroEPO post-treatment with glutamate significantly reduced the expression of caspase-3 compared to the levels observed in cells treated only with glutamate (1.82±0.17 versus 2.21±0.20) (Fig. 7A). More important, the caspase-3 activity, μM pNA, was significant increased (*p≤0.001) in cells exposed to glutamate compared with control (0.058±0.003 versus 0.031±0.001) (Fig. 7B). This effect of glutamate was significantly (#p≤0.05) reduced when NeuroEPO was added after the removal of the glutamate (0.036±0.001). NeuroEPO reduced both the activity as well as expression of caspase-3 compared with cells exposed to glutamate.
Glutamate increase the expression and activity of caspase-3. Cortical neurons were treated with 100μM glutamate for 5 min, followed without or with NeuroEPO (100 ng/mL) for 24 h. A) Glutamate induced significant increases of the caspase-3 expression, while that treatment with NeuroEPO prevented the increases of caspase-3 induced by glutamate. The levels of the proteins were evaluated by analysis of data obtained from the western blot method where β-actin was used as a loading control. The graphic represent densitometric western blot analyzes of at least three independent experiments (±SEM) n = 9. *p < 0.05). B) Glutamate induced a significant increase of the caspase-3 activity, while that treatment with NeuroEPO prevented the increases of caspase-3 induced by glutamate. Data indicates the amount of pNA products that were released. The results are from one representative experiment of three performed that showed similar patterns. Each point represents the means±SEM of three independent experiments. *p < 0.05.
DISCUSSION
The main findings of the study are: 1) NeuroEPO can substantially attenuate the morphological changes caused by glutamate-induced excitotoxicity in neurons; 2) NeuroEPO exposure reduces the apoptotic mechanisms involved in mitochondrial (intrinsic) pathway that are triggered when the excitotoxicity causes mitochondrial dysfunction; 3) NeuroEPO exerts its neuroprotective function acting directly on neurons; and 4) NeuroEPO can exert its neuroprotective activity once the glutamate excitotoxicity has been induced.
We found that NeuroEPO protects the survival of the primary culture of cortical cells from glutamate-induced death. We observed a decrease in the length and number of dendrites in cultures exposed to glutamate, as has been reported by previous studies [47, 52]. In contrast, treatment with NeuroEPO, after exposure of the neurons to glutamate, mitigates the morphological changes induced by glutamate. We also found modifications in the content and distribution of DNA from neurons exposed to glutamate. We found a greater number of nuclei with condensation of the chromatin in the cells exposed to glutamate while the treatment with NeuroEPO reduces the number of the nuclei with chromatin condensation. The morphological changes together with alterations in nuclear DNA are widely accepted as indicative of apoptotic and necrotic death [53, 54]. Since the results presented here have been obtained in a neuron-enriched cultures as was demonstrated by the expression of MAP-2 and the very low expression of GFAP, our results show that NeuroEPO can exert its neuroprotective action acting directly on the neurons. Taken together, these data indicate that glutamate excitotoxicity induce morphological changes, increase neuron death, and apoptosis in cortical cells culture and that NeuroEPO significantly attenuates the neurotoxic effects caused by excitotoxicity.
It has been suggested that the accumulation of calcium by the mitochondria, in response to the excessive influx of calcium induced by glutamate, promotes the loss of mitochondrial membrane potential owing to the alteration of the mechanisms that regulate the integrity of the outer mitochondrial membrane [55, 56]. Maintaining the mitochondrial membrane potential is essential for the mitochondria to perform its functions and prevent the activation of the intrinsic apoptotic pathway [57, 58]. The alteration of the mechanisms that regulate the mitochondrial permeability transition pore (MPTP) opening is considered to be the primary and important event leading mitochondrial dysfunction. Briefly, under physiological conditions, the mitochondrial membrane permeabilization is controlled by the B-cell lymphoma-2 (Bcl-2), a family of proteins with pro-apoptotic and anti-apoptotic activity that, through their complex interplay between with each other, function as the major regulators of the integrity of the mitochondrial outer membrane in face of apoptotic insults [59, 60]. The primary role of Bcl-2 family members is the regulation of apoptosis acting directly on the mitochondrial outer membrane. The anti-apoptotic proteins, mainly Bcl-2 and Bcl-xl, act on mitochondria by stabilizing membrane integrity and to prevent opening of the MPTP, whereas that various pro-apoptotic members (Bax and Bak) promote it. Neuronal apoptotic processes are initiated when anti-apoptotic Bcl-2 activity decrease and pro-apoptotic Bax activity increases. If this imbalance between anti-apoptotic and pro-apoptotic proteins is in favor on the second, causes the opening of the MPTP in the mitochondrial outer membrane, facilitating the release of cytochrome c from the mitochondria into the cytosol. Once in the cytosol, cytochrome c in the presence of (d)ATP binds with apoptotic protease-activating factor 1 (Apaf-1) forming an “apoptosome”, that recruits and activates caspase 9, which in turn activates caspase 3, the effector caspase of the intrinsic apoptotic pathway, ensuring cell death [58]. We found that glutamate excitotoxicity decreased expression of Bcl-2 and increased expression Bax resulting a significant change in the Bcl-2/Bax ratio. There is also increase of cytochrome c in the cytosol and increase of activity of caspase-3. All these are hallmark of apoptosis. The results that show that excitotoxicity causes the death of neurons by apoptosis have also been reported by others [28, 60]. We found that excitotoxic apoptosis was attenuated by treatment with NeuroEPO applied to neurons that had been exposed to glutamate. NeuroEPO mitigated glutamate-induced apoptosis by increasing Bcl-2 expression and reducing Bax expression keeping the Bcl-2/Bax ratio at levels similar to those found in neurons not treated with glutamate. Consequently, treatment with NeuroEPO reduced to normal levels the presence of cytochrome c in the cytoplasm and the activation of the executioner of apoptosis, caspase-3.
