
Abstract
Clarifying the relationships between neuropsychiatric symptoms and Alzheimer’s disease (AD)-related pathology may open avenues for effective treatments. Here, we investigate the odds of developing neuropsychiatric symptoms across increasing burdens of neurofibrillary tangle and amyloid-β pathology. Participants who passed away between 2004 and 2014 underwent comprehensive neuropathologic evaluation at the Biobank for Aging Studies from the Faculty of Medicine at the University of São Paulo. Postmortem interviews with reliable informants were used to collect information regarding neuropsychiatric and cognitive status. Of 1,092 cases collected, those with any non-Alzheimer pathology were excluded, bringing the cohort to 455 cases. Braak staging was used to evaluate neurofibrillary tangle burden, and the CERAD neuropathology score was used to evaluate amyloid-β burden. The 12-item neuropsychiatric inventory was used to evaluate neuropsychiatric symptoms and CDR-SOB score was used to evaluate dementia status. In Braak I/II, significantly increased odds were detected for agitation, anxiety, appetite changes, depression, and sleep disturbances, compared to controls. Increased odds of agitation continue into Braak III/IV. Braak V/VI is associated with higher odds for delusions. No increased odds for neuropsychiatric symptoms were found to correlate with amyloid-β pathology. Increased odds of neuropsychiatric symptoms are associated with early neurofibrillary tangle pathology, suggesting that subcortical neurofibrillary tangle accumulation with minimal cortical pathology is sufficient to impact quality of life and that neuropsychiatric symptoms are a manifestation of AD biological processes.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) features a long prodrome during which pathology accumulates without causing neurocognitive symptoms [1]. AD is characterized by the accumulation of neurofibrillary tangles (NFT) and amyloid-β plaques, as well as neuron loss [2]. Neuron loss is the best predictor of cognitive decline, followed by NFT burden [3, 4]. The Braak staging system is a highly reproducible scheme capturing NFT spread in cortical regions [5]. PET-tau imaging studies corroborate the validity of Braak staging in vivo [6]. In Braak stages I/II, cortical NFT are confined to the entorhinal cortex and parts of hippocampus. NFT spread to paralimbic cortices in Braak stages III/IV and finally reach the higher-order association and primary neocortex in Braak stages V/VI [5]. On average, the first signs of cognitive decline occur during Braak stage III, meanwhile most cases at Braak stages I/II remain cognitively normal [5, 7].
Longitudinal studies suggest that a higher prevalence of neuropsychiatric symptoms (NPS) exist years preceding cognitive decline in individuals who later develop AD-type dementia, compared to age-matched controls [8–22]. Some suggest that NPS such as depression and anxiety are risk factors for AD [23–27]. However, a growing number of investigations using biomarkers suggest that NPS are driven by AD-related neuropathological changes [28–36]. Nevertheless, the question of whether NPS are part of the AD spectrum or exist independently or as epiphenomena is still open, especially in precognitive stages.
In addition to the cortex, NFT are abundant in subcortical nuclei. In fact, NFT pathology onset in key brainstem and hypothalamic nuclei consistently precedes cortical NFT pathology, correlates with neuronal death, and increases in severity along Braak stages [12, 37–45]. In 2011, Braak and colleagues revised their staging system to include the onset of NFT pathology in subcortical nuclei as a precortical stage [42, 46]. Among the subcortical nuclei accumulating NFT at Braak stage 0 are the locus coeruleus, dorsal raphe nucleus, and perifornical nucleus of the hypothalamus, which modulate anxiety, depression, and sleep disturbances, respectively. This early subcortical involvement corroborates claims that NPS may be part of the constellation of early clinical symptoms, corresponding to stage 2 of the NIA-AA neurocognitive staging system, and that effective treatment of these symptoms will require targeting AD pathology [47]. However, direct validation of this hypothesis is missing. Determining if AD pathology underlie specific NPS at pre-cognitive stages has important implications, including the possibility of using specific NPS as outcomes for clinical trials targeting prodromal AD phases, or designing effective treatment for NPS associated with AD.
