
Abstract
Microglia constitute the majority of innate immune cells in the brain, and their dysfunction is associated with various central nervous system diseases. Human microglia are extremely difficult to obtain experimentally, thereby limiting studies on their role in complex diseases. Microglia derived from human stem cells provide new tools to assess the pathogenesis of complex diseases and to develop effective treatment methods. This study aimed to develop a reliable method to derive human microglial-like cells (iMGLs) from induced pluripotent stem cells (iPSCs) expressing microglia-specific markers IBA1 and TMEM119 and respond to lipopolysaccharide (LPS) stimulation. Thereafter, we compared iMGL functions from Alzheimer’s disease (AD) patients and cognitive normal controls (CNCs). AD-iMGLs displayed stronger phagocytic ability with or without stimulation. High LPS concentrations (>2μg/ml) caused death in CNC-iMGLs, while AD-iMGLs did not display significant cell death. Cytokine analysis revealed that TNF-α, IL-6, and IL-10 secreted by AD-iMGLs were significantly increased upon LPS stimulation compared to those in CNC-iMGLs. The present results indicate that AD-iMGLs exhibit significant inflammatory characteristics and can reflect some pathological changes in microglia in AD, thereby providing new valuable tools to screen candidate drugs for AD and to elucidate the mechanisms underlying AD pathogenesis.
INTRODUCTION
Microglia are mononuclear phagocytes in the central nervous system (CNS) [1, 2]. They are widely distributed in the brain and spinal cord and play an important role in maintaining CNS homeostasis [3]. In the resting state, microglia carry out immune surveillance. They can be activated during brain diseases and injuries, and they carry out phagocytosis and secrete cytokines [4, 5]. Microglial dysfunction is associated with multiple neurodegenerative diseases, such as Alzheimer’s disease (AD) [6, 7]. Although microglial function and dysfunction are closely associated with numerous neurodegenerative diseases, their exact contributions to disease pathogenesis and progression are yet unclear [8].
Since the applications of primary human microglia are severely limited, most of our studies on microglia involve animal models and immortalized cell lines, both quite different from primary human microglia [9, 10]. The development of a reproducible source of human microglia via induced pluripotent stem cells (iPSCs) may hence present a viable method to resolve this issue [11–19]. Muffat et al. first induced iPSCs as an embryoid body (EB) in a component-determined serum-free neural differentiation medium mimicking human cerebrospinal fluid, supplemented with IL-34 and colony-stimulating factor 1 (CSF1). Cellular differentiation lasted 8 weeks [12]. Abud et al. and Amos et al. differentiated microglia from iPSC-derived hematopoietic progenitor cells or myeloid progenitor cells [14, 19]. Certain protocols also involve the co-culturing of neurons, astrocytes, etc., with microglial progenitor cells to mimic the in vivo environment, thereby further enhancing the phenotypic and functional characteristics of microglia. Pandya et al. first generated myeloid-like intermediate cells, and microglia were obtained after co-culture with human astrocytes [11]. Haenseler et al. and Takata et al. generated primitive hematopoietic progenitor cells and co-cultured these cells with iPSC-derived neurons to generate yolk sac macrophage-like cells with certain morphological and functional characteristics of microglia [15, 17].
AD is the most common neurodegenerative disease clinically characterized by progressive memory disorders [20]. The primary pathological changes in the brain in AD patients are the formation of numerous neurofibrillary tangles in neurons and the formation of extracellular senile plaques, with diffuse brain atrophy [21, 22]. Microglial activation reportedly plays an important role in AD progression [23]. Activated microglia can phagocytose and clear Aβ or Aβ–Aβ antibody complexes, thereby reducing Aβ-mediated neuro-cytotoxicity [24]; however, activated microglia can produce numerous biologically active substances, such as inflammatory cytokines, proteases, complements, oxygen free radicals, excitatory amino acids, nitric oxide, etc., to promote inflammatory reactions in the brain, resulting in neuronal damage and accelerating AD progression [25]. The present study aimed to optimize previously reported methods to stably and reproducibly obtain microglia differentiated from iPSCs-derived hematopoietic progenitor cells, which were further used to analyze AD-related pathological characterization. To elucidate phenotypic and functional changes in human microglia in AD, we used iPSCs from AD patients to induce microglia-like cells and used healthy human iPSCs as the control. Our results may provide a new and effective tool to analyze the pathomechanism and treatment of AD.