According to our results, NeuroEPO reduces the apoptotic effect induced by excitotoxicity through the mitochondrial intrinsic apoptotic pathway. Although NeuroEPO shows a reduction in the expression of the main markers of the pathway, including Bax, cytochrome c, and caspase-3, these effects may result from increased expression of anti-apoptotic protein Bcl-2. It is noteworthy that the increase in the expression of Bcl-2 induced by NeuroEPO in neurons treated with glutamate is also observed in untreated neurons. It is well known that Bcl-2 plays a pivotal role in maintaining the integrity of the outer membrane structure of the mitochondria [59, 60] counteracting the activation of pro-apoptotic proteins by regulating the permeabilization of mitochondrial outer membrane and consequently the release of cytochrome c into cytoplasm and the mitochondrial intrinsic apoptotic pathway. The integrity of the mitochondria implies the maintenance of its redox state and in this situation the mitochondria would cease to be the main source of ROS, necessary for the appearance of a state of oxidative stress. This would justify the hypothesis that the neuroprotective effect of NeuroEPO against situations of excitotoxicity would be through the induction of increased expression of Bcl-2. By preserving the integrity of mitochondria, Bcl-2 cancels the main source of ROS induced by glutamate.
In response to the overload of calcium in the mitochondria, there is an increase of expression of Bax that can to lead to mitochondrial dysfunction. In this situation, an increase in superoxide (O2
Our results have been obtained by using an in vitro model. Experimental in vitro models are often used to study morphological and biochemical changes related to degeneration processes because they allow an easier and precise control of the extracellular environment compared to in vivo models. However, as it happens with all studies carried out with cell cultures, it has a series of limitations that prevent the extrapolation of the results to in vivo conditions. First, we used primary cortical neurons culture with very low presence of glial cells. This condition has the advantage that it allows to associate the results to a direct effect of NeuroEPO on neurons, but it has the disadvantage that the interaction between neurons and glial cells is lacking. Second, NeuroEPO is immediately added after inducing the excitotoxicity and its effect is limited to 24 h. Without a doubt, with a view to a potential therapeutic application it would be interesting to know the therapeutic window effect of NeuroEPO. Another limitation refers to the efficacy of the use of cell cultures in the study of the mechanisms of chronic excitotoxicity because there are multiple and complex pathways involved in the chronic excitotoxicity that induce delayed death. On the other hand, although it is undoubted that EPO and its derivatives act on the cells through its interaction with the EPO receptor (EPOR), a study regarding the interaction NeuroEPO with EPOR expressed in neurons and the impact on signaling pathways must therefore be considered. The brain has mechanisms to maintain its functionality against factors that cause neurological damage. It is known that in response to brain injury are activated survival mechanisms, mainly antioxidants, anti-inflammatory cytokines, antiapoptotics, and growth factors, which tend to counteract the action of mechanisms that promote neuronal death: excitotoxicity, oxidative stress, inflammation, and apoptosis (Fig. 8). In this framework, after previous studies of the effect of NeuroEPO in animal models of stroke and Alzheimer’s disease with very promising results [20, 23], we considered it necessary to know the direct effect of NeuroEPO on neuronal damage in an excitotoxicity model, since excitotoxicity is a common pathophysiological mechanism that plays an important role in neurodegenerative diseases. The results show that NeuroEPO preserves neurons by activating survival mechanisms. In a previous study [47], we found that the neuroprotective effect of NeuroEPO against the oxidative stress induced by glutamate excitotoxicity was associated with increases of antioxidant activity without significant changes in the oxidant activity. This demonstrates that, like EPO [67, 69], NeuroEPO acts as powerful antioxidant keeping the cellular antioxidant activity, which allows neurons to regulate their intracellular redox condition. In this work we show that NeuroEPO participates in the mechanisms of survival inducing the expression of the anti-apoptotic protein Bcl-2 that leads to preserve the activation of the apoptotic pathway induced by glutamate excitotoxicity.
