
Abstract
Empirical evidence indicates a strong association between insulin resistance and pathological alterations related to Alzheimer’s disease (AD) in different cerebral regions. While cerebral insulin resistance is not essentially parallel with systemic metabolic derangements, type 2 diabetes mellitus (T2DM) has been established as a risk factor for AD. The circulating “toxic metabolites” emerging in metabolic syndrome may engage several biochemical pathways to promote oxidative stress and neuroinflammation leading to impair insulin function in the brain or “type 3 diabetes”. Thioredoxin-interacting protein (TXNIP) as an intracellular amplifier of oxidative stress and inflammasome activation may presumably mediate central insulin resistance. Emerging data including those from our recent studies has demonstrated a sharp TXNIP upregulation in stroke, aging and AD and well underlining the significance of this hypothesis. With the main interest to illustrate TXNIP place in type 3 diabetes, the present review primarily briefs the potential mechanisms contributing to cerebral insulin resistance in a metabolically deranged environment. Then with a particular focus on plausible TXNIP functions to drive and associate with AD pathology, we present the most recent evidence supporting TXNIP as a promising therapeutic target in AD as an age-associated dementia.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of age-associated dementia, inevitably growing with life expectancy in developed countries [1]. While an eminent control on cardiovascular events mortality has emerged over the past few years [2], AD is increasingly identified as the cause of elderly deaths in the United States [3]. According to the latest Alzheimer’s Association report, the illness stands as the fifth-leading cause of death for those aged above 65, of those one in 10 (10%) has AD [4]. Still, however, people who expire from complications like pneumonia associated with AD-induced swallowing disorders may not be counted in these statistics [5]. Beyond age as the greatest risk factor [6, 7], several other elements may contribute to AD development and progress. People with history of AD in first-degree relatives are more likely to get AD. Although such vulnerability might be partly described based on the inherited apolipoprotein ɛ4 allele (APOE ɛ4) risk gene [8, 9], heredity may not entirely explain familial AD and shared cultural, environmental and lifestyle factors might be arguably involved. In view of occupational risk factors, people at high risk of repeated head injuries (such as boxers) are more prone to neurodegenerative diseases and dementia [10, 11]. Not surprisingly, higher social and cognitive engagements [12, 13] or more years of formal education [14, 15] are known to lower the risks for developing dementias.
With such findings grounding cognitive rehabilitation therapies in AD patients, an established link between systemic organ diseases and AD development has to be constantly considered in AD patients, either in preventive or therapeutic approaches. As a typical instance, health of the hemodynamic system is closely monitored as it influences cerebrovascular integrity and brain function. That is, many risk factors for cardiovascular disease are also associated with increased risk of AD and dementia. In this line, smoking [16, 17], obesity [18, 19], hyperlipidemia [20, 21] and hypertension [22, 23] have been frequently addressed as risk factors for either cardiovascular disorders or AD. Standing on statistics, it has been shown all type 2 diabetes mellitus (T2DM) patients do not develop AD though, they present similar pathological changes and amyloid plaque formation at cerebral tissue [24]. Existing evidence also postulates T2DM and AD share some etiological features, of which increased circulating glucose and lipids or the progressive insulin resistance may directly instigate deteriorating effectors escalating risks for dementia and AD development [25, 26]. In this connection, undoubtedly, many hypotheses center on those effectors involved in inflammation or oxidative stress as a common ground in both T2DM and AD.
Oxidative stress as an imbalanced redox state is especially of great influence in integrity of brain where high oxygen demand and abundance lipid cells predispose it to profound peroxidation. An accumulating body of evidence is increasingly holding oxidative stress as a common detrimental pathway in several neurodegenerative diseases such as AD or Parkinson’s disease [27]. Thioredoxin binding protein (TXNIP) is a multi-functional protein which is directly induced by reactive oxygen species (ROS) generated in divergent conditions to restrain the antioxidant activity of thioredoxin (TRX) system and enable oxidative stress to induce inflammation and apoptosis [28, 29]. In a metabolically deranged environment, TXNIP may get recruited by circulating toxic metabolites either directly in response to hyperglycemia or indirectly by the ROS overload. Upon stimulation, TXNIP escalates insulin secretion from pancreatic cells and glucose uptake in peripheral tissue. More importantly, as here we are to discuss, it provides a unique link for systemic metabolic derangements to enforce insulin resistance in the brain “type 3 diabetes” with its long history link to AD.
Assimilating the most recent updates, this review primarily provides an updated evidence-based view on effectors downstream to systemic metabolic deregulation, which may contribute to AD development/progress. Of note, this review does not speak about the slowly progressing atherosclerotic changes associated with hyperlipidemia, a hallmark of metabolic derangement in diabetic bodies, which is still a major risk factor for associated vasculopathies in AD brains. Alternatively, we are more concentrated on circulating mediators and their immediate established effectors to contribute to type 3 diabetes in the milieu of metabolic syndrome. In this connection, besides the biochemical interference with cerebral insulin signaling, oxidative stress or systemic inflammation would be more detailed as the major links. Then with a particular focus on TXNIP, we would put effort to picture the plausibly central TXNIP place to link these mediators to central impairment of insulin signaling and AD molecular pathology per se. This gathering, we hope, might pave the way to define practical therapeutic targets for AD or control T2DM as a pivotal modifiable risk factor to manage the accelerated emergence of AD and its lifelong burdens.
ASSOCIATION BETWEEN AD AND T2DM
Vascular dementia caused by impaired blood supply to parts of the brain is the typical form of dementia associated with T2DM, mostly following endothelial damage or blood vessel blockage. With more controversies in early investigations, risk of AD development has been shown to increase in T2DM patients. This might be well exemplified by some findings supporting the link. In a population-based study in 2002 on individuals aged 80 years and older showing no association with AD in 31 T2DM patients out of 187 patients diagnosed with dementia, there was an elevated risk to develop vascular dementia (RR = 2.54, 95% CI = 1.35–4.78) [30]. In a parallel study, 5,574 Canadian subjects participated in 5-year follow-up and T2DM was shown to be associated with vascular dementia (RR: 2.03; 95% CI: 1.15–3.57), while no risk was identified for AD [31]. The later Framingham study with a 12 year-long follow up on 2,210 subjects also did not indicate any increased risk in AD development in 202 patients with T2DM [32], which is well consistent with a recent cross-sectional study on 1,037 Brazilian subjects where no association has been detected between AD and T2DM (217 patients) when corrected for the effect of APOE ɛ4 allele [33].
With numerous cohort studies addressing the association, several confounding factors appear to affect the conclusions. Undoubtedly, the sample size shortage may work as the main factor to harm the power of the analysis. More importantly, T2DM is also closely associated with established risk factors for AD, including cardiovascular disease and the APOE ɛ4 allele, which are not the mere comorbidities in the studied individuals. In fact, the association between T2DM and AD seemingly is influenced by the APOE ɛ4 allele, ethnicity or severity and duration of hyperglycemia. This necessitates to consider the inclusion criteria which is well practiced in meta-analyses.
The first comprehensive meta-analysis in 2012 quantitatively pooled 19 final papers meeting the required qualifications. Accordingly, with a total of 6,184 people with diabetes, the collective relative risk of AD for people with diabetes was indicated as 1.5 (95% CI = 1.2–1.8) suggestive of increased AD risk in T2DM patient [34]. The study also supported the earlier belief that T2DM patients are at high risk for vascular dementia (RR: 2.48, 95% CI: 2.08–2.96). A year later, another meta-analysis considering a total of 28 studies reported a pooled RR of 1.56 (95% CI = 1.41–1.73) for developing AD and 2.3 for developing vascular dementia 2.27 (95% CI = 1.94–2.66) in patients with T2DM [35]. The most recent meta-analysis published in early 2017 pooled data from 1,746,777 individuals from 17 qualified studies and showed a significant higher incidence of AD in individuals with T2DM (RR: 1.53, 95% CI: 1.42–1.63). Interestingly, when stratified by ethnicity, the calculation indicated that the RR of AD incidence in T2DM patients were 1.36 (1.18–1.53) and 1.62 (1.49–1.75) in Western and Eastern populations, respectively [36], indicating more vulnerability in Eastern people.
METABOLIC SYNDROME AND INSULIN RESISTANCE IN THE BRAIN: BIOCHEMICAL LINKS
The state of over-nutrition and a sedentary lifestyle have been long proven to promote a clustering of metabolic and physiological components recently described as metabolic syndrome. The condition earlier defied as “insulin resistance syndrome”, is a varying composition of some generally agreed components including central obesity, insulin resistance, dyslipidemia, and hypertension [37]. Metabolic syndrome is associated with a doubling risk of cardiovascular disease and stroke [38], a variety of cancers, i.e., in colorectal and liver tissue [39], and a five-fold increase in incidence of T2DM [40, 41]. For a better understanding of the involved pathologic pathways, we would first brief how the metabolically deranged systemic environment may impair insulin signaling in the brain through biochemical interactions. To provide a clear view, the likely circulating mediators are discussed separately.