Previous investigations have used postmortem evaluation to examine the relationships between AD pathology and several NPS [12, 48–53], but they either were limited by a relatively small sample [48, 49], were enriched for individuals at moderate or severe disease stages with cognitive decline, rarely excluded cases on the basis of co-existing pathologies which are confounders, or focused on one or few NPS.
Studies focused on determining the neurobiological basis of prodromal NPS in AD are lacking, and the relationships between pathology and NPS remain unclear. Despite recent improvements, neuroimaging and fluid-biomarker studies remain insensitive to subcortical AD pathology, likely critical loci for studies of this nature. To examine our hypothesis that AD pathology constitutes a neurobiological basis of NPS, even at prodromal stages, we analyze a large, population-based clinicopathological series to examine the relationships between the burdens of protein hallmarks of AD— NFT and amyloid-β— and domains from the neuropsychiatric inventory (NPI) [54].
MATERIALS AND METHODS
Participants
The local ethical committees approved this study, and all informants signed an informed consent. Cases were sourced from the Biobank for Aging Studies from the Faculty of Medicine at the University of São Paulo (BBAS-USP), formerly known as the Brain Bank of the Brazilian Aging Brain Study Group [7, 55]. Cases were collected between 2004 and 2014 by a city autopsy service. The inclusion criteria of the BBAS-USP includes age at death of 50 years or older and availability of a knowledgeable informant with at least weekly contact with the deceased in the six months before death. Cases with inconsistent data or brain tissue incompatible for neuropathological analyses were excluded [55]. Out of the 1,092 available cases, we excluded all cases with any level of non-AD pathology, including Lewy bodies, TDP-43 inclusions, primary tauopathies, and cerebrovascular changes, bringing the sample to 455 cases with a broad gradient of AD-type pathology across a range of clinical statuses.
Evaluation of symptoms
Scores from the 12-item NPI were collected in semi-structured postmortem informant interviews and reflect the participant’s status three months before death to avoid influence of peri-agonal events [54]. The NPI evaluates 12 domains: agitation, apathy, anxiety, appetite, delusions, depression, disinhibition, elation, hallucinations, irritability, motor, and sleep. Scores are typically calculated by multiplying frequency (1–4) and severity (1–3) for domains with any disturbance. As the median domain scores of our participants are zero, this was set as the cutoff for a negative diagnosis, with any score above zero receiving a positive diagnosis. Functional cognitive status was collected using the informant version of the Clinical Dementia Rating Sum of Boxes score (CDR-SOB) [56, 57]. The informant version of the CDR-SOB has been validated in the Brazilian population [58].
Neuropathological assessment
The BBAS-USP uses a 14-region immunohistochemistry panel to detect neurodegeneration and universally-accepted criteria to stage and diagnose cases [7, 55]. NFT pathology was assessed in formalin-fixed paraffin-embedded sections by immunostaining for phospho-Ser396/Ser404 tau (PHF-1, 1:2000; gift from Peter Davies, New York), scored by Braak stage, encoded into four groups following the conventional categorization [5, 59]: Braak stages 0, I/II, III/IV, and V/VI. Amyloid-β pathology was scored using CERAD-NP for the density of neuritic plaques, scored as none, sparse, moderate, or frequent [60].
Statistics
One-way ANOVA and Chi-squared tests were used to compare demographic and clinical metrics across groups. NPS were assessed across groups using logistic regression. The independent variables were the four Braak groups and CERAD-NP, using “Braak stage 0” and “CERAD-NP None” as the reference groups. The dependent variables were the NPI score and each of the domains (binary: 0/>0). Logistic regression models assessed the odds of having a NPS for given pathologic groups compared to reference groups. Models were adjusted for CDR-SOB. We further adjusted models for age, sex, years of education, and the other hallmark (e.g., adjusting for CERAD-NP in the Braak stage model). Multicollinearity was examined using variation inflation factors, with factors less than ten accepted. Statistics were conducted using RStudio. The α-level was set at 0.05 for two-tailed tests.