Diagram simplified of survival and death mechanisms in response to excitotoxic insults in neurons. Excessive glutamate activates mechanisms that mainly results in mitochondrial damage, cell membrane disruption, oxidative stress which act synergistically causing apoptotic or necrotic neuron death. These effects can be counteracted by NeuroEPO, activating survival promoters’ mechanisms, mainly antioxidants and antiapoptotic proteins.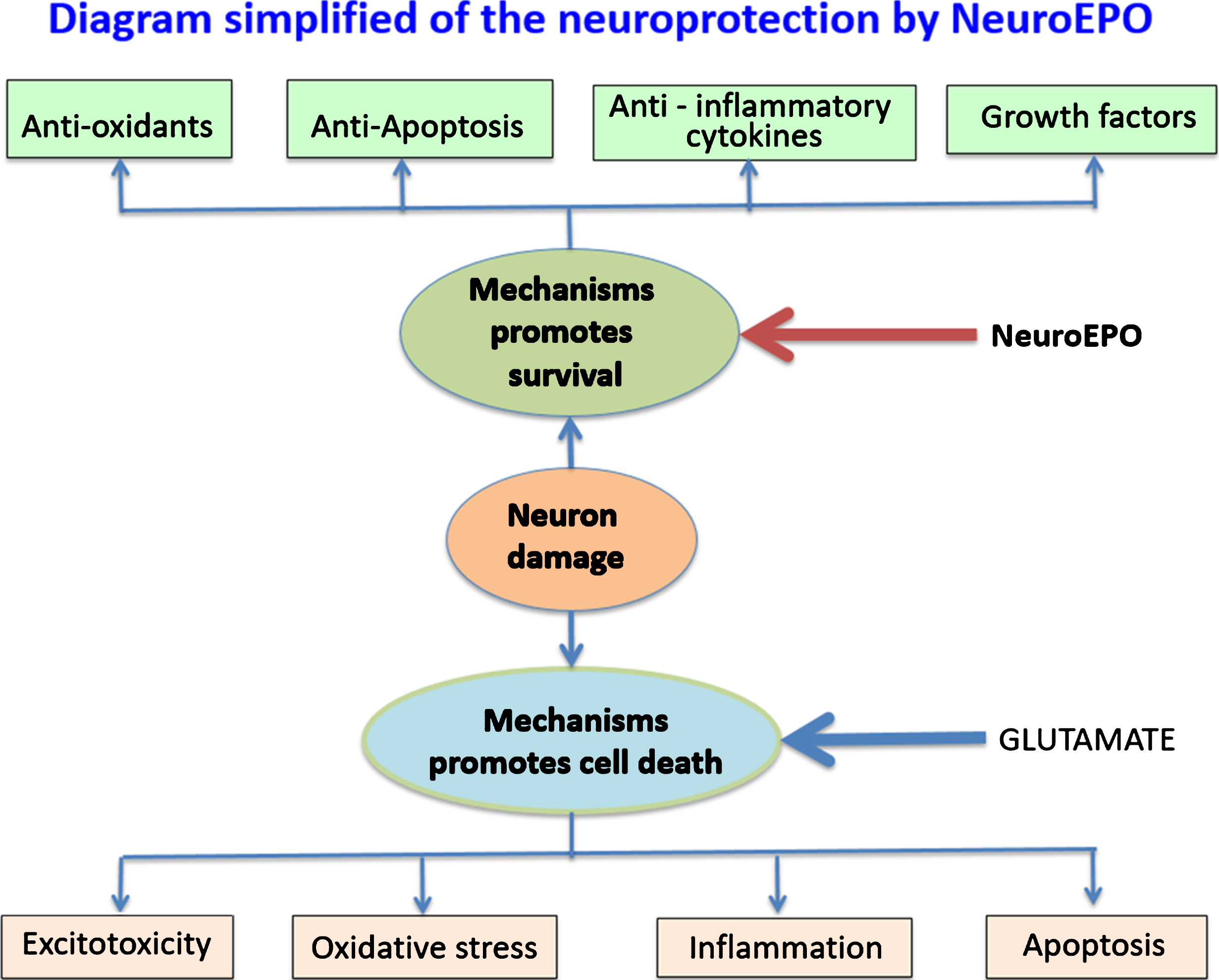
EPOR is a transmembrane receptor member of the type I cytokine superfamily, which is pre-formed as homodimers on the cell surface. The binding of EPO to its receptor induces conformational changes that cause phosphorylation by Janus kinase 2 (JAK-2) and activation of signaling pathways that include signal transducer and activator of transcription 5 (STAT-5), phosphoinositol 3′-kinase (PI3K), mitogen-activated protein kinase (MAPK), and protein kinase c (PKC) that eventually lead to protein regulation anti-apoptotic. However, the EPOR expressed in non-erythroid tissues is structurally different from the classical EPOR because it is a heterodimer composed of a monomer of the canonical EpoR and another subunit of the “β-common receptor”, also known as CD131 (EPOR-β comm), which is identical to the beta region of cytokine receptors such as interleukin-3 (IL3) receptors or granulocyte macrophage colony-stimulating factor (GMSCF) receptors [62]. Glycosylation of rhEPO affects the biological activity, immunogenicity, pharmacokinetics and in vivo clearance rate of rhEPO. Differences in glycosylation cause changes in the binding kinetics of the EPO receptor [63]. NeuroEPO and rhEPO differ markedly in their glycosylation. Surprisingly, this difference does not seem to affect the neuroprotective effects of NeuroEPO against neuronal damage induced by excitotoxicity.
Our results about of the effect of glutamate excitotoxicity on apoptosis in neurons culture are like to those already described by others [53, 64]. However, this study is so far the only one about the effect of NeuroEPO on apoptosis induced by glutamate excitototoxicity in primary cultured neurons. The effect of NeuroEPO found in the present study confirmed those reported in other studies. In animals, using an Alzheimer’s disease model, NeuroEPO mitigate apoptosis in the APPSwe [22, 23]. In that study, NeuroEPO prevents the induction of oxidative stress, the increase of Bax levels and of Bax/Bcl-2 ratio of mice not treated with NeuroEPO. As a consequence, the