Fatty acids and lipid metabolites
Dyslipidemia (defined as serum HDL <40 mg/dL in men and <50 mg/dL in women, and serum triglycerides >150 mg/dL) is associated with elevated circulating fatty acids either form enlarged adipocytes and ectopic fat deposits (liver or muscle) or from augmented de novo hepatic lipogenesis, may play a pivotal role in modulating insulin sensitivity [42, 43]. In fact, with their immediate passage from blood-brain barrier (BBB) fatty acids have different ways to induce insulin resistance in the brain. Accumulating body of evidence indicates that fatty acids specially saturated derivatives act on immune cells to elevate several cytokine transcripts which are characterized as the main inducers of insulin resistance [44]. It is of note, even a few hours elevation in human plasma non-esterified fatty acids (NEFA) is efficient enough to initiate resistance to insulin [45, 46]. According to the existing findings, some biochemical hypotheses have been posited to explain such a drastic effect. The first experimentations implied that intracellular NEFAs may compete with glucose for substrate oxidation and thereby inhibiting pyruvate dehydrogenase and phosphofructokinase activity and less glucose uptake [47]. Nevertheless, the hypothesis was challenged with later findings proposing intracellular metabolites of particularly saturated fatty acids are the pivotal effectors to impair insulin signaling. That is the intracellular content of fatty acid metabolites like diacylglycerol (DAG), long chain acyl-coenzyme A (LCCoAs), and ceramides may activate a kinase cascade leading to serine/threonine phosphorylation of insulin receptor substrate-1 and 2 (IRS-1 and IRS-2) and reduced PI(3)K activity [48, 49]. However, besides such direct effects, saturated fatty acids (SFA) have been shown to activate TLRs dimerization possibly by interaction with hydrophobic pockets present in the extracellular domain and their movement into the region of lipid rafts [50, 51], which then might stimulate the proinflammatory axis IKKβ-NF-κB [52, 53]. Interestingly, activation of the IKKβ-NF-κB pathway during obesity is demonstrated to increase cognitive impairment [54, 55].
Particular lipid metabolites have established strong effect to drive insulin resistance. Importantly, upon entrance to intracellular space, ceramides might go through a distinct pathway to contribute to IL-1β overproduction as a common feature in obesity. Sensed as a “damage signal” by the intracellular complex NOD-like receptor pyrin domain-containing-3 (NLRP3), ceramides drive the formation of NLRP3 inflammasome and subsequent caspase-1 activation which is responsible for the maturation of pro-IL-1β, pro-IL-18, and IL-6 [56]. Inflammatory cytokines like TNF-α, IL-6, and IL-1β, as described in the following sections; are all potent inducers of insulin resistance in the brain. While many of lipid metabolites like sphingosines (ceramides precursors) have similar effects, significant anti-inflammatory effects have been substantiated for others. According to our previous studies for instance, sphingosine-1-phosphate may remarkably prevent neuroinflammation in the brain through S1P-1 receptor modulation [57, 58]. This might support the alleviating effect of some controlling lipid metabolites on insulin signaling. Interestingly, fingolimod (S1P analogue) treatment has been also shown to reduce amyloid-β either in neural cell cultures or in APP transgenic mice [59].
Hyperglycemia
Hyperglycemia may go through multiple pathways to escalate insulin resistance, some of which are expectedly similar to those for lipotoxins. While cerebral tissue is very likely to be exposed to far lower glucose levels in CSF [60], the due values drastically rise in a metabolic disease and would translate to hyperglycemia for CNS cells [61]. Impaired activation of Akt linked with glucose-induced intracellular ceramide accumulation is of the primarily proposed mechanisms [62]. Under normal conditions, insulin requires activation of IRS-1 that is essential for further activation of PI3K in response to insulin followed by phosphorylation of AKT to mediate biological effects of insulin. In a pattern similar to fatty acids, increased glucose metabolism may also improve intracellular diacylglycerols (DAG) leading to activation of PKC, decreased insulin-stimulated IRS-1/IRS-2 tyrosine phosphorylation and then PI3K activation. In the muscles, this results in reduced insulin-stimulated GLUT4 translocation and decreased glycogen synthesis [63]. Besides glucose intracellular metabolites like xylulose-5-phosphate, advance glycation end-products (AGEs) are of important effectors to mediate insulin resistance in hyperglycemic conditions through enhancing TXNIP transcripts. TXNIP as a regulator of cellular oxidative status in response to nutrients, is a major effector of hyperglycemia to cause insulin resistance [64], and is overexpressed instantly in response to hyperglycemia [65].
Adipokines
With metabolic syndrome and T2DM often associated with obesity, adipose tissue has been always in critical focus in insulin resistance development and has been reviewed elsewhere [66]. As an active metabolic tissue, adipose tissue is known to secrete multiple active proteins known as ‘adipokines’ [67, 68], some playing significant role in insulin resistance associated with visceral obesity in particular. However, there is not a general agreement; some believe the term adipokine is inclusive for any excretion from fat tissue like inflammatory cytokines with other resources might have been identified for. Retinol-binding protein-4 (RBP4) is an instance of adipokines bringing insulin resistance through reducing PI3K signaling in muscle and inducing the gluconeogenic enzyme phosphoenolpyruvate carboxykinase in the liver through a retinol-dependent mechanism [69].
On the other hand, adiponectin is downregulated in obese patients with insulin resistance. It reduces hepatic glucose production by decreasing transcripts of two essential gluconeogenesis enzymes: glucose-6-phosphatase and phosphoenolpyruvate carboxykinase [70] and has been characterized to improve insulin sensitivity through activation of AMPK [71]. Omentin represents another intriguing instance of the differential harmonic insulin regulation by adipocytes residing in an obese or lean body mass with different genes transcripts regulation. That is omentin transcripts are strongly expressed in visceral adipose tissue and acquire higher plasma levels in lean subjects [72]. Like adiponectin, omentin enhances insulin sensitivity and improves Akt phosphorylation [73]. Whether peptide constructs of adipokines may cross BBB through constitutional carriers has not yet been fully answered yet though, a few specific transporters have been identified to make the brain accessible for adipokines namely for the appetite controlling leptin [74, 75]. Still, however, modulating inflammatory signals may also provide a major alternate pathway for adipokines like leptin [76, 77] to alter insulin signaling as described in the following sections.
Circulating cytokines
Several concrete evidences support the emergence of subtle persisting inflammation along with metabolic syndrome. With many penetrating the BBB, the circulating cytokines may alternatively induce CNS inflammatory reactions by signaling through vagal fibers [78]. Increased content of inflammatory cytokines in plasma, roots from at least two main resources in such a metabolic disorder. First, the circulating lipotoxins (e.g., ceramides) associated with hyperlipidemia may act on immune cells to enhance the cytokine transcripts and activate damage signal sensor NLRP3. Secondly, the other likely component of metabolic syndrome, central obesity (routinely indexed with waist to hip ratio), might work as an endocrine source of pro-inflammatory cytokines. This phenomena is explained according to the spatial constraints imposed by visceral extracellular matrix (ECM) which is essentially rich in collagen fibrils and fibronectin to contribute to the structural integrity of adipocytes and for pre-adipocyte differentiation [79]. This works to restrict blood flow and induce hypoxia inducible factor 1 (HIF-1α). The existing evidence suggests the transcriptional activity of HIF-1α contributes to ECM fibrosis and remodeling, indicating a feedforward loop to limit blood flow and escalating the existing inflammation. Lysyl oxidase (LOX) is hypothesized to mediate this HIF-1α effect through cross-linking collagen I and III to form the fibrillary collagen fibers [80] in presence of enough levels of secreted protein acidic and rich in cysteine (SPARC) to the ECM [81]. Interestingly, high-fat feeding has been shown to develop looser ECM structure, enlarged adipocyte and fat pad size, and reduced inflammation either in SPARC–/–mice [82] as well as obesity-prone ob/ob mice with silenced collagen VI gene [83]. In state of obesity or lipotoxicity, the increased circulatory TNF-α activates c-Jun N-terminal kinase (JNK) [81], protein kinase C (PKC), and inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ) [84, 85] which along with inducing notable NF-κB expression phosphorylates serine residues of the IRS protein blocking downstream activation of the insulin signaling. TNF-α and IL-6 as well, are higher in visceral adipose which might explain part of the association of cardiovascular disease and metabolic syndrome with central obesity [86]. Similar to TNF-α, IL-6 overproduction has also been shown inhibiting IRS-1 and IRS-2 phosphorylation which abrogates insulin signaling beside its effect to block the synthesis of hepatic glycogen IL-1β on the other side, has been reported to repress the transcripts of insulin signaling constituents. That is in primary human adipocytes and 3T3-L1 mice adipocytes as well, a significant reduction in expression of IRS-1, p85 PI3K, and AKT have been determined by IL-1β overexpression [87]. For the very vast effect of pro-inflammatory cytokines to drive insulin resistance, other effectors might be involved. For instance, suppression of cytokine signaling (SOCS) proteins has been also postulated to interact with the phosphorylated insulin receptor and prevent activation of the IRS proteins [88, 89], making a way for cytokines to induce insulin resistance.