A secondary analysis using conditional logistic regression in age-matched pairs of Braak stage 0 and Braak stage I-II cases (n = 198) was done to examine these relationships free of possibly confounding age-related effects. This model was corrected for sex, years of education, CDR-SOB, and CERAD-NP.
RESULTS
Demographic and clinical statistics for the study population by Braak group (n = 455)
p value determined using chi-square test unless otherwise stated;
p value determined using one-way ANOVA;
CERAD-NP scores were assigned as 0 for none, 1 for sparse, 2 for moderate, and 3 for frequent;
Agitation, delusions, and depression have one missing case each
Demographic and clinical statistics for the study population by CERAD-NP (n = 455)
p value determined using chi-square test unless otherwise stated;
p value determined using one-way ANOVA;
Agitation, delusions, and depression have one missing case each.
Odds ratios and 95% confidence intervals for neuropsychiatric symptoms by Braak stage group (n = 455)
Models adjusted for age, sex, years of education, CERAD-NP, and CDR-SOB. Reference group: Braak stage = None;
Agitation, delusions, and depression have one missing case each.
Odds ratios and 95% confidence intervals for neuropsychiatric symptoms by CERAD-NP score (n = 455)
Models adjusted for age, sex, years of education, Braak stage, and CDR-SOB. Reference group: CERAD-NP = None;
Agitation, delusions, and depression have one missing case each.
To examine these relationships free of potential age-related effects, we used a conditional logistic regression model in age-matched subgroups of the Braak 0 (controls) and Braak I-II cases corrected for sex, education, CDR-SOB, and CERAD-NP. In agreement with our primary analysis, increased odds were detected for agitation (OR, [95% CI] = 17.6, [1.61, 192.4]; p = 0.019), anxiety (OR, [95% CI] = 2.79, [1.08, 7.14]; p = 0.033), appetite changes (OR, [95% CI] = 2.87, [1.34, 6.17]; p = 0.006), and sleep dysfunction (OR, [95% CI] = 2.95, [1.13, 7.71]; p = 0.028). Although increased odds for depression was not significantly correlated with Braak stages I-II in this secondary analysis, a tendency for increased odds of depression remained (OR, [95% CI] = 3.81 [0.78, 18.5]; p = 0.098). Braak stage I-II did not correlate with higher odds for any of the other seven NPI domains, aligning with the primary analysis.
DISCUSSION
NPS are frequent across all AD stages, adding substantial burden for caregivers and patients [8–21]. By employing a large population-based postmortem sample of cases over 50 years of age, encompassing healthy controls and individuals across the AD spectrum, this study is well-positioned to investigate how NPS correlate with AD pathological hallmarks, particularly in precognitive stages. All cases were free of non-AD pathology, minimizing confounders introduced by co-occurring pathology. We found significant positive associations between NFT burden and several NPS, as early as Braak stages I/II, at which time the NFT are confined to subcortical and allocortical areas and, in most cases, the burden of amyloid-β plaques is very low (Fig. 1) [42, 61]. These results support the hypothesis that NFT-related neurodegeneration constitutes the biological basis of NPS in AD, beginning in pre-cognitive stages.