MATERIALS AND METHODS
Maintenance and culture of human pluripotent stem cells
Experiments including stem cells were performed with approval from the Ethical Committee. Human peripheral blood mononuclear cells (PBMCs) were used to for differentiation of iPSCs. Blood samples from AD patients meeting the eligibility criteria were collected from the Department of Neurology of Chinese People’s Liberation Army (PLA) General Hospital and Beijing Geriatric Hospital. AD patients and cognitively normal controls have been clinically diagnosed by neurologists and neuropsychologists in accordance with the NINCDS/ADRDA criteria. Sporadic AD was determined on the basis of family history and genetics. All donors provided informed consent. Human iPSC cell lines were generated by Shanghai IxCell Biotechnology Co, Ltd. (IxCell Co, Ltd, Shanghai). They transfected four transcription factors (Oct3/4, Sox2, c-Myc, and Klf4) into PBMCs in accordance with previously reported methods [26]. Pluripotency of all cell lines was confirmed. The iPSCs lines used in this study are enlisted in Table 1.
iPSCs lines used in this study
Table summarizing demographic information with each line. sAD, sporadic AD; —, No mutation detected. PSEN2 C.C261T, P.H87H, There is no clear report that the mutation increases the risk of the disease.
List of Antibodies uesed for immunofluorescent and flow cytometry analyses
iPSCs were cultured in 6-well plates (Corning) in feeder-free conditions, using hESC-qualified Matrix Matrigel (BD Bioscience, # 354277) in complete mTeSR™1 medium (STEMCELL Technologies, # 05850) in a humidified incubator (5% CO2, 37°C). iPSCs were cultured in fresh medium every day, and when the cells approached 70–80% confluence, they were passaged at a ratio of 1:3.
Immunofluorescence staining
After washing cells with PBS, they were fixed with 4% paraformaldehyde (PFA) for 30 min at 4°C. Prior to immunofluorescence staining, endogenous peroxidase of the cells was quenched in 1% hydrogen peroxide (H2O2) for 30 min and blocked and permeabilized with blocking buffer (PBS with 3% BSA, 0.1% Triton X-100) for 1 h at room temperature (RT). Thereafter, cells were stained for 2 h at RT with primary antibodies. After three repeated washes in TBST, cells were stained for 60 min with secondary antibodies. Cells were incubated with Cy3-Tyramide (Ty-Cy3) or fluorescein isothiocyanate (FITC)-Tyramide (Ty-FITC) for 15 min at RT. The nuclei were finally stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min at RT, followed by laser-scanning confocal microscopic observation (Nikon A1). Primary antibodies and secondary antibodies are enlisted in Table 2.
iPSCs karyotyping
The iPSCs were karyotyped using G-banded metaphase chromosomes with an approximate resolution of 300–400 bands per haploid genome, using standard procedures (Zhejiang Dian Diagnostics Co, Ltd. Zhejiang, China). The iPSCs were confirmed to display a normal karyotype (46, XX or XY).
RNA extraction and reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from iPSCs, using TRIzol® Reagent (Thermo Fisher Scientific, # 15596026) in accordance with the manufacturer’s instructions. Contamination in the genome was eliminated using the TURBO DNA-free™ Kit (Thermo Fisher Scientific, # AM1907). cDNA was synthesized using the SuperScript® VILO™ cDNA Synthesis Kit (Thermo Fisher Scientific, # 11754050). Target genes were amplified using Jump Start TMRED Taq® Ready Mix TM Reaction Mix (Sigma, # P2893-100RXN). The primers used for PCR were as follows: KOS F (5’-ATG CAC CGC TAC GAC GTG AGC GC -3’) and R (5’- ACC TTG ACA ATC CTG ATG TGG-3’); KLF4 F (5’- TTC CTG CAT GCC AGA GGA GCC C -3’) and R (5’- AAT GTA TCG AAG GTG CTC AA -3’); c-MYC F (5’- TAA CTG ACT AGC AGG CTT GTC G -3’) and R (5’- TCC ACA TAC AGT CCT GGA TGA TGA TG -3’).
EB assay in vitro
Cell aggregates were cultured at a density of 105 cells/ml onto a 60-mm bacterial grade culture dish containing 5 ml of EB Formation Medium (STEMCELL, # 05893). The culture dish was incubated, taken out twice a day, and gently agitated to suspend the cells. Half of the medium was changed every two days. Usually, on the third day of culturing, cell aggregates began to stratify, with an external layer of larger endoderm-like cells and an internal core comprising ectodermal-like cells and stem cells. After an additional 8 d of culture, stratification increased and a cavity was observed, and the entire embryo was spherical, which was a saccular embryo. Immunofluorescence was performed to identify ectodermal (TUJ-1), mesodermal (SMA), and endodermal (AFP) markers.
