
Abstract
We report the case of two monozygotic twins with Thr272fs mutation in progranulin gene. Both patients developed frontotemporal dementia with 5 years difference in age at onset (Twin 1:73 years, Twin 2:68 years), with early behavioral, language, dysexecutive, and memory problems. They had the same formal education (5 years), but while Twin 1 dedicated more to social and leisure activity, Twin 2 worked all her life. At neuroimaging (MRI for Twin 1 and CT for Twin 2), they both showed asymmetric atrophy with left predominance. The two were discordant for total tau levels in cerebrospinal fluid, neuropsychological testing, and smoking habits. The description of the twins can help identify environmental factors that influence the onset and phenotype of frontotemporal dementia.
INTRODUCTION
Frontotemporal dementia (FTD) due to mutations in progranulin gene (GRN) is a heterogeneous disease with a wide spectrum of phenotypic variability and age at onset, even within the same family [1–3]. The most frequent clinical conditions are behavioral variant frontotemporal dementia (bvFTD) and primary progressive aphasia with some cases presenting with corticobasal syndrome [4], Alzheimer’s disease, and Lewy body dementia. Neuroradiologically it is characterized by white matter hyperintensities [5, 6] and asymmetric atrophy, which can even be identified using visual rating scales [7]. It is known from literature that cognitive reserve, calculated with level of education and occupation, modulates functional connectivity in GRN carriers [8] and recently it has been demonstrated that leisure time activities mitigate the burden in FTD [9]. Higher occupation levels were associated with a more severe hypometabolism in prefrontal cortex [10].
We report the clinical, neuropsychological, and imaging findings in a monozygotic twin pair of bvFTD due to GRN mutation.
CASE REPORTS
Twin 1
The proband presented at the age of 74 with a one-year history of short-term memory complaints, mild apathy, and difficulties taking drugs and following conversations. Her medical history revealed only hypothyroidism. She had 5 years education. She worked in a cotton mill for 15 years until she married at 30 years. She had two daughters and one miscarriage and became a widow at the age of 64. She never smoked or drank alcohol and had minimal physical activity. She never had head trauma. She regularly read newspapers and increased her book reading and social activities after the death of her husband. She always lived in the north of Italy. General and neurological examination were normal. Routine blood testing was unremarkable. Neuropsychological testing highlighted deficiencies in language and executive function, with spared memory and visuospatial functioning (see Table 1).
Results of the neuropsychological test at presentation. The symbol *means results below the normal range
MMSE, Mini-Mental Score Examination; TMT A, Trail Making Test A; TMT B, Trail Making Test B; VOSP, Visual Object Space Perception.
MRI showed subcortical white matter hyperintensities (Fazekas score 1) and mild left frontal atrophy, while FDG PET showed cortical hypometabolism in the frontal and temporal lobes on the left side (see Fig. 1).

MRI and FDG-PET of Twin 1. Axial T1 showing left frontal atrophy, axial T2 showing bilateral white matter hyperintensities, axial and coronal FDG-PET showing left frontal and temporal hypometabolism. Images presented in radiological orientation (left hemisphere on the right side of the figure).
She underwent lumbar puncture at that moment, which revealed normal cerebrospinal fluid (CSF) biomarker levels: amyloid-β 1096 pg/mL (normal value >600), total-tau 342 pg/mL (normal value <400), and phospho-tau 37 pg/mL (normal value <60).
After one year from presentation, she became disinhibited, obsessive in her habits, lost good manners, and developed hyperphagia and compulsive hoarding. Language disorders worsened as well, with anomia, interrupted speech, and echolalia.