METABOLIC SYNDROME TO IMPAIR CEREBRAL INSULIN SIGNALING: IS OXIDATIVE STRESS THE MAIN ROAD?
Numerous concrete findings imply metabolic syndrome results in persisting production of reactive oxygen species which act as strong effectors to either a progressive impaired insulin signaling in peripheral tissues (muscles and liver) or insulin production in the organ of origin (β islets). At the emergence of metabolic derangements (e.g., hyperlipidemia), some circulating toxic metabolites (e.g., AGE or ceramides) may readily pass the BBB to act as the main ROS instigators, while the main forms of ROS (superoxide and peroxide ions) formed throughout the body directly affect the cerebral insulin signaling if not degraded before brain access. The chronic rise in circulating glucose and lipids appear to play the eminent role in metabolic syndrome. The influence of the other accompanying endocrine (e.g., glucocorticoids) and paracrine agents like obesity associated adipokines and inflammatory cytokines might be regarded as adjuvant co-factors leading to ROS propagation in the emerging inflammatory damage to sub-cellular compartments. In brief, two mechanisms are speculated to explain massive ROS generation in the state of metabolic syndrome.
Firstly, energy overload and the consequent mitochondrial dysfunction take a significant role to drive leaking ROS from electron transport chain. High concentration of blood glucose, NEFA, and diverse lipotoxins as the typical feature of metabolic stress increase the burden of mitochondrial oxidative phosphorylation. With a massive flow of glucose and fatty acids to TCA cycle and the overflowing products to the mitochondria, the electron transport chain has to accept much more electron donors (NADH and FADH2) than the normal conditions. With the transmembrane proton gradient reaching a critical value, the electron transport chain would stop at Complex III and ensue with the reverse electron flow to coenzyme Q (CoQ). The leaking electrons then are recruited to the one-electron oxygen resulting in formation of superoxide anion radicals and H2O2 [90, 91].
The other leading pathway to directly escalate cellular redox status in T2DM or the preceding metabolic derangements is PKC activation, which is known to stimulate the transcription factor NF-κB that finally increases ROS generation [90]. In this connection, membrane-associated NADPH oxidases play a central role as demonstrated in vascular cells [92]. Different components of metabolic syndrome have been identified to mediate this PKC activation. Hyperglycemia has been recognized as an established indirect activator of PKC. This might take place through the enhanced AGE binding to its receptor on the cell surface [93]. PKC-α and PKC-δ have been shown to activate NADPH oxidase and induce toll-like receptors in human monocytes (TLR-2 and TLR-4) [94] which in turn contribute to intracellular inflammatory and oxidative signals. Furthermore, angiotensin II acts as another stimulus for PKC [95, 96] or NADPH oxidase [97] and it is likely to increase in metabolic syndrome either as an adipokine by the likely associated central obesity or deranged renin-angiotensin accompanying the concurrent hypertension.
MAIN ROS EFFECTORS TO INDUCE INSULIN RESISTANCE: SERINE/THREONINE KINASES OR INFLAMMASOMES?
The plausible extra ROS generation during metabolic stress is presumed to directly activate several serine/threonine kinases including MAPK (mitogen-activated protein kinase), GSK-3 (glycogen synthase kinase-3), PKC, Akt, and mTOR (mammalian target of rapamycin), all of which may decrease responsiveness to insulin by selective phosphorylation of IRS serine and threonine residues so that they cannot interact with the insulin receptor [98, 99]. Among these kinases, MAPK signaling appears to play the prominent role. P38 MAPK is a member of MAPK family, the family of mitogen-stimulated serine/threonine protein kinases, which might be induced in response to many biological and physical factors. ROS and inflammatory cytokines both are efficiently upregulated in response to high circulating glucose, and lipids are among the well characterized P38 MAPK activators [100, 101]. JNK, the other serine kinase of MAPK family, might be involved in oxidant-induced insulin resistance in adipocytes [102]. JNK1 isoform has been shown to be activated by ROS and plays a central role in loss of insulin functionality in obesity [103]. The serine protein kinase IκB Kinase β (IKKβ) is also an efficient effector of ROS and selective inhibition of IKKβ has been shown to improve insulin sensitivity [104]. IKKβ controls transcription factor NF-κB functioning by phosphorylating the inhibitory IkB subunit. The NF-κB/IkB/IKKβ activation has been found to correlate with oxidative state and impairs insulin signaling at IRS-1 [105]. Pharmacological or genetic blockade of the NF-κB signaling disrupts insulin resistance development in high-fat diet fed animals [104]. However, NF-κB is of remarkable importance to mediate ROS effect on IRS; as noted earlier in this review, inflammatory cytokines are also strong agonists of the TLR/NF-κB pathway to induce insulin resistance [106]. Given the fact that oxidative stress is associated with inflammatory responses particularly in resident immune cells, it is literally hard to differentiate whether ROS or inflammatory cytokines are providing the main path for metabolically diseased circulation to impair cerebral insulin signaling. Such skepticism comes to more significance while considering the immediate ROS association with the cytoplasmic multiprotein complex, NLRP3 inflammasome.
Sensing several damage signals, the cytosolic pattern recognition receptor NLRP3 is known as a sensor for metabolic disease. Upon several stimuli including ROS, NLRP3 recruits the adapter protein the apoptosis-associated speck-like (ASC) and pro-caspase-1 leading to caspase-1 production and subsequent IL-1β maturation and release [107]. Besides IL-1β maturation, caspase-1 drives pyroptosis featured with intramembranous hole formations in microglia and macrophages and thereby resulting in a massive release of inflammatory cytokines [108], each of which may contribute to insulin resistance as described earlier.
Inflammasome activation has been shown by concrete evidence to impair insulin signaling to reduce insulin sensitivity in several target tissues namely hematopoietic cells [109]. Namely, lipotoxins-induced assembly of inflammasome NLRP3 in macrophages has been shown to mediate insulin resistance and early T2DM. That is, free fatty acids and subsequent generation of lipid molecule ceramides has been suggested to activate NLRP3/IL-1β cascade in fat tissue macrophages, and mice deficient in NLRP3 and caspase-1 have been demonstrated protected against insulin resistance induction by HFD [56, 110]. This might somehow explain the observation that weight loss in human is associated with decreased NLRP3/IL-1β expression in adipose tissue [111]. The close interplay between ROS and NLRP3 inflammasome supports the hypothesis that circulating toxins associated with metabolic syndrome, either circulating toxic metabolites or their ROS offspring, might converge on NLRP3 inflammasome to amplify the initial stimuli to more massive inflammatory and oxidative reaction, all of which disrupt insulin signaling via intracellular kinases and mitochondrial dysregulation.
INSULIN SIGNALING IN THE BRAIN: CONTRIBUTIONS TO FUNCTION AND INTEGRITY
While insulin resistance is defined as “reduced responsiveness of body tissues to insulin” meaning reduced glucose utilization in periphery, it basically points to impaired insulin function in cerebral tissue in divergent biological functions other than the rudimentary glucose uptake [112]. A brief look on the described different roles of insulin signaling in CNS cell populations [113] would provide a better understanding on the likely cerebral dysfunctions that would raise by lack of insulin signaling.
Insulin signaling in endothelial cells
The fact that glucose uptake in neural cell is not insulin-mediated never undermines the insulin dependent glucose transport from BBB endothelial cells, making the required CSF pool available for the CNS cells. The CSF to plasma glucose ratio is far below one in normal subjects. Seemingly with endothelial insulin resistance, the BBB controlling gate goes out of work, producing a hyperglycemic CSF space in the diabetic patient [113, 114], which in turn may impair the insulin signaling in the brain parenchyma. Appropriate vasoreactivity is another significant insulin effect in the vascular bed throughout the body. Insulin is known to induce vasodilation through the PI3K signaling pathway and consequent nitric oxide (NO) production in skeletal muscle endothelial cells leading to enhanced cyclic guanosine-monophosphate (cGMP) in vascular smooth muscle cells [115]. Still, however, insulin possesses vasoconstrictor effect through MAPK-endothelin-1 production [116], which explains part of impaired subcutaneous microvascular perfusion in diabetic patients. In brief, the net effect of IR in endothelial cells might be pictured as deficient vasodilation and improper vasoconstriction in cerebral vasculature [117].