Increased odds for neuropsychiatric symptoms across Alzheimer’s disease neurofibrillary tangle progression. There are significantly higher odds of developing agitation, anxiety, appetite dysfunction, depression, and sleep disturbances during Braak stages I/II, when pathology is restricted to subcortical regions and the transentorhinal cortex, after correcting for age, sex, years of education, Braak stage, and CDR-SOB. After Braak stages I/II, the odds of anxiety, appetite dysfunction, depression, and sleep disturbances are not significant when correcting for CDR-SOB. Agitation remains at significantly increased odds during Braak stages III/IV. Increased odds of delusions are associated with Braak stages V/VI. This illustrates that neuropsychiatric symptoms could possibly be used as a clinical marker in stages preceding the onset cognitive decline. The trajectory for cognitive decline represents median CDR-SOB score (Table 1).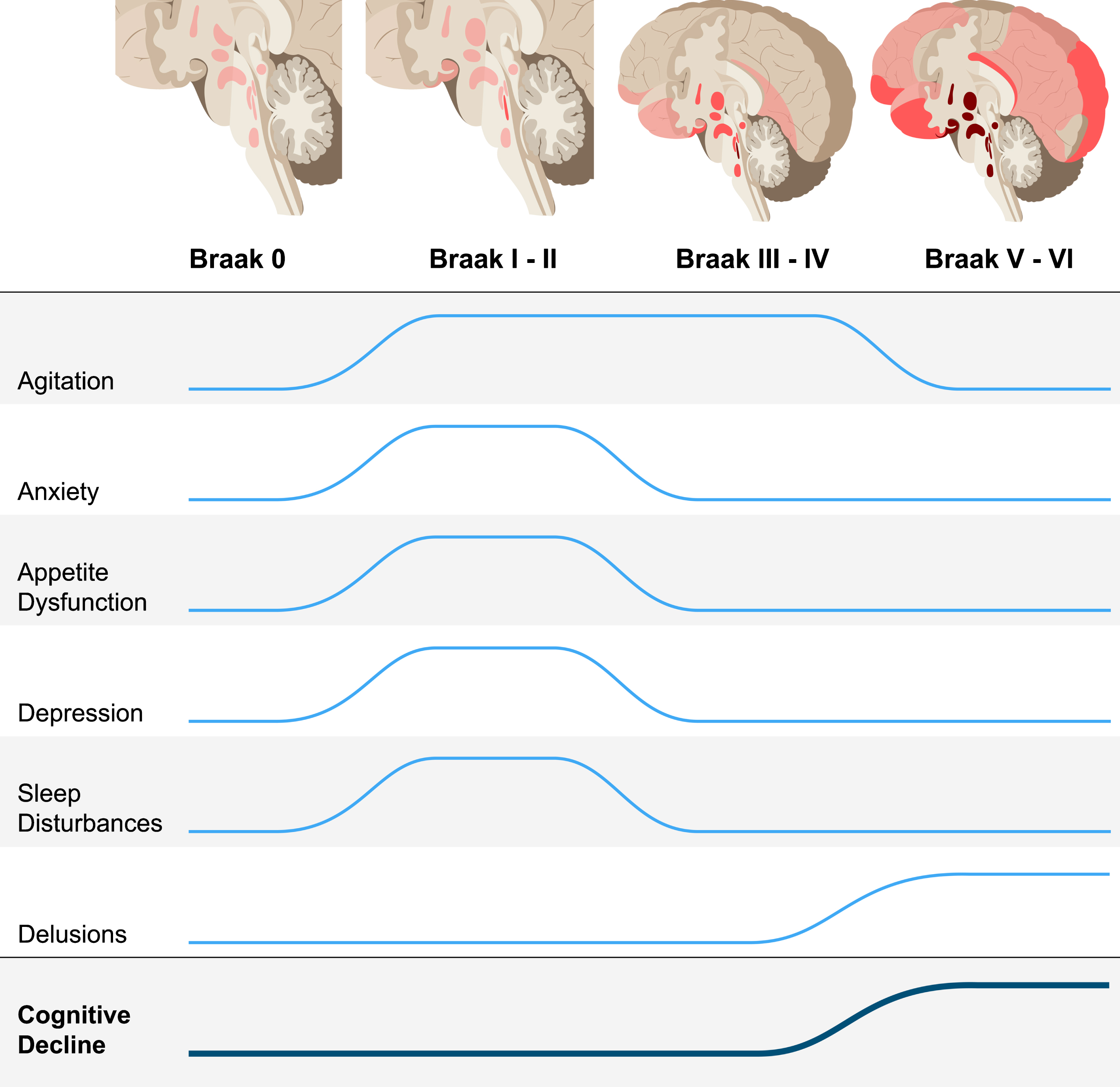
An increasing number of studies highlight that early vulnerability of brainstem and hypothalamic nuclei to NFT pathology occurs before involvement of the entorhinal cortex in AD [12, 38–44]. This burden of NFT pathology in subcortical nuclei increases with Braak stage. Taken together with the literature documenting the functional anatomy of these regions, our results showing that AD pathology may manifest first as NPS— agitation, anxiety, appetite dysfunction, depression, and sleep disturbances— are not surprising [12, 38–45]. Given the functional correlates of these regions affected by early NFT pathology, we can develop testable hypotheses to guide further research on mechanisms that drive early NPS in AD.