Twin 2
Past medical history was unremarkable. She smoked 20 cigarettes a day for 40 years. She never drank or had head trauma. During her life, she had 5 years education and performed minimal physical activity. She worked in the same cotton mill as her sister for 12 years until the age of 27 when she got married. She moved to Germany for three years and later came back to north of Italy always near the sister. She had two sons. She never read books or newspaper and has not been involved in social activities. She worked as a waiter for 35 years until the age of 67 when her family noticed language disorders, in particular word finding and impaired functional communication. Moreover, she showed memory impairment and difficulties using money and in her housework. Her family also noticed difficulties in driving. The symptoms gradually worsened and at the age of 68, she presented with apathy, loss of interest, social withdrawal, and daytime sleepiness. Subsequently, she showed mild disinhibition and she changed her food preferences, with a sweet tooth and episodes of binge eating. At that time, general and neurological examination were unremarkable.
Her brain CT scan revealed mild bilateral hypodensities interpreted as of vascular origin and atrophy especially in the left hemisphere (images not available). Neuropsychological testing at that point demonstrated deficiencies in executive function and visuospatial with sparing of language and memory (see Table 1).
She underwent lumbar puncture at the age of 68, one year after symptom onset, that revealed normal amyloid-β (680 pg/mL) with high total-tau (713 pg/mL) and p-tau (64 pg/mL).
She went through a phase of restlessness and agitation which was followed by a rapid progression of speech reduction until she became mute. She developed orofacial apraxia with swallowing problems and is now fed through a nasogastric tube and gradually became bedridden.
Family history
The mother of the twins developed dementia at 90 years old and died at 93. The mother’s sister developed dementia at 70 with changes in behavior and hallucinations and died at the age of 72. The father died at 82 years old without cognitive decline (See Fig. 2).
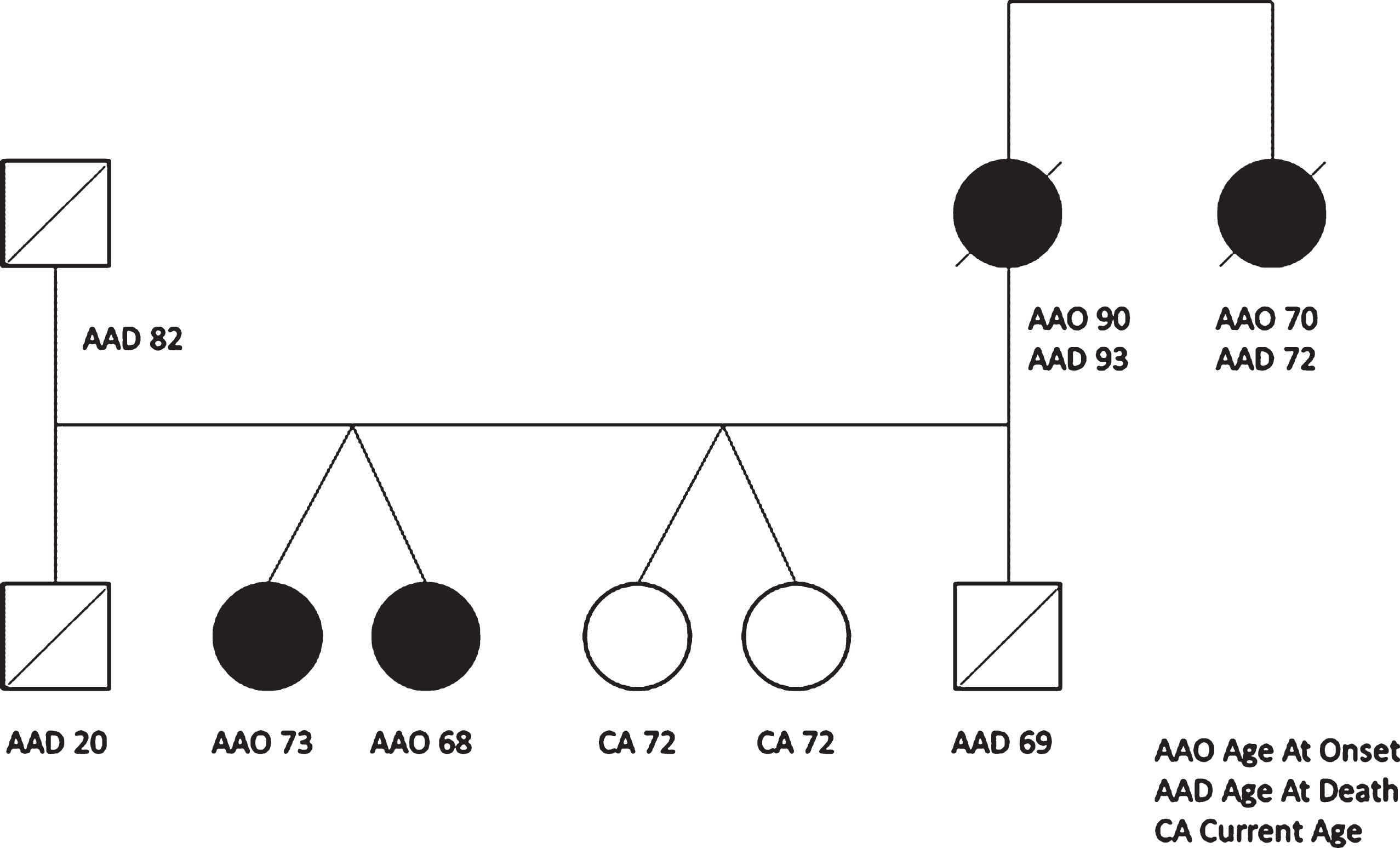
Genetic tree of the family with age at onset and age at death. Full circle means cognitive decline.
METHODS AND RESULTS