Insulin signaling in glial cells
Astrocytes composing 20–40% of all glia in the human brain [118, 119] are also capable to take up glucose via insulin independent GLUT-1 transportation and contribute to glycogen synthesis and glucose transport via BBB. Noteworthy, via the so-called process “astrocyte–neuron lactate shuttle”, astrocytes are capable of glycolytic processing of glucose and transporting lactate to neurons as an alternative fuel source, to exceptionally back neurons in hypoglycemic condition [120, 121]. Astrocytes insulin signaling is also required for the hemostatic inflammatory responses adjacent to microglia, the main resident immune cells within the brain. In fact, in vitro evidence demonstrates insulin receptors may modulate microglial inflammatory responses in a complex manner [122]. That is insulin can enhance the secretion of certain inflammatory cytokines while inhibiting others. In this connection, either in cats infected with feline immunodeficiency virus [123] or in human primary microglia obtained from HIV-1-infected fetal tissue, insulin shows selective anti-inflammatory and antiviral actions [124].
Insulin signaling in neurons
While glucose works as the major energy source for neural cells, its uptake is only indirectly influenced by insulin [125, 126]. Except for a sub-population of widely scattered GLUT-4 expressing neurons [127], neural cell populations expressing GLUT-1 and GLUT-3, are dependent to insulin in many vital biological processes other than glucose uptake. Insulin receptors highly enriched in synapses [128] are residing at both presynaptic axon terminals and the postsynaptic plates [129, 130]. Insulin plays numerous important roles in neurosynaptic functioning. Through its two major effector pathways, IRS/AKT or MAPK signaling [131, 132], insulin enhances neurite outgrowth [133], modulates catecholamine release and uptake [134] and activity-dependent synaptic plasticity [135]. Intriguingly, insulin plays a critical role in memory and cognition by tuning long-term potentiation (LTP) and long-term depression (LTD) [135] as well as regulating the expression and localization of GABA [136], N-methyl-d-aspartate (NMDA) [137], and α-amino— 3— hydroxy— 5— methyl— 4— isoxazole propionic acid (AMPA) receptors [138].
IMPAIRED INSULIN SIGNALING CONTRIBUTION TO NEURODEGENERATION AND AD
The predominant roles of insulin in different CNS cell types provide a compelling feature of several impaired biological responses in an insulin non-responsive brain, either developed intrinsically or contributed by systemic metabolic derangement. Insulin plays a fundamental controlling role in appetite, memory, and activity level [139]. Besides, considering its regional impacts, as briefly described, insulin downstream effectors are pivotal regulators of protein synthesis, cell survival, and inflammation and hence an “insulin deficient brain” is not surprisingly prone to many inflammatory and degenerative disorders. Beyond this, as demonstrated in Fig. 1, insulin inefficiency has been found to directly contribute to molecular pathology specific to AD.

Insulin resistance potential contributions to AD brain. Insulin irresponsiveness is associated with malfunctions in the brain to drive Alzheimer’s disease (AD) pathology as described in boxes. Normal insulin signaling (white arrows) is required in endothelial cells to produce enough NO to support insulin entrance to the brain parenchyma besides its role in GLUT-4 glucose transportation. Insulin resistance disturbs the normal insulin uptake and glucose hemostasis generally leading to increased CSF to blood ratio of glucose. Impaired insulin signaling associates with reduced ACh levels either via ChAT repression or enhanced AChE activity. In neural cell reduced ACh detrimentally impact synaptic events involved in memory and cognition, whereas in glial cells; reduced ACh functionality frees inflammatory cytokine from 7nAChR control, aggravating the preexisting neuroinflammation in AD brain. Insulin dysfunction may also directly contribute to AD pathology by increasing Tau phosphorylation and aggregates’ buildup typically by inducing MAPK family. Aβ production may also increase as a result of either higher APP transcription or improved BACE cleavage activity. This may lead to Aβ accumulation in extracellular space which further is augmented by the deficient IDE degrading activity associated with insulin resistance. Ins, insulin; Glu, glucose; ACh, acetylcholine; AChE, acetylcholinesterase; ChAT, choline acetyl transferase; IDE, insulin degrading enzyme; Aβ, amyloid-beta; NFT, neurofibrillary tangles; 7nAChR, alpha-7 nicotinic acetylcholine receptor; APP, amyloid-beta protein precursor; BACE, β-secretase; GSK, glycogen synthase kinase; CDK, cyclin-dependent kinases.
Cerebral insulin resistance and neurodegeneration
Insulin dysfunction may contribute to neurodegenerative disorders in several ways either by disturbing optimal metabolism or by triggering apoptotic signals. The intracellular substrate of Akt AS160 (or TBC1D4), a Rab GTPase-activating protein, is among the essential insulin signaling constituents involved in insulin dependent glucose uptake. Loss of insulin function has been described to reduce AS160 activity to mediate GLUT-4 translocation to the cell membrane [140, 141]. At first sight, metabolic consequences of insulin resistance arguably are not discernible in terms of “glucose uptake” in the brain where the high GLUT-1 and GLUT-3 expression renders glucose intake approximately irresponsive to insulin signaling. Nevertheless, with cerebrovascular endothelial cells and some neural population living on the insulin dependent carrier GLUT-4, resistance to insulin effects may compromise the efficient glucose uptake and metabolism. The other important insulin effect is AKT-mediated activation of mammalian target of rapamycin (mTOR) which triggers versatile downstream effectors and numerous processes. Noteworthy, mTOR provides feedback autoregulation through site-specific serine phosphorylation of IRS-1 and IRS-2 [142]. Therefore, inhibiting insulin signaling, mTOR signal is not always downregulated but rather dysregulated in insulin resistance [143]. mTOR implication in degeneration or neural integrity may not be simply concluded, however; the effects on many aspects of cell metabolism, survival, and autophagy are expected to play critical and divergent roles. Instantly 4EBP proteins are pivotal mTOR effectors to control eIf4E, the main driver of cap-dependent translation serving to produce essential proteins to proliferation and repair. On the other hand, mTOR may interfere with normal clearance of debris and toxic tangles by inhibiting the process of autophagy by blocking ULK-1 activity. Cerebral insulin irresponsiveness may also recruit pro-apoptotic molecules, particularly BAD, FoxO1, and Caspase-9, which have demonstrated enough power to drive neurodegeneration in type 3 diabetes. In fact, insulin augments pro-survival signals and improves synthesis of constitutional protein through mTOR as well as MAPK, which is especially important for regulating the innate and adaptive immune function [144, 145]. Simultaneously, insulin-induced Akt activity may abrogate apoptotic signals by inhibiting the key elements like BAD and FoxO1 [146]. Coupled with the phosphorylation control of caspase-9 [147, 148], impairment of Akt signaling in insulin resistance may leave CNS unprotected and exposed to any detrimental stimuli which may instantly proceed to progressive neural damage.
Cerebral insulin resistance and AD pathology
Investigations on the role of insulin in memory formation has a long history with most findings indicating a synaptic base for insulin to modify neurotransmitter release and modulate the activities of both excitatory and inhibitory postsynaptic receptors [149]. In particular, a crosstalk between NMDA and insulin receptor has been proposed to modulate insulin effect on plasticity and memory [150]. However, the role of insulin in memory formation in unnegotiable; it is not simply possible to conclude whether AD or insulin resistance has a causative role to induce the other. Based on the existing evidences, IRS phosphorylation and brain insulin resistance are early features of AD [151]. The following mechanisms are postulated to explain the contribution of type 3 diabetes to AD pathology as hypothetically roots from consequent extracellular Aβ and intracellular tau accumulates or aberrant cholinergic transmission.
Tau phosphorylation by particular kinases
Glycogen synthase kinase-3 beta (GSK3β) is a multifunctional serine/threonine kinase that was originally defined as a regulator of glycogen metabolism and was later found to enhance abnormal hyperphosphorylation of tau. GSK3β phosphorylation by Akt inhibits the activity of this key kinase. GSK3β has several protein substrates like microtubule-associated proteins and cAMP-responsive element-binding protein (CREB). Impaired insulin signaling would leave GSK3β signaling uncontrolled ending with notable effects on glucose metabolism, apoptosis, and neuroplasticity. Regarding the specific AD pathology, solid evidence indicates phosphorylation by GSK3β is efficient enough to make tau filaments form tangle-like aggregates in vitro and correlates with the presence of NFTs like those observed in AD [152, 153] or Parkinson’s disease brains [153]. The highly expressed protein kinase in neurons, cyclin-dependent kinase 5 (CDK5), is also a critical kinase with an initially defined key role in cell cycle and sensory pathways. Any CDK5 trivial deregulation is strongly linked to hyperphosphorylation of cytoskeletal substrates like tau and AD pathology [154, 155].