Differentiation of iPSCs to iMGLs
Human iPSC-derived hematopoietic progenitors (iHPCs) were generated using the STEMdiff™ Hematopoietic Kit (STEMCELL, # 05310). iMGLs were generated under defined conditions with several modifications in accordance with previous protocols [14].
iHPCs were harvested using MACS (Miltenyi, # 130-091-333) in accordance with the manufacturer’s instructions. Isolated CD43+ iHPCs were cultured at 2.0×105 cells onto Matrigel-coated 12-well plates in 1 mL of differentiation media (Microglia Medium (ScienCell Research, # 1901), 2% B27 (Thermo Fisher Scientific, # 17504044), 0.5% N2 (Thermo Fisher Scientific, # 17502048), 1X Glutamax (Thermo Fisher Scientific, # 35050061), 1X NEAA (Thermo Fisher Scientific, # 11140050), 25 ng/ml GM-CSF (R&D Systems, # 215-GM), 25 ng/ml M-CSF (R&D Systems, # 216-MC), 50 ng/ml IL-34 (R&D Systems, # 5265-IL), 50 ng/ml TGF-β1 (R&D Systems, # 240-B), and 25 ng/ml IGF-1 (R&D Systems, # 291-G1). Each well was supplemented with 0.5 mL of differentiation media every 2 d.
Flow cytometry
iHPCs were suspended in buffer (1X DPBS, 2% BSA, and 0.05 mM EDTA) and washed with buffer (300× g for 5 min). Thereafter, cells were stained with APC-anti-human CD43 and PE/Cy7-anti-human CD34 antibodies along with their corresponding isotype controls for 1 h at 4°C. After staining, iHPCs were washed once and suspended using 500μl buffer and analyzed using the BD FACSARIA Fusion system (BD Biosciences).
Time-lapse microscopic imaging and the cell growth curve
Cells were plated in well plates and incubated in the IncuCyte Zoom Live Cell Imager (Essen Bioscience) to obtain real-time images of cells, and the basic analysis software (Essen Bioscience) was used to automatically determine cell confluence, thus reflecting the number of cells.
Phagocytosis assays
iMGLs were plated onto 96-wells at a density of 3.0×105 cells/ml in Microglia Medium in a 37°C and 5% CO2 incubator. After 24 h, media were aspirated and 100μl of Microglia Medium containing different concentrations of LPS was added in each well. LPS concentrations ranged from 0.5μg/ml to 2.0μg/ml. After 24 h, media were aspirated and 100μl of PBS containing 0.05% neutral red was added and cells were cultured for an additional 30 min. Thereafter, the supernatant was aspirated, cells were washed thrice with PBS, 100μl of cell lysate (V (glacial acetic acid): V (ethanol) = l: 1) was added to each well, and the plates were incubated at 4°C for 2–3 h, followed by measurement of the optical density at 540 nm using a microplate reader after cell lysis.
Enzyme-linked immunosorbent assay (ELISA) for TNF-α
iMGLs were incubated in 96-well plates in Microglia Medium for 24 h and activated with LPS for 24 h. Cell culture supernatants were probed using the Human TNF-α ELISA Kit (NEOBioscience, # EHC103) in accordance with the manufacturer’s instructions.
Luminex analysis of cytokines
Levels of human IL-17A, IL-2, IL-6, TNF-α, IL-10, IL-1β, IFN-γ, and IFN-α2 were determined using Luminex assay kits (Millipore, #HCYTOMAG-60K) in accordance with the manufacturer’s instructions after stimulation with LPS.
RESULTS
iPSCs displayed stem cell-like characteristics
We investigated the characteristics of iPSCs from all subjects, all displaying typical characteristics of pluripotent stem cells, being morphologically similar to embryonic stem cells and expressing embryonic stem cell markers such as Nanog, Oct4, Sox2, and SSEA4 (Fig. 1a). iPSCs had a normal karyotype (Fig. 1b). Programmed iPSCs harboring exogenous transcription factors were also completely degraded (Fig. 1c). In vitro analysis of EB differentiation revealed that all cell lines can differentiate into the three germ layers, i.e., the ectoderm, mesoderm, and endoderm (Fig. 1d).