For example, dysregulation of norepinephrine due to early NFT accumulation in the locus coeruleus may contribute to the increased odds for agitation in Braak I–IV [62–64]. Several experimental studies have demonstrated increased agitation in response to dysfunction of norepinephrine-producing neurons, including those of the locus coeruleus [65–67]. In a review of neuroanatomical correlates of NPS in AD, Rosenberg and colleagues highlight studies correlating agitation with volumetric loss in the frontal cortex, anterior and posterior cingulate cortices, insula, amygdala, and hippocampus, rather than subcortical pathology [63]. Other studies detected a correlation between agitation and NFT burden in the orbitofrontal and anterior cingulate cortices in postmortem samples [48, 52]. While these studies suggest that NFT in these regions contribute to agitation, they were confined to examination of cortical regions in symptomatic individuals. Thus, it is challenging to tease out the contribution of early subcortical and cortical pathology with the patients at more severe disease stages with possible co-occurring pathology.
In fairly large clinicopathological studies, depression has been correlated with an increased burden of cortical plaques and tangles [50, 51]. Similar to agitation, NFT burden in the orbitofrontal and anterior cingulate cortices has also been correlated with depression, but this effect was only seen in later stages [48, 52]. Instead, early NFT-induced dysfunction of neurons in the locus coeruleus and dorsal raphe nucleus is likely driving our findings of increased odds for depression in Braak stages I/II, in line with the monoamine hypothesis of depression [53, 68–70].
Anxiety has been described as an early symptom in AD and a risk factor for dementia [26, 72]. Historically, the norepinephrine-producing locus coeruleus has been viewed as central in modulating networks involved in anxiety [73, 74]. So-called “anxiety cells” have also been identified in CA1 of the hippocampus, an early cortical site to accumulate NFT (Braak stage II) [5, 75]. Thus, both locus coeruleus and hippocampal involvement in Braak stages I/II align with our findings of increased anxiety at these stages [38, 41]. Donovan and colleagues have recently shown that anxiety is an early clinical manifestation of AD [36]. While this investigation was based on neuroimaging measurements of amyloid-β, the findings may be driven by concurrent subcortical NFT pathology, which could not be measured in vivo. Other neuroimaging studies correlate changes in the amygdala, insular cortex, and anterior cingulate cortex (Braak stage III) with several anxiety disorders in symptomatic patients, suggesting that these regions may also contribute to anxiety, in addition to earlier subcortical pathology [76, 77].
The ventromedial and lateral hypothalamus modulate satiety and hunger, respectively, as well as the locus coeruleus [78–83]. These nuclei are known to develop NFT early in AD, which may be driving the higher odds of appetite dysfunction we detect in Braak stages I/II, suggesting that there is a neuropathological basis driving dysfunction related to eating habits and preferences that has been associated with Alzheimer-type dementia [38, 84]. Further work is needed to refine the features of these appetite changes.
Many AD patients experience sleep disturbances even preceding cognitive decline [85–87]. Wakefulness is governed by high firing rates of wake-promoting neurons including neurons of the locus coeruleus, dorsal raphe nucleus, tuberomammillary nucleus, and lateral hypothalamus, all of which are susceptible to NFT in early AD [38, 88–90]. Recent evidence linking poor sleep to exacerbation of cortical pathology suggests existence of a positive-feedback loop leading to an overall increase in pathology [91, 92].