Aβ production and clearance
As demonstrated in a transgenic mouse model of AD, insulin deficiency may upregulate β-secretase enzyme and its substrate, amyloid-β protein precursor, leading to accelerated Aβ accumulation [156]. Another potential mechanism for Aβ accumulation is deterred extracellular proteolytic Aβ degradation by insulin-degrading enzyme (IDE). IDE is involved in the clearance of Aβ plaques in the brain as well as degrading insulin [157]. In fact IDE’s activity, concentrations, or transcripts have been observed to be reduced in AD brain tissue, while reduced degradation of Aβ and insulin have been also reported in brains of knockout mice that lack the enzyme [158, 159]. Due to IDE’s high affinity for insulin, cerebral hyperinsulinemia, whether caused by perfused insulin from periphery or intrinsically induced within the cerebral parenchyma, would leave Aβ to accumulate and contribute to AD.
Cerebral cholinergic system repression
AD is associated with the reduced level of choline acetyl transferase (ChAT) expression and the insulin resistance is thought to be involved [160]. The idea is in line with evidence indicating main insulin signaling-related proteins like insulin receptor (InsR), IRS-1, and GSK3β are co-located with ChAT in CA1 region [161]. Important findings in this regard indicate that rats treated with intracerebroventricular (ICV) STZ develop memory impairment and abnormalities in the central cholinergic system, with a significant decrease in choline acetyltransferase ChAT activity [162, 163] and remarkable increase in acetylcholinesterase activity [164]. Therefore, enough concrete evidence is available to conclude that insufficient insulin signaling may decrease the production of acetylcholine providing a plausible biochemical link between metabolic syndrome and AD by impairing the cognitive abilities [165]. Inadequate acetylcholine signaling impair normal memory and cognition, and critically impair alpha-7 nicotinic acetylcholine receptor to control immense inflammatory cytokines from microglial cell which may potentially aggravate the neuroinflammation associated with AD [166, 167].
TXNIP THE PLAUSIBLE CORE IN TYPE 3 DIABETES
Although expressed at low levels in cerebral tissue [168], TXNIP, a 46-kDa ubiquitously expressed protein consisting of 391 amino acids residues [169], critically governs the body energy metabolism either through its energy sensing role in thalamus and brain stem [170] or via intracellular interplay with mitochondria and metabolism in the periphery [171]. As a precise switch point in energy distress, TXNIP is instantly upregulated in metabolic diseases including obesity and diabetes [65, 172]. In fact, even slight hyperglycemia as the mainstay hallmark in metabolic syndrome strongly induces TXNIP expression which in turn readily limits more glucose uptake through the main constitutional transmembrane transporters GLUT1 (insulin independent) and GLUT4 (insulin dependent). This may result in elevated plasma glucose levels which are possibly further contributed to by increased hepatic gluconeogenesis subsequent to transcriptional activity of TXNIP [173]. Still, however, this established TXNIP overexpression has a variety of other effectors to drive/potentiate irresponsiveness to the described vital insulin functionality in the brain (type 3 diabetes). Beyond its central place in glucose metabolism, TXNIP plays significant role in oxidative stress as well as inflammation which may in turn escalate cerebrovascular and neurodegenerative diseases as recently described in detail [29]. TXNIP nomenclature roots from its direct controlling effect on TRX system. TRX works as an efficient ROS scavenger whose insufficiency has been reported in subjects’ suffering from AD [174]. However, it is hard to distinguish whether TXNIP “redox-independent roles” do not take the upper hand to drive insulin resistance and thereby contribute to AD pathology. Importantly, the emerging data about TXNIP role on other proteins’ transcripts either through direct DNA interactions or by microRNAs are opening new insights to the mechanisms whereby TXNIP regulates several pathways contributing to spread oxidative stress and inflammation and eventually pave the way for cerebral insulin resistance and subsequent memory abnormalities.
TXNIP regulation: Transcriptional and post transcriptional
TXNIP readily responds to environmental signals at both protein and mRNA levels in a highly dynamic manner. First identified as vitamin D3 upregulated protein (VDUP1) in HL-60 cells [169], TXNIP was later described under precise control by many factors other than vitamin D3, at transcriptional and post-translational levels (Fig. 2). Above all, the intricate signals involved in TXNIP regulation, AMPK and Akt signaling appear to converge on TXNIP to create a harmonic adaptive system in response to nutritional stress and growth inputs.
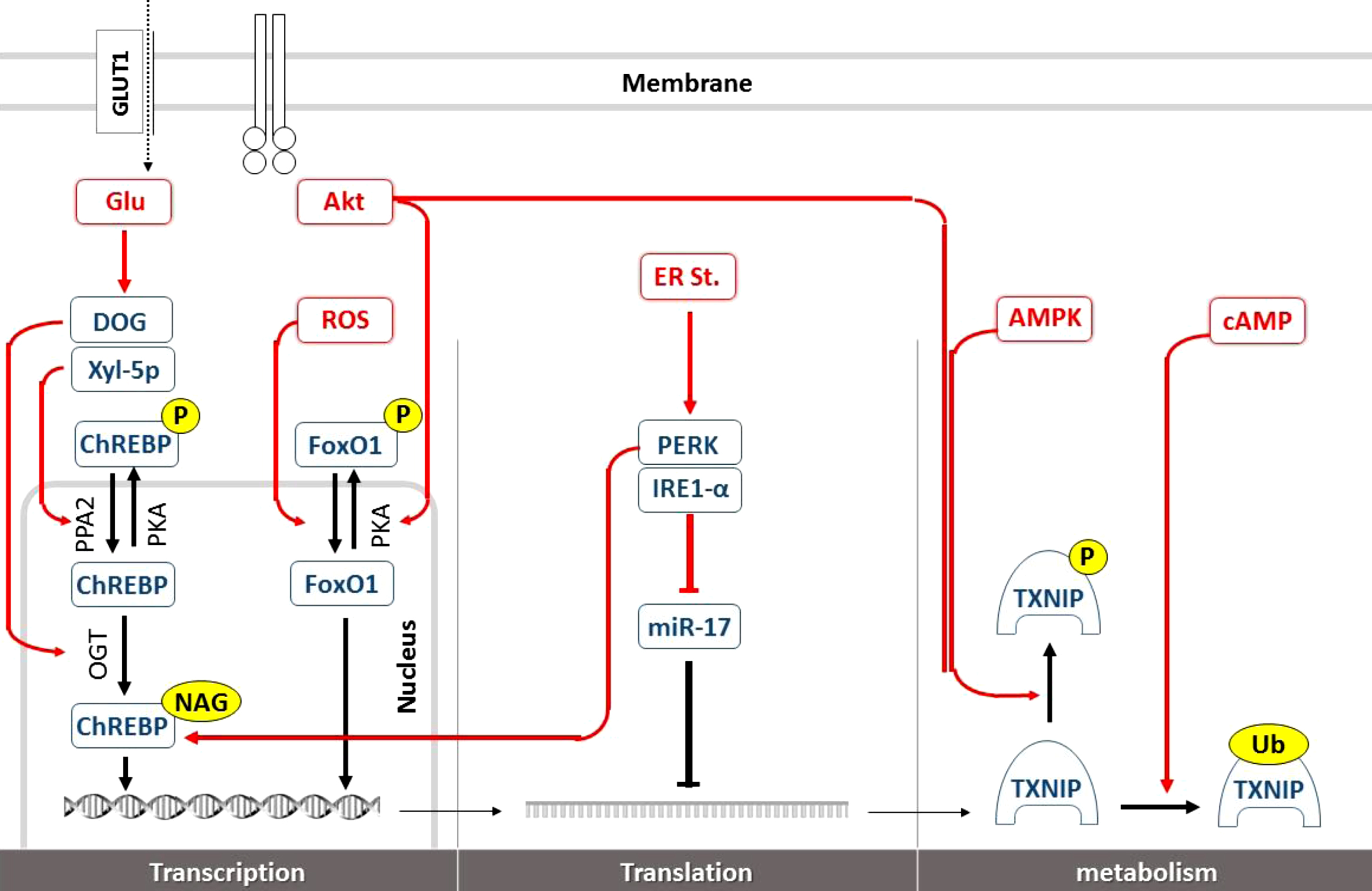
Brain TXNIP regulation at transcriptional and posttranslational levels. Glucose as the principal TXNIP transcripts inducer, is metabolized to Xyl-5p and DOG in neural cytosolic milieu to promote nuclear ChREBP transcriptional activity on TXNIP promoter. Stimulating PPA2 and OGT enzymes, Xyl-5p and DOG may respectively enhance ChREBP dephosphorylation and N-acetylglucosamine attachment increasing ChREBP stability and activity on ChREs. ROS accumulation further enhances TXNIP transcription via FoxO1 DNA interaction. Growth signals like that of insulin improve Akt kinase activity and FoxO1 phosphorylation opposing to TXNIP transcription and allowing for more glucose available for anabolic activities. In normal physiologic conditions, part of TXNIP production is controlled by miR-17 in cytosol where it interferes with TXNIP mRNA translation. Such normal quiescence is interrupted by ER stress blocking miR-17 through IRE-1α, while it is still enhancing ChREBP transcriptional activity on TXNIP through PERK. Following enough translation, TXNIP protein is subject to inactivating phosphorylation which is typically induced by both Akt and AMPK activation working as sensors of growth signals and nutrition deficiency respectively, to improve neural glucose access. More specific to G-protein intracellular second messenger, cytosolic cAMP overload may also help some particular ligands to restrain TXNIP activity by ubiquination which predispose TXNIP to endosomal degradation. GLUT, glucose transferase; Glu, glucose; DOG, 2-Deoxyglucose; Xyl-5P, xylulose-5-phosphate; ChREBP, carbohydrate response element-binding protein; ChREs, carbohydrate response element; PPA2, phospholipase A2; PKA, protein kinase A; OGT, O-linked β-N-acetylglucosamine transferase; NAG, N-acetylglucosamine; ROS, reactive oxygen species; FoxO1, Forkhead box protein O1; PKA, protein kinase A; ER St, endoplasmic reticulum; PERK, protein kinase R-like ER kinase; IRE, serine/threonine-protein kinase/endoribonuclease; miR-17, micro RNA-17; cAMP, cyclic adenosine monophosphate; AMPK, cyclic adenosine monophosphate-activate kinase; TXNIP, thioredoxin interacting protein; Ub, ubiquinone.