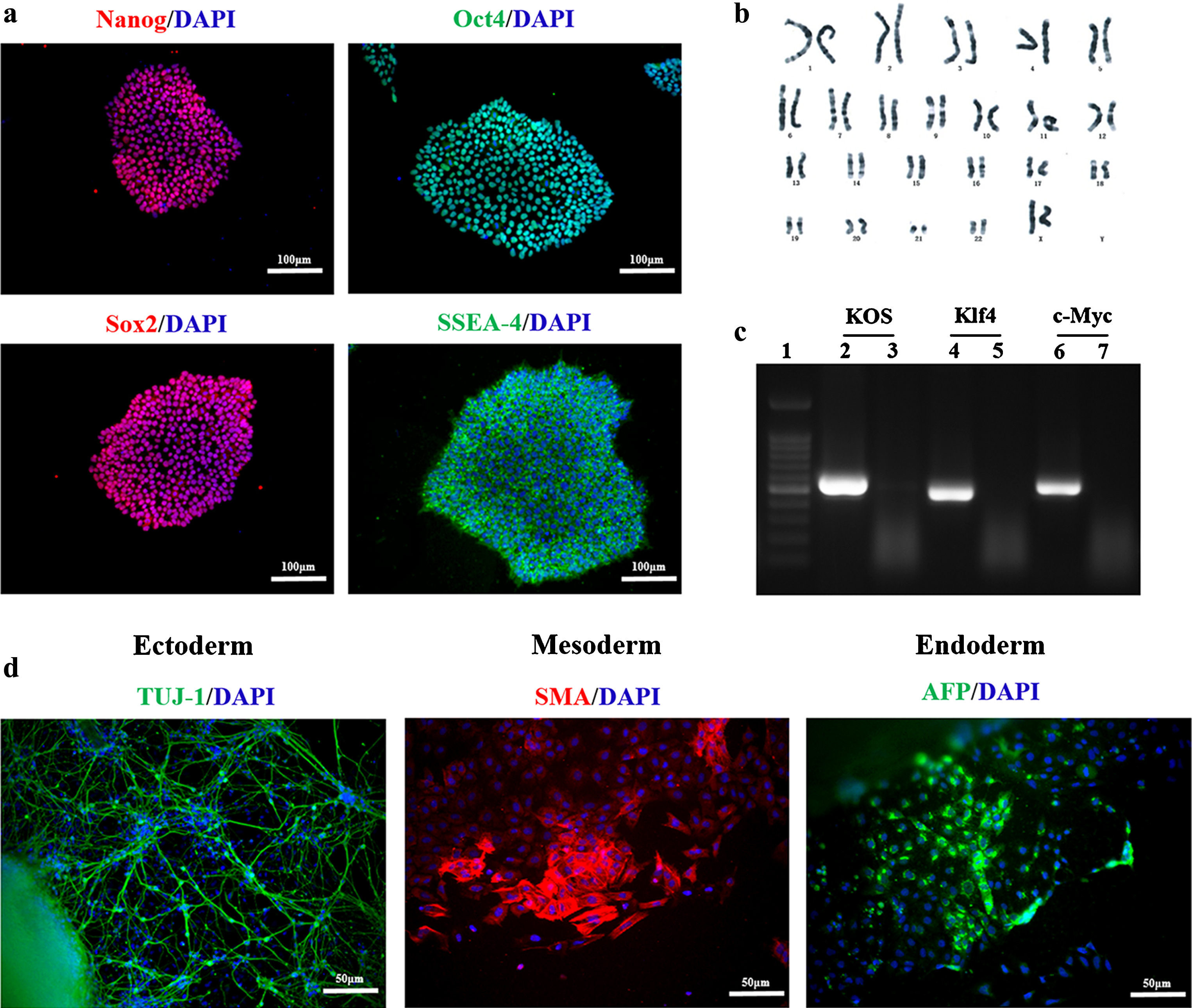
iPSCs expressing stem cell markers and displaying differentiation potential. Immunofluorescence staining for the stem cell markers NANOG (red), OCT4 (green), SOX2 (red), and SSEA4 (green) (a). Karyotype analysis confirmed a normal karyotype for iPSCs (b). Transcript levels of endogenously expressed pluripotency-associated genes in iPSCs determined via reverse-transcription polymerase chain reaction analysis. 1) DNA Ladder; 2) KOS positive control; 3) KOS disappeared in the 12th generation; 4) Klf4 positive control; 5) Klf4 disappeared in the 12th generation; 6) c-Myc positive control; 7) c-Myc disappeared in the 12th generation (c). Immunofluorescence labeling for TUJ-1 (ectoderm, green), SMA (mesoderm, red), AFP (endoderm, green) (d). (a, d-cell nuclei labeled with DAPI(blue). Scale bar for a-50μm, d-100μm).
iMGLs can be derived from human iPSCs
We used the iPSCs from young subjects to establish a method of induction. We initially used hematopoietic progenitor cells for microglial differentiation. To ensure the stability of induction, we used a commercial hematopoietic progenitor induction kit to induce differentiation of iPSCs into hematopoietic progenitor cells (Fig. 2a). After induction for 12 d, CD43+ cells approached 68% confluence (Fig. 2b). After magnetic bead sorting, CD43+ cells were induced to differentiate into microglia in media supplemented with GM-CSF, M-CSF, IL-34, IGF-1, and TGFβ-1, and iMGLs could be harvested after approximately 25 d of induction. On day 25, iMGLs stably expressed IBA1 and TMEM119 microglial markers (Fig. 2c). Cells were immunostained for IBA1 and TMEM119 on days 10 and 20 during induction. On day 10, microglia expressed IBA1, but not TMEM119; day 20, both IBA1 and TMEM119 (Fig. 2d).