Both patients met the criteria for probable bvFTD with more than 3 typical symptoms (apathy, disinhibition, hyperorality, and executive functions deficit), clinical progression, neuroimaging confirmation, and exclusion of biomarkers indicative of Alzheimer’s disease [11]. In Twin 1, progranulin plasma levels, evaluated through an ELISA kit (Adipogene Inc., Seoul, Korea) after the ethical committee approval and patients’ consent, were below the cut off (28 ng/mL, normal value <61.55 ng/ml). Genetic analysis was performed in DNA from whole blood for Twin 1 and in DNA from sputum for Twin 2. Direct sequencing of the causative pathogenic genes demonstrated the presence of GRN Thr272fs mutation. Conversely, no mutations in MAPT and C9ORF72 genes were found.
The Cognitive Reserve Index Questionnaire (CRIq) [12] was filled with the help of the relatives of the twins and revealed similar CRIq total scores (Twin 1 80, Twin 2 76) and education (both twins 87) but different working activity (Twin 1 75, Twin 2 88) and leisure time (Twin 1 91, Twin 2 70) sub scores (Supplementary Table 1).
The other living relatives did not give informed consent for genetic analysis and blood collection.
DISCUSSION
The differences found between the two twins, such as age at onset or neuropsychological profile, can be caused by environmental or epigenetic factors while the similarities are possibly due to genetic conditions, not only the GRN mutation but also other genes may influence the phenotype [13]. It has already been demonstrated that the genotypes of TMEM106B and more recently GFRA2 [15] influence the disease in progranulin mutation carriers, but there were no differences between the twins in terms of these two genes. Since the mutation is present from conception and low plasma level of progranulin are detectable many years before onset [16], it is reasonable to hypothesize that still unknown environmental or epigenetic factors can play a role in the development of the disease [17]. In this context, the different cognitive reserve between the two support the hypothesis that social and leisure time activities can play a retarding role [9]. The benefit from working is known to provide some compensation for low educational attainment but is possible via a complex occupation later in life [18]. Both twins had only 5 years of formal education but Twin 2 maintained a low-level occupation while Twin 1 stopped working early.