At the transcriptional level, TXNIP is regulated thoroughly by at least two seemingly opposing transcription factors: the transcription complexes carbohydrate-responsive element-binding protein (chREBP)/max-like protein (Mlx) and Forkhead box protein O1 (FoxO1) transcription factor. With more nutrients and glucose entering the cells, the increased intermediate of pentose phosphate pathway xylulose-5-phosphate (Xyl-5p) would selectively activate a particular isoform of protein phosphatase 2A (PP2A). PP2A is responsible for cytosolic and nuclear ChREBP activation through its dephosphorylation at Ser196 and Thr666. Upon efficient DNA binding, ChREBP would control a myriad of transcripts particularly those critical in energy metabolism like TXNIP [175]. 2-Deoxyglucose is another recognized effector of glucose metabolites to play a role in the nutrient sensing mechanism and is described to improve TXNIP transcription through inducing O-GlcNAc transferase (OGT) [176], adding N-acetylglucosamine to ChREBP improving its stability and activity [177, 178]. Besides the well characterized glucose overload as a TXNIP inducer, endoplasmic reticulum (ER) stress is known as a potent TXNIP inducer which attains significance in metabolic derangements. One of the established effectors of ER stress, protein kinase R-like ER kinase (PERK) signaling may also enhance ChREBP expression and nuclear translocation leading to increased TXNIP transcription [179].
As the more recently defined transcription factor, FoxO1 also upregulates TXNIP expression in neural and endothelial cells [180]. FoxO1 as one of the predominant forms of the four existing FoxOs in neural cells has been shown to mediate ROS induced TXNIP expression via direct interaction with its promoter [181]. Additionally, FoxO1 is a unique switch point for growth signals to promote glucose uptake via TXNIP suppression. AKT activation by growth factors (e.g., insulin) leads to FoxO1 phosphorylation and subsequent sequestration in the cytoplasm which ends its transcriptional activity and TXNIP overexpression as described in vitro and in vivo [182, 183]. That is while in pancreatic and liver cells, FoxO1 downregulates TXNIP either via competing with the strong inducer ChREBP or inhibiting OGT activity [184].
Furthermore, Akt abolishes TXNIP transcription by FoxO1 phosphorylation and cytosolic translocation resulting to more persisting TXNIP getaway and guarantee the energy required for anabolic processes in a long run [185].
Not surprisingly, TXNIP protein is expected to undergo posttranscriptional modifications to meet immediate metabolic needs and serve as a central gatekeeper for glucose. Following efficient transcription, TXNIP mRNAs are assigned to translation, which provides another checkpoint for intracellular TXNIP hemostasis. Instantly the TXNIP-destabilizing microRNA, miR-17 has been shown to target the 3-UTR of TXNIP mRNA and inhibits the translation process [186]. Intriguingly, in addition to PERK, ER stress has another downstream effector, serine/threonine-protein kinase/endoribonuclease (IRE1-α), which may augment TXNIP protein by reducing miR-17 levels [187, 188]. The fact that glucose uptake may be induced acutely by insulin or energy stress in just minutes, implies TXNIP prompt posttranslational modifications. The TXNIP protein phosphorylation at S308 is among the best characterized posttranslational alterations rendering TXNIP susceptible to degradation and thereby halting its effects including those dealing with glucose transport [189]. Both AMPK and AKT regulating opposing metabolic pathways in context of starvation and growth signals, respectively, have been shown to induce TXNIP S308 phosphorylation and enhance glucose uptake into the non-neural cells. That is while these two signals keep their contrast at TXNIP gene transcription, with AMPK signaling inducing but AKT repressing TXNIP mRNA transcription. With a particular focus on TXNIP transcriptional control by signals attributing to growth stimuli or nutrition deprivation, a recent study by Nagaraj et al. has specified insulin or insulin like growth factor downregulates TXNIP mRNA expression in prostate cancer cell lines. In human embryonic kidney HEK293 cells, serum deprivation was shown to induce a time-dependent TXNIP overexpression which was interestingly reversible by insulin treatment [190].
A logistic adaptive behavior may explain AMPK contrasting effects on TXNIP transcriptional and posttranslational processes, as a hemostatic regulation. Conceivably, in response to lasting energy stress, AMPK stimulates TXNIP gene transcription to adapt the low glucose environment requiring less transporters. In an acute severe nutritional famine, however, endangered cells craving for any available nutrition may benefit from immediate TXNIP inactivation by AMPK-induced phosphorylation/degradation [191]. On the contrary, while cells are exposed to growth signal and Akt signaling is on, Akt abolishes TXNIP transcription by FoxO1 phosphorylation and cytosolic translocation resulting in persisting TXNIP getaway and guarantee the energy required for anabolic processes in a long run [185]. This is well potentiated with the immediate phosphorylation-induced paralysis of TXNIP making the pre-existing TXNIP protein out of work and glucose available for acute influx [192].
Regulated with elaborated mechanisms, both TXNIP mRNA and protein are known as short-lived molecules for rapid catabolism, which is still subject to strong controllers. Interestingly, cAMP signaling involved in many biological functions have been suggested to affect proteasome-mediated TXNIP degradation [193]. This has been postulated to explain the remarkable protective effect of glucagon-like peptide 1 (GLP-1) receptor agonist, exenatide, to repress TXNIP ubiquitination and degradation in β cells of mouse and human reducing apoptotic events [194].
TXNIP function: Associating oxidative stress, inflammation, and type 3 diabetes
According to immense evidence, oxidative stress might be considered as the main path linking peripheral insulin irresponsiveness to central insulin resistance. All along the redox drive, inflammation is also considered as a potent non-negotiable effector in response to toxic metabolites (e.g., high levels of fatty acids and ceramides) to impair insulin signaling. In a simplified view, the feed-forward loop between ROS and inflammatory cytokines seemingly acts as the power of metabolic derangement to drive and efficiently escalate insulin ineffectiveness. In such a picture simplified in Fig. 3, we are to conclude how TXNIP may work as a strong ROS amplifier and a potential driving force.