iPSCs differentiated into iPSC-derived microglia-like cells (iMGLs) from hematopoietic progenitor cells. A diagrammatic representation of the major steps in microglial differentiation from human iPSCs (a). Changes in CD34 and CD43 expression between days 0 and 12 during the hematopoietic progenitor induction stage (b). Immunofluorescence labeling for IBA1 (red) and TMEM119 (green) on day 25 (c). Immunofluorescence labeling for IBA1 (red) and TMEM119 (red) on days 10 and 20, respectively (d). Data were from young healthy individuals. White boxes indicated the areas of the magnified insets. c, d-cell nuclei labeled with DAPI (blue). Scale bar for c, d-50μm.
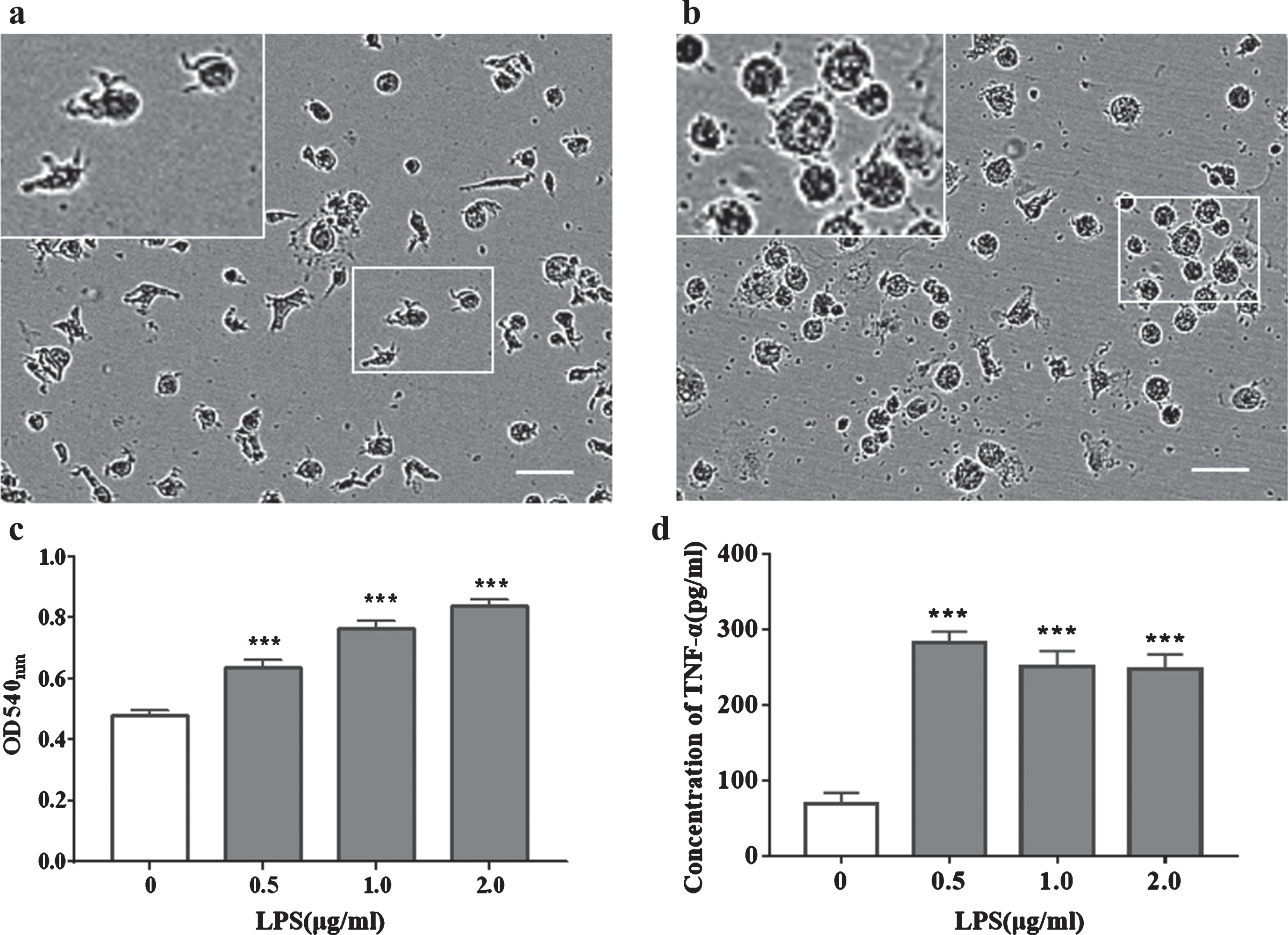
LPS-induced activation of iMGLs with morphological changes, increased phagocytosis, and increased TNF-α secretion. Bright-field microscopic image of iMGLs in the resting state (a). Bright-field microscopic image of iMGLs after 24 h of treatment with 1μg/ml LPS (b). Phagocytic ability of iMGLs, and LPS treatment increased phagocytosis. The ordinate shows the absorbance of neutral red (c). iMGLs secreted TNF-α, and LPS treatment increased TNF-α secretion (d). Data were obtained from young healthy individuals. White boxes indicate areas of the magnified insets. Scale bar for a, b-100μm. c, d-bar graphs presented as mean±SEM values. Data were analyzed using one-way ANOVA, followed by Dunnett’s multiple comparisons test (The experiments performed in triplicate); ***p < 0.001, compared with 0μg/ml LPS group.