Finally, we detected high odds of delusions in subjects at Braak stages V/VI. Delusions manifest in several neurodegenerative conditions including Lewy body disease and frontotemporal dementia, and their anatomical correlates are complex [63, 93–95]. In AD, delusions have been associated with degeneration in the right inferior frontal gyrus, inferior parietal lobe, and claustrum, aligning with our findings of delusions beginning late in Braak stages V/VI [96]. As delusions are infrequent in healthy individuals and remain low until late stages, the odds ratio for delusions at Braak stages V/VI is very high, even after controlling for possible confounders (Table 1). Delusions in AD patients are often considered to be an indication of overlapping Lewy body disease [97]. However, in this series, any cases with α-synuclein positivity were excluded, suggesting that at late stages, delusions may be driven by cortical NFT pathology itself.
Clinical and epidemiological studies suggest a correlation between apathy and Alzheimer-type dementia [8–11, 19–21], but we failed to find such correlations here. Most studies of apathy in AD have been conducted in vivo, without autopsy verification. For this reason, they likely included mixed pathologies, such as Lewy body disease, which can increase the risk of apathy [31, 99]. Inflammatory dysregulation, which is not captured by Braak staging or CERAD-NP, is another possible underpinning of apathy [100–103]. The neuroanatomical basis of apathy in AD has also been suggested to include dysfunction of the anterior cingulate, dorsolateral prefrontal cortex, and left medial frontal cortex [48, 104]. Atrophy in the caudate, temporoparietal junction, temporal gyri, and frontal operculum-anterior insula region, detected by neuroimaging, has been associated with apathy in frontotemporal dementia [105]. Studies of apathy in AD focus on cortical changes and typically do not correct for cognitive status or co-pathologies which may explain the differences with our findings. Further, protein hallmarks such as NFT may only be related to mild apathy symptoms, to which the NPI-informant report may be insensitive, compared to direct clinical assessment.
The negative associations between amyloid-β severity, measured by CERAD-NP, and NPS have been observed previously [48, 106]. There are several possible explanations for this finding. In more advanced neuropathological AD stages, the severe cognitive impairment may overshadow NPS detection by informants. Additionally, in early AD stages, amyloid-β pathology is confined to neocortical regions that are less likely to modulate NPS than the subcortical regions already carrying substantial NFT burden, making possible correlations between amyloid-β and NPS very weak and the results artificial. Evidence that soluble amyloid-β pathology is a better predictor of clinical symptoms than plaques could also explain the negative relationships [107]. It is also possible that our correction of each model for Braak, due to the collinearity between CERAD-NP and Braak stage, could drive the negative relationships we are finding. Nonetheless, these findings add to mounting evidence that NFT, rather than amyloid-β plaque pathology drives the clinical phenotype of AD.