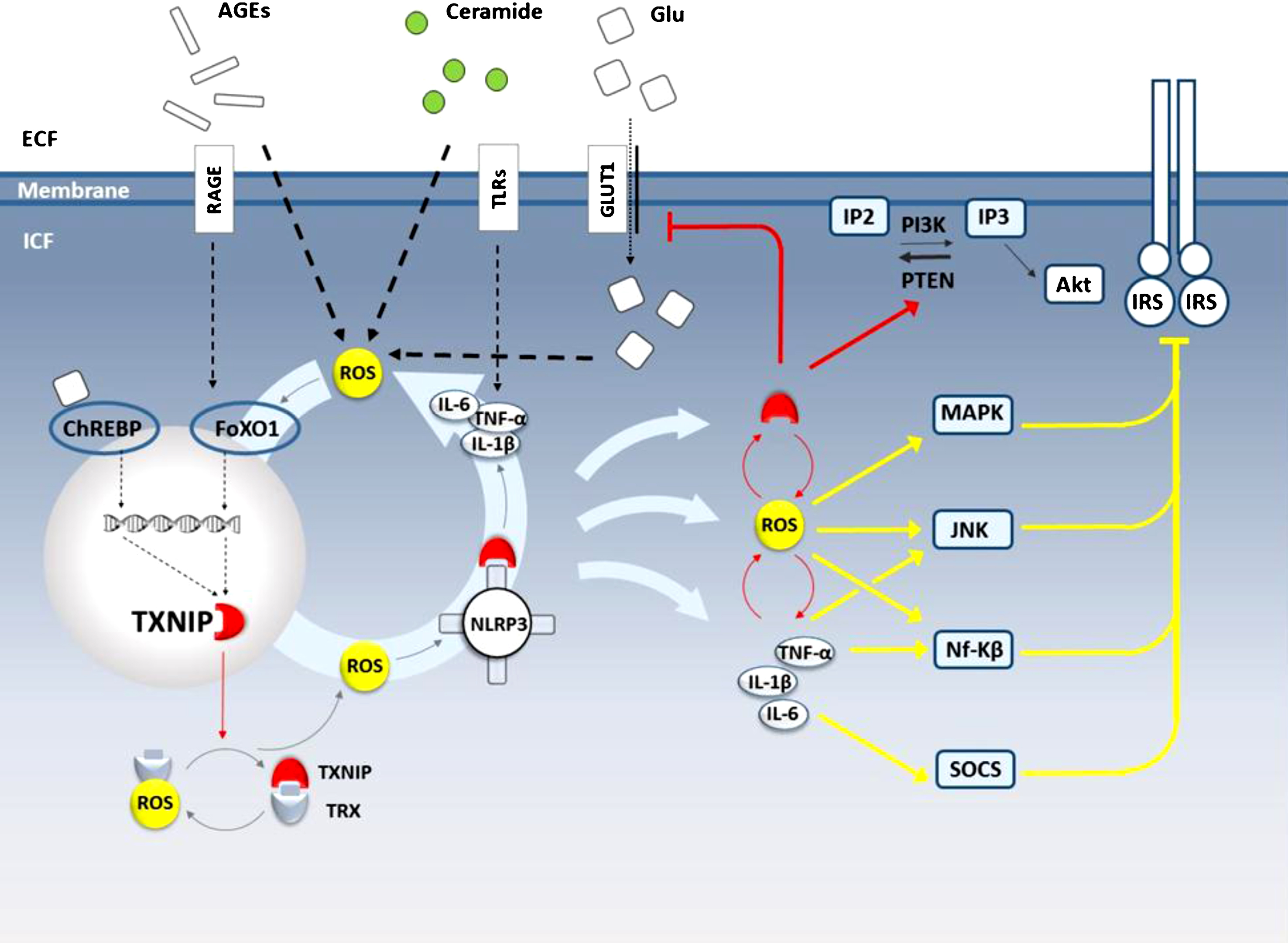
Metabolic syndrome and TXNIP contribution to in insulin resistance in the brain. Following penetration through the BBB, circulating lipids and glucose or their metabolites like AGEs and ceramides converge on ROS generation to improve TXNIP transcript through FoxO1 activation. Through paralyzing TRX antioxidant activity, TXNIP further amplifies ROS liberation which in turn recruits TXNIP to NLRP3 inflammsome activation followed by inflammatory cytokine release. Driving the feedforward loop, TXNIP intensifies the initial ROS, inflammation and its own transcription. Lipid or glucose metabolites may also fuel such amplifying circuit through TLR/NF-κB to increase inflammatory cytokines or via ChREBP activation enhancing TXNIP transcripts. The intracellular ROS and cytokine accumulation overtly trigger intracellular kinases leading to IRS hyperphosphorylation and deactivation whereas particular cytokines reduce the constitutional transcripts act through SOCS activation. Such insulin irresponsiveness might get escalated by TXNIP-induced endocytosis of GLUT-1. Importantly, TXNIP may directly activate PTEN to reduce intracellular IP3 pool leading to Akt inactivation and thus inhibiting much of insulin receptor functionality. AGE, advance glycation product; RAGE, receptor for advance glycation product; TLRs, tool like receptors; Glu, glucose; GLUT, glucose transporter; IP3, inositol three phosphate; IP2, inositol two phosphate; IP3K, inositol three phosphate kinase; PTEN, phosphatase and tensin homolog; IRS, insulin receptor substrate; ROS, reactive oxygen species; TRX, thioredoxin; TXNIP, thioredoxin interacting protein; ChRE, carbohydrate-responsive element; ChREBP, carbohydrate-responsive element-binding protein; FoxO1, forkhead boxO1; NLRP3, NOD-like receptor pyrin domain-containing-3; MAPK, mitogen-activated protein kinase; JNK, c-Jun N-terminal kinases; Nf-KB, Nuclear factor-κB; SOCS, suppressors of cytokine signaling.
ROS has been described as a strong TXNIP inducer with comparable potency to glucose. High glucose exposure in rat mesangial cells was primarily shown to improve TXNIP mRNA and protein levels; the phenomena was then suggested to take place via ROS generation [195]. With the emergence of further findings described in both in vivo and in vitro models of ischemic reperfusion injury, it was later confirmed oxidative stress can induce TXNIP expression with or without high glucose exposure [196, 197]. As a common effector for many deteriorating signals, ROS is now known to direct the message of a variety of danger signals to go through TXNIP signaling. However, the exact mechanism of ROS/TXNIP interaction is not yet characterized, enhanced FoxO1 transcriptional activity is likely to be involved [180].
Enhanced TXNIP protein levels and activity produce divergent effects in different cellular compartments [198]. The TXNIP protein mostly residing in nucleus in normal conditions is responsible for regulating transcripts genes (including its own) namely by improving dephosphorylation and activation of the transcription factor ChREBP [199]. Many of the effects of TXNIP on metabolism instantly take place through its transcriptional effects as described later in the pertinent section. Besides inducing overall TXNIP expression, oxidative stress makes TXNIP translocate to the mitochondria, oxidizes the antioxidant molecule thioredoxin type II (TRX2), and makes it useless to scavenge more ROS [200]. Occupied by TXNIP, TRX2 then has to leave ASK1 to drive cytochrome C release and apoptosis, as a significant event in β cells contributing to lessen insulin reservoirs [198]. Coupled with the later discovery of TXNIP shuttling to plasma membrane to eliminate GLUT1 [191], this might suppose the significance of TXNIP contribution to metabolic syndrome as a systemic malaise, independent of ROS or inflammation.
While dealing with insulin resistance in the brain, the role of TXNIP in insulin dependent glucose uptake is almost indiscernible. Alternatively, the cytosolic TXNIP besides the shuffled TXNIP to the mitochondria, may act as a key amplifier for ROS and inflammation to invade cerebral insulin signaling constituents. As pointed above, TXNIP has a prominent effect to demolish TRX antioxidant activity, which has well described TXNIP as a core place in redox biology, where ROS are both up and downstream molecules. That is while ROS act as upstream TXNIP inducers, genetic silencing of TXNIP remarkably reduces glucose-induced ROS generation in human aortic endothelial cells [180]. In fact, the redox-related protein complex TRX/TXNIP, named “redoxisome” [201], is a core regulator for ROS signalling and has been defined as a critical point in propagation and pathogenesis of degenerative diseases [202]. Regarding the imminent role of ROS to modify several constituents of insulin signalling, ROS propagation is definitely an immediate path for cerebral TXNIP to impair brain insulin functions.
The other conceivably stronger effector of TXNIP in type 3 diabetes is the cytosolic multi-compartment complex NLRP3 inflammasome mediating eminent inflammatory responses in face to ROS overload, as described earlier. NLRP3 inflammasome is the main component of sterile inflammation and thus has a remarkable role to escalate insulin signalling while it has an established role in animal models of acute brain injury [203] and degenerative disorders as well [204]. Noteworthy, TXNIP has been described as the main link between ROS and subsequent NLRP3 inflammasome activation, IL-1β secretion, and spread of inflammatory cytokines like TNF-α through pyroptosis. According to the initial examinations on macrophages exposed to different NLRP3-inflammasome activators (e.g., ATP or urate crystals), TXNIP binding has been demonstrated as an essential step for NLRP3 inflammasome activation [205]. Empirical evidence firmly supports a direct TXNIP/NLRP3 interaction with details remain to be unravelled. According to the molecular modelling estimates, TXNIP/NLRP3 association cause a conformational change in the NLRP3 protein pyrin domain [206] or S-nitrosylated sites in NLRP3 [207], which suggestively are responsible for more IL-1β maturation. Following enough instant NLRP3 activation by ROS, the inflammatory responses besides the following release of cell debrides accelerate ROS generation and bring more damage associated molecules ready to induce TXNIP/NLRP3 association and cytokine release. Within such elaborated loop, TXNIP represent a sophisticated point to enforce, empower, and amplify the initially imposed redox and immune signals invading to the brain by circulating toxic metabolites (either glucose, fatty acids, or their metabolites), to successfully endanger insulin functionality in a brain that appears “open” to circulatory metabolic pollutants.