iMGLs can respond to LPS stimulation
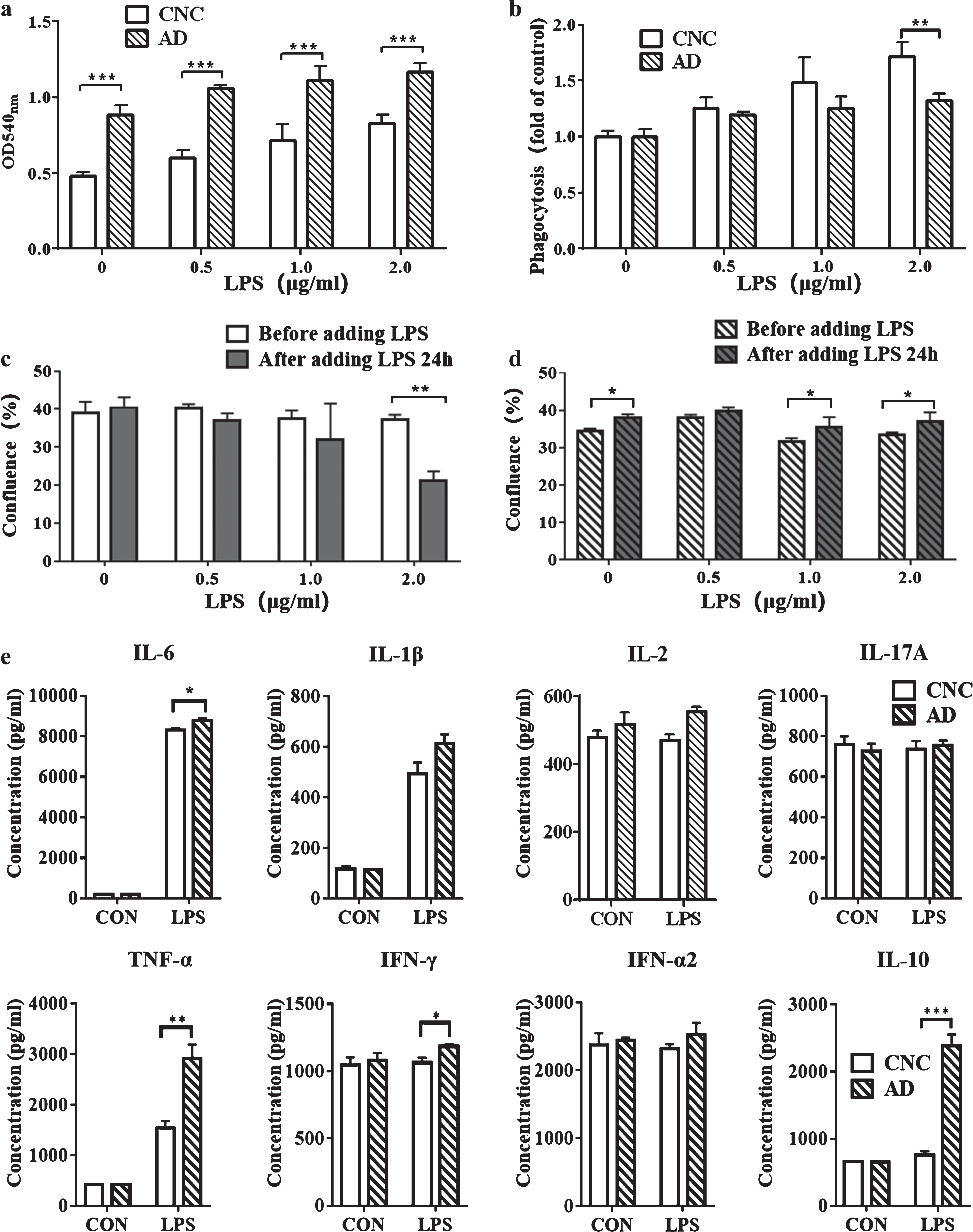
AD-iMGLs showed more pronounced inflammatory characteristics than CNC-iMGLs. Comparison of phagocytic ability between AD-iMGLs and CNC-iMGLs (a). The degree of enhancement of phagocytosis of AD-iMGLs and CNC-iMGLs after LPS treatment. The ordinate was the ratio of absorbance after stimulation to absorbance without stimulation (b). The confluence rate of CNC-iMGLs before LPS stimulation and after 24 h of LPS treatment (c). Confluence rate of AD-iMGLs before LPS stimulation and after 24 h of LPS treatment (d). Cytokines were secreted by CNC-iMGLs and AD-iMGLs after treatment with 1μg/ml LPS (e). Bar graphs display data as mean±SEM values. Data were analyzed using two-way ANOVA, followed by Sidak’s multiple comparisons test (The experiments performed in triplicate); *p < 0.05, **p < 0.01, ***p < 0.001.