In the literature, only two papers describe monozygotic twin pairs with GRN mutation [19, 20]. In both cases, as well as our patients, there is a striking asymmetric pattern of atrophy/hypometabolism, typical of GRN mutations [21]. Moreover, all the couples show the same hemisphere more atrophic, curiously the left. The phenotypic heterogeneity described in families with the same mutation also involve the most atrophic side [3], as well as the clinical syndrome. So far, no answer on why subjects with the same mutation develop atrophy on different sides but, since homozygous twins share the same pattern, it is reasonable to think that a still unknown genetic factor, different from GRN, can play a role in the laterality. This genetic factor can, for example, influence cortical susceptibility leading to neuronal death and atrophy. Nevertheless, further studies comparing the GRN-right to GRN-left would be needed to test this hypothesis.
White matter hyperintensities, as often described in GRN mutation carriers, were present in Twin 1 in a small amount (Fazekas score 1), while there were bilateral hypodensities in CT for Twin 2. Despite being considered of vascular origin, these lesions are present also in GRN patients without vascular risk factor or history of stroke. Recently it has been demonstrated that in a patient with GRN mutation, neuropathology of brain regions with significant white matter hyperintensities at MRI displayed severe cortical and white matter pathology with prominent white matter microglial activation and microglial dystrophy, but only mild axonal loss and minimal vascular pathology [22].
Regarding age at onset, the previous published cases developed the disease at an interval of 2 years (Twin 1 61 years, Twin 2 59 years for McDade et al. [19], Twin 1 75 years, Twin 2 77 years for Redaelli et al. [20]), while in the cases that we describe, there is a 5-year difference. It is also important to emphasize that while the mean age at onset for the Thr272fs is 59.6±5.9 [23], the mother of the twins, who was probably a carrier as her sister (see Fig. 2), died at 93 years old after 3 years of cognitive decline, potentially making her case one of the latest age at onset of GRN mutation described.
When looking at the neuropsychological profile, it is important to highlight that at onset, while both our patients show evidence of executive function deficits and preserved verbal memory, Twin 1 presented language impairment, whereas Twin 2 revealed a slightly more impaired attentional deficit associated with a severe constructional apraxia. Moreover, while earlier both show behavioral changes with disinhibition, altered food preferences, and social impairment, later in the disease, both developed important language disorders, reflecting the greater involvement of the left hemisphere. It could be possible that the higher cognitive reserve of Twin 1, particularly her reading attitude, could play a role as a retarding factor in the development of neuropsychological deficits.
The twins, in terms of lifestyle, were discordant for smoke habits: Twin 1 not smoking while Twin 2 was a heavy smoker. The studies that analyzed the role of tobacco smoking in FTD did not find differences in age at onset or disease duration between FTD smoker and non-smoker [24] and determined that smoking is not a risk factor for the development of FTD [24, 25]. Neither of the patients had alcohol consumption, thyroid disease, or traumatic brain injury that were reported previously as risk factors for the development of FTD [26].
It has been previously demonstrated that CSF total tau in FTD due to GRN was lower than sporadic FTD but higher than controls [27]. Of note, Twin 2 had higher levels of CSF tau and a more aggressive disease despite lumbar puncture being performed at a younger age. Moreover, both twins underwent lumbar puncture one year after symptom onset. Since it was already demonstrated that elevated total tau values in CSF predict prognosis in sporadic FTD [28], the difference observed in the CSF values of the twins may be related to the severity of the disease at the time of lumbar puncture or alternatively to epigenetic or environmental factors to be further investigated.
The main limitation of the study is the lack of examinations for Twin 2 that were not done at time of onset, and due to the severity of the condition at present (no FDG-PET, MRI, and progranulin dosage). We could not provide evidence of her left sided atrophy with images, but a CT report and the rapid development of language problems made us infer a greater involvement of the left hemisphere.
Conclusion
In conclusion, these monozygotic twins with a mutation in GRN, who share the brain atrophy distribution, were discordant for age at onset, neuropsychology, smoking habit, and total tau in the CSF. Environmental and epigenetic factors such as cognitive reserve can influence age at onset and clinical phenotype in GRN mutation carriers whereas the most affected hemisphere can have a genetic yet unknown origin.