activity of caspase 3 was reduced. In a recent study with EpoL, an in silico derivative EPO also with low content of sialic acid as NeuroEPO, using PC12 cells as a neuronal model and carbonyl cyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP, a decoupling agent of the respiratory chain of electrons, which induces oxidative stress) or H2O2 instead of glutamate, as a model of oxidative stress, it was founded that EpoL has a neuroprotective effect, reduces oxidative stress and increases the expression of Bcl-2 [65].
The neuroprotective mechanisms described here for NeuroEPO have already been described for EPO [66, 67]. Like NeuroEPO, EPO protected cultured cortical neurons from glutamate-induced death [68]. Moreover, it has been already reported that EPO prevents cellular apoptosis through preserve mitochondrial function by activation of anti-apoptotic pathway through the up-regulation of Bcl-2 [11, 70].
Previous reports showed that neuroprotection by EPO against excitotoxicity in neuronal cultures requires pre-treatment with EPO at least several hours before exposure to glutamate [69, 72]. However, our results show that post-treatment with NeuroEPO has neuroprotective effects on neuronal damage induced by glutamate. This property of NeuroEPO is very important for its therapeutic application.
Although results obtained in cell cultures must be extrapolated with caution, our findings reported here support the abundant existing results that argue that EPO and derivatives contribute to neuronal survival against neuronal injury caused by excitotoxicity, a mechanism described as common in the most relevant neurodegenerative pathologies, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Taken together, our results indicate a possible use of NeuroEPO as a biosimilar to treat CNS diseases in which excitotoxicity plays an important role in its pathogenesis.
Footnotes
ACKNOWLEDGMENTS
We thank Drs. Daniel Amaro and Teresa Rodriguez (CIMAB (Habana, Cuba) for providing NeuroEPO and to Dr. E. Capilla for manuscript review and Dr. Pinto, T.C.C. Department of Neuropsychiatry and Behavioral Sciences, Federal University of Pernambuco (UFPE) Brasil for his help in the statistical analysis of the results. This work is a scientific collaboration between University of Barcelona and the University of Medical Sciences of Havana, Institute of Basic and Preclinical Sciences “Victoria de Girón”. This work was funded by University of Barcelona (RR) and by grant SAF2016-7426-R (MINECO) (ES).