We examined the function of iMGLs from young subjects and observed changes in iMGLs upon LPS-induced activation. Normally, most iMGLs exhibited a ramified morphology (Fig. 3a). After 24 h of LPS addition, almost all cell branches were shortened and showed an amoeboid activation pattern (Fig. 3b). iMGLs could perform phagocytosis, and LPS stimulation enhanced phagocytosis in a dose-dependent manner (Fig. 3c). Simultaneously, iMGLs also secreted the proinflammatory cytokine TNF-α. Upon LPS stimulation, TNF-α secretion increased albeit in a non-dose-dependent manner (Fig. 3d).
Pathological changes in AD-derived iMGLs
We compared the phagocytic ability of CNC-iMGLs with that of AD-iMGLs via natural red imaging of phagocytic cells, and observed changes in iMGLs undergoing phagocytosis under different concentrations of LPS stimulation (Fig. 4a). In the absence of LPS, phagocytosis was significantly increased in AD-iMGLs rather than in CNC-iMGLs. LPS stimulation enhanced phagocytosis in both AD-iMGLs and CNC-iMGLs in a dose-dependent manner, phagocytosis being more pronounced in AD-iMGLs upon stimulation. However, by comparing the enhancement in microglial phagocytosis upon LPS stimulation, phagocytosis was enhanced to a lesser extent in AD-iMGLs than in CNC-iMGLs (Fig. 4b). We recorded the confluence rates of cells via IncuCyte before and after 24 h of LPS stimulation. Interestingly, with an increase of LPS concentration, CNC-iMGLs underwent increased cell death. However, after LPS stimulation, the number of AD-iMGLs cells increased and demonstrated greater tolerance to high concentrations of LPS stimulation (Fig. 4c, d). Furthermore, cytokine secretion by iMGLs was quantified after stimulation with 1μg/ml LPS. Compared with the control group, AD-iMGLs secreted more IFN-γ, TNF-α, IL-6, and IL-10 after LPS stimulation (Fig. 4e). These factors are closely associated with the inflammatory response, suggesting that AD-iMGLs have a more pronounced inflammatory phenotype.
DISCUSSION
Microglia are central to neuronal viability, maturation, and activity, pruning and formation of synapses, and development and maintenance of neuronal circuits in the brain [27]. Microglial dysfunction has been implicated in various neurodegenerative disorders, such as AD. Difficulties in isolating primary microglia have led to attempts to generate microglia-like cells from hematopoietic stem cells or circulating monocytes [28, 29]; however, the lack of a reliable method of their maintenance in a microglia-like state characterized by the expression of endogenous microglial markers and displaying endogenous microglial function. The use of human embryonic stem cells or iPSCs to generate the desired differentiated microglia in vitro may resolve these issues. Microglia originate in the yolk sac and mature in the early stages of development from the primitive hematopoietic process, unlike many other types of macrophages that develop from definitive hematopoiesis [30]. Microglial induction has always been challenging, and the lack of tools to distinguish microglia from other monocytes has limited the generation of microglia from iPSCs. In 2016, Bennet et al. reported transmembrane protein 119 (Tmem119) [31], which encodes a cell-surface protein of unknown function. As a unique microglial marker in mouse and humans, this enabled microglial generation from stem cells.
On summarizing the existing differentiation protocols, some iPSCs first differentiated into CD235+ primitive hematopoietic cells and were then used as a starting point for microglia induction. Other microglia were derived from defined hematopoietic progenitor cells. On comparing the duration of induction, microglial differentiation required at least 4 weeks, usually 5–8 weeks. On comparing the yield of induced microglia, other than the protocol devised by Abud et al., who amplified microglia 10-fold from 1×106 iPSCs, other reported protocols started with 1×106 iPSCs, and microglial yield was mostly between 1×106 and 4×106. Induced microglia exhibit limited proliferative potential.