Despite our efforts to minimize weaknesses in the study design, remaining shortcomings should be noted. First, as neuroimaging studies have shown more robust associations between NPS and pathology longitudinally than cross-sectionally, the inherent cross-sectional design of clinicopathological studies prevents us from tracking domain-specific changes and longitudinal relationships among NPS. However, neuropathology remains the gold standard for diagnosing neurodegenerative disease; despite recent advances, methods for staging AD pathological markers in vivo fail to reach the same level of sensitivity and specific pathological prediction. This discrepancy is particularly prominent at early AD stages when tau burden is primarily subcortical. Second, clinicopathological studies such as this one are often criticized for being descriptive or correlational rather than mechanistic. However, clinicopathological studies play an important role in the advancement of medicine by providing the foundation and context for mechanistic studies. Third, despite universal use in clinical and research settings to evaluate NPS, NPI informant reports may not be sensitive to mild symptoms such as dysthymia. Furthermore, NPI scores are informant-dependent and thus, may be imprecise. To minimize potential bias related to this limitation, the BBAS-USP requires informants to have had close weekly contact with the deceased in the six months prior to death. Fourth, some NPS (i.e., hallucinations and elation) were infrequent in our sample, possibly yielding imprecise estimates for the association of these symptoms with neuropathology. Finally, a lack of correlation between amyloid-β plaque scores and NPS may be derived from bias caused by the metric used to assess amyloid-β burden. CERAD-NP measures the highest cortical density of amyloid-β neuritic plaques, which represents a different, albeit related, quality of amyloid-β than the anatomically-defined Braak stage represents for NFT. CERAD-NP scores have a strong ceiling effect and estimate plaque burden in only a small fraction of the brain. Despite this shortcoming, CERAD-NP is sensitive to the earliest brain area to develop neuritic plaques. In our sample, 63% of individuals lack any amyloid neuritic plaques, and the remaining cases represent all CERAD-NP scores. Thus, we would expect to find positive association with amyloid-β burden and NPS in early AD stages if existing. Moreover, extensive literature including clinicopathological and biomarker studies suggests little impact of amyloid-β pathology and several other cognitive metrics [7]. Thal phase is an alternative and perhaps more appropriate metric to use for measuring amyloid-β pathology; however, as this metric was only introduced in 2011, it is unavailable for the majority of our cases. Nevertheless, a need to investigate possible contributions of amyloid-β to NPS further remains.
This study also offers several considerable strengths. We studied a unique population-based clinicopathological series free of biases typically found in convenience samples that are usually enriched for later AD stages and oldest-old individuals. 28% of our large series of individuals over 50 years of age lacked cortical AD changes and another 44% had AD-tau changes limited to the limbic areas and low amyloid-β burden, which is unprecedented. Moreover, due to the comprehensive postmortem examination, we were able to exclude all cases with non-AD pathology to isolate the independent effects of AD-tau and AD-amyloid-β deposition, a feat not achievable by other available methods. Additionally, in the population sampled, the rates of antipsychotic and antidepressant use are low, which could otherwise mask NPS [108].
In this study, we identify increased risk for five NPS associated with NFT pathology, four of which were associated with early stages (Fig. 1). Our results strongly support the hypothesis that NPS are part of the AD clinical spectrum and are a manifestation of NFT-related neurodegeneration. Further research is needed to clarify the relationships between subcortical pathology and early NPS, as well as determine the prevalence of specific NPS subtypes. By understanding the pathophysiology driving early symptoms, better strategies can be developed to diagnose, target, and effectively treat AD before significant cognitive decline in advanced stages. In similar fashion, neuropathological evidence that Parkinson’s disease develops in non-dopaminergic nuclei, preceding substantia nigra involvement, recharacterized Parkinson’s disease as a multi-systemic disease with important non-motor components that precede motor symptoms. Such a change in paradigm opened room for improved management of non-motor symptoms and encouraged basic and clinical research to properly treat these symptoms. The positive outcomes from this revised view of Parkinson’s disease illustrate the importance of understanding the relationships between NPS and AD pathology and will hopefully encourage the field to follow suit.
Footnotes
ACKNOWLEDGMENTS
This study was supported financially by NIH Grant #R01AG040311 and the John Douglas French Alzheimer’s Foundation. Additional financial support was provided by FMUSP LIM-22, FAPESP, CAPES, and Hospital Israelita Albert Einstein, São Paulo. Dr. Grinberg mentoring effort is supported by NIH Grant #K24AG053435. Dr. Resende is an Atlantic Fellow for Equity in Brain Health at the Global Brain Health Institute and thanks the program for their support on this study. Dr. Gatchel has received funding from the BrightFocus Foundation and the Alzheimer’s Association and thanks them for their support.
We thank Shayna Herns (University of California, San Diego School of Medicine) for helpful editorial feedback, the families of the brain donors, the physicians, and staff of the São Paulo Autopsy service for their unconditional support, and the Brazilian Aging Brain Study Group for their assistance with data collection.