Considering the yield and differentiation time of microglia, we used the protocol of Abud et al. to generate iMGLs, with certain modifications. We used commercial hematopoietic progenitor cell induction medium to induce iPSCs differentiation into iHPCs, which in turn induced iPSC differentiation without the formation of EBs. We ensured that the differentiated iHPCs were stable and homogeneous. Stable and functional iHPCs were more likely to differentiate into iMGLs. Furthermore, we added growth factors GM-CSF and IGF-1, replaced DMEM/F12 with Microglia Medium as the base culture. Microglia Medium contained serum and could maintain primary microglial growth without the need for additional factors. Serum provided greater nutritional support for cellular growth. GM-CSF promoted microglial proliferation [32]. IGF-1 was considered a pro-survival factor for glial progenitor cells and prevents apoptosis [33]. iMGLs exhibited microglial morphology, expressed microglial surface proteins, and displayed microglia-like cellular behavior; furthermore, they displayed stable growth for more than 1 month. We observed changes in the microglial phenotype and function in AD-iMGLs by using iMGLs from sporadic AD patients. AD-iMGLs exhibited typical inflammatory characteristics. Aβ in the brain of AD patients activates the nucleotide binding and oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome in microglia [34], which is necessary for the secretion of pro-inflammatory cytokines. Therefore, microglia in the brain of AD patients secretes more pro-inflammatory factors [35, 36], concurrent with the present results.
Phagocytic ability is a typical microglial characteristic. When the central nervous system is stimulated by factors such as inflammation, infection, and trauma, microglia are rapidly activated and they phagocytose apoptotic neurons, synapses, and cellular debris. As the brain’s primary immune cells, microglia are activated in AD [37, 38]. Microglia have different phagocytic ability at different stages of AD. The role of microglial phagocytosis in different stages of AD in yet unclear. In the early stages of AD, microglia actively play a role in phagocytosis, thereby promoting AD pathogenesis because activated microglia can phagocytose live neurons [39, 40]. However, in the later stages of AD, microglial phagocytosis is significantly reduced [41]. In the present study, the phagocytic ability of AD-iMGLs was significantly higher than that of normal controls, suggesting that AD-iMGLs exhibit early-stage AD; this warrants further experimental verification. Furthermore, enhancement of phagocytosis in AD-iMGLs was less than that in CNC-iMGLs, indicating that AD-iMGLs may already be active, thereby displaying a less pronounced response to LPS.
Furthermore, in the present study, upon high-dose LPS stimulation, CNC-iMGLs underwent cell death, while the number of AD-iMGLs increased. Microglia secrete proinflammatory cytokines after inflammatory injury. Intracerebral LPS injection reportedly leads to microglial death [42]. However, microglia secrete growth factors such as nerve growth factor (NGF), neurotrophin-3 (NT-3), and brain-derived nerve growth factor (BDNF) before death [43, 44], thereby facilitating the survival of injured neurons. Therefore, microglial death can protect neurons. Apoptotic death in activated microglia is a suggested mechanism for resolving brain inflammation; however, methods of regulating microglial death after activation are unknown. P2X7 activation is reportedly associated with the induction of microglial death [45, 46]. Furthermore, numerous studies have reported the up-regulation of P2X7 in AD [47–49]; however, it is unclear whether this up-regulation is due to P2X7 dysfunction or stress upregulation in AD. Apoptosis-related disorders that activate microglia in the brain of AD patients may be associated with P2X7 function; nonetheless, further experimental verification is required. AD-iMGLs survive under high-dose LPS stimulation and can continuously secrete pro-inflammatory cytokines, chemokines, reactive oxygen species, and iNOS, and may further lead to chronic inflammation in the nervous system, thereby aggravating AD progression.
We generated iMGLs and sued them to further characterize AD. To obtain the desired number and state of iMGLs, the experimental conditions must be strictly controlled. Increasing the number of experimental samples (not just the cell lines listed herein), inter-individual differences in cells yield, survival time, and cell state were observed. These differences may be associated with various factors, such as personal genetic background, disease status, sex, age, methods for reprogramming iPSCs, etc. [50, 51]. iPSCs from different individuals reportedly exhibited distinct gene expression patterns, proliferation rates, and hematopoietic developmental potential [52]. The major advantage of iPSCs is that they harbor genetic information of the patient, and each cell is representative of one patient; hence, iPSCs have received increasing attention as a tool for assessing complex diseases.
The limitation of this study is that it is unclear whether changes in cellular phenotype and function observed herein are pathological or genetic; this warrants further investigation in future studies.
In conclusion, iPSCs have great potential in organismal development, healthcare, and disease research, opening novel avenues and interesting challenges. We believe that renewable resources of this type will help further resolve these issues.