TXNIP CONTRIBUTING LINKS TO AD
TXNIP co-expression with amyloid polypeptides in islet β-cells provides a view on the causative role between AD and key components of metabolism [208]. Aβ or NFTs are definite TXNIP/NLRP3 inflammasome activators as they drive the inflammatory deteriorating signals [209–211]. Therefore, at least part of TXNIP overexpression associated with AD brains is conceivably the consequence of the disease itself. Recently, we and others have determined TXNIP upregulation in diabetes [212], stroke [197], and AD [202]. TXNIP has been recognized as one the most overexpressed genes in the hippocampus of both AD patients and 3Tg transgenic AD mice [213]. Our preliminary data coupled with a recently published study further postulate an important role for TXNIP in AD [211]. TXNIP is required for NLRP3 inflammasome activation [214], a recently discovered mechanism mediating inflammatory response in several diseases including AD [200, 215]. Regarding the data indicating blocking NLRP3 signaling rescues cognitive function in AD mouse [216], these findings suggest that TXNIP may play a causative role in AD pathology and serve as an upstream molecular link between brain insulin resistance and AD. Therefore, to deliberately picture TXNIP in a causative point, here we come up with our conclusion around all TXNIP presumable effects to link insulin resistance (i.e., in metabolic syndrome, T2DM or type 3 diabetes) to AD progress.
TXNIP and insulin level
While cytosolic and mitochondrial TXNIP primarily control oxidative state, inflammation, and metabolism, much of TXNIP is localized in the nucleus where it downregulates many target genes like PPAR-γ. According to the emerging data on a wide range of proteins appeared to be impressed, TXNIP may regulate insulin transcripts at different levels. First, miR-204 was shown to mediate TXNIP-induced inhibition of insulin production [217]. Shortly afterwards, it was shown that miR-200 expression is upregulated by TXNIP in pancreatic β–cells leading to downregulation of the transcription factor zinc finger Ebox-binding homeobox 1 (Zeb1) and β-cell apoptosis [218, 219].
TXNIP also has repressive effects on miR-124a expression known to enhance expression of islet amyloid polypeptide protein (IAPP) [208, 209] aggregating into amyloid fibrils and driving β-cell cytotoxicity [209, 220]. According to a recent study, β–cells proliferation and the subsequent insulin production could also get inhibited by TXNIP transfection in islet cells in vitro [221]. Such suppressive effects, however, demolish insulin production reservoirs and may not readily affect CSF insulin content.
More importantly, TXNIP may affect the insulin available to the brain tissue by perturbing endothelial cell (EC) nitric oxide production. In fact nitric oxide (NO) signaling directly improve EC insulin transport by enhancing protein S-nitrosylation [222]. In this line, TXNIP–/–macrophages have been shown to produce meaningfully higher levels of NO and inducible nitric oxide synthase (iNOS) in response to LPS [223]. Further in vivo studies then implied blocking the TXNIP/NLRP3 inflammasome activation is associated with reduced ROS but increased NO generation in vascular endothelium [224] and corroborated with more recent evidence indicating TXNIP/NLRP3 inflammasome inhibits endothelial nitric oxide synthase (eNOS) and NO production in HUVECs [225] or in mice kidney exposed to long-term administration of carbonyl-AGE [226]. Given NO represses the transcription of TXNIP by itself, this may conceivably produce a control feedback loop [227].
TXNIP and cerebral insulin resistance
As illustrated in Fig. 3, the established TXNIP overexpression in conditions associated with lipotoxicity and nutrition overload produces major pathways to impair insulin receptor sensitivity in the brain [228]. Noteworthy, the influence of TXNIP in insulin production has not been included. As described in details, demolishing TRX antioxidant activity and amplification of inflammation and oxidative stress provides TXNIP a broad path to direct the abrogating message of many ROS generating stimuli to turn down insulin signaling in the brain (e.g., via IKKβ or GSK3 activation). However, alternate pathways may also explain TXNIP-induced insulin resistance in the brain. The redox sensitive TXNIP effector, phosphatase and tension homolog (PTEN) was later shown to render TXNIP capable of Akt inhibition [229, 230]. PTEN is a lipid phosphatase that confines the pool of PIP3 by catalyzing dephosphorylation of PIP3 to phosphatidylinositol and thus limits subsequent Akt activation [231]. Thus, maintaining PTEN in a reductive (i.e., active) state, TXNIP negatively regulates and deters much of insulin signaling effects. It is important to note that solid evidence implies at least part of TXNIP effects on insulin resistance is redox independent. Accordingly, the redox insensitive Cys-247-Ser mutant of TXNIP with no TRX interaction has been characterized as an effective glucose uptake inhibitor similar to wild-type TXNIP [232].
TXNIP and metabolism
Glucose utilization has been shown to remarkably decrease in CNS cells of AD brains. This is a hallmark of AD brain and plays a pivotal causative role in cognitive dysfunction [233, 234]. Such hypothesis is in line with evidences showing association between impaired ATP production [235], starvation and insulin-induced hypoglycemia with sever cognitive impairment. Specifically dealing with feature of AD brains, the decline in glucose utilization in parieto-temporal regions precedes the reduction in blood flow and oxygen consumption, becoming similar at the later stages of AD [236]. In fact, with such evidence prioritizing disrupted brain metabolism as a fundamental event to encourage Aβ and tau pathology, there is an emerging interest on pertinent effectors. TXNIP as a critical switch in glucose utilization has much to offer to prohibit glucose metabolism as is extensively reviewed elsewhere [171]. In brief, TXNIP looks to work as a fine-tuning switch for cellular metabolism and glucose consumption through regulating glucose uptake, glycolysis, and OXPHOS process in the brain tissue.
TXNIP has been shown to provide a baseline GLUT1 endocytosis via clathrin-coated in almost all body cell types and acutely increase glucose influx. Loss of TXNIP expression in HepG2 cells has been also shown to dramatically decrease GLUT1 mRNA leading to lasting GLUT1 protein downregulation. In addition to regulating GLUT1, TXNIP has been proven to facilitate GLUT4 endocytosis and thus confine insulin-dependent glucose uptake under basal conditions [191]. While reducing glucose influx, TXNIP also appears as a key point in glucose metabolism via different ways [237]. As an instance, TXNIP presumably may inhibit glycolysis as in murine embryo’s lung, it has been shown to inhibit hypoxia-induced transcription factor (HIF1α) expression which is known to induce the expression of key glycolytic enzymes [238]. In the mechanistic basis, in vivo and in vitro examinations have demonstrated TXNIP may assemble with von Hippel–Lindau protein (pVHL) and the beta-domain of byprolyl-hydroxylases to interact with HIF-1α and enhance the ubiquitin-induced degradation of HIF-1α. This would end with nuclear export of HIF-1α and cytosolic degradation [239]. The repressed glycolysis by TXNIP is supposed to reduce pyruvate production required for citric acid cycle leading to less NADH fed to oxidative phosphorylation into mitochondria
However, this may suggest TXNIP shuts down the intracellular glucose consumption; such a conclusion is not easy for the contradictory evidence. That is HIF1α also inhibits OXPHOS to reduce mitochondrial biogenesis as a part of adaptation to hypoxic conditions. With studies on cancer cells, this is thought to take place through inhibiting pyruvate dehydrogenase (PDH) complex and interfering with tricarboxylic acid (TCA) cycle though [240, 241] and recently has been supposed to go through AMPK suppression [242]. Therefore, TXNIP repressive effects on HIF1α may not necessarily conclude reduced glucose consumption while the decreased glucose uptake still is a strong basis.
Above reducing glucose uptake in almost all cell types including neural cells, hypothalamic TXNIP might play a sophisticated unique role in tuning body metabolism and energy expenditure. In fact, hypothalamic TXNIP has been shown to affect the whole-body nutrient availability, utilization, and partitioning. HcB-19 as well as TXNIP-null mice have been shown hypoglycemic and hyperinsulinemic with a 40% increase in fat to muscle ratio [229, 243]. Hypothalamic TXNIP overexpression in particular, has been shown to increase body fat and decrease energy expenditure, as valid indexes for reduced metabolism. That is while hypothalamic TXNIP known as a metabolic sensor is induced by acute nutrient excess in mice [170].
It is of note, although the real mechanism of disrupted glucose metabolism in AD brains is surrounded by skepticism, some findings imply the established early sharp synchronous discharges in cortical and hippocampal circuits [244, 245] act as the main driving force to impair intracellular glucose metabolism. Intracellular Ca2 + overload and glycolytic enzymes have been proposed to act as the main mediators [246]. In this line, in a recent study confirming acute hyperglycemia increases Aβ production [247, 248], induction of ATP-activated potassium (
TXNIP and tau pathology
As described earlier, P38 MAPK activity is of established value in bridging oxidative stress to insulin resistance. There is no surprise therefore, TXNIP as a fundamental component of oxidative stress is an activator of this kinase. Once it comes to AD pathology, p38 MAPK phosphorylation activity is in rather high significance as it plays a discreet role in tau pathology. Oxidative stress has been empirically shown to stimulate tau phosphorylation by inducing p38 MAPK activation. In cultured neurons, Aβ has been shown to activate p38 MAPK (p38) and tau hyperphosphorylation, which was prevented by p38 MAPK inhibition. Interestingly the in vitro findings paralleled in vivo results in APP/PS1 transgenic mice exhibiting a high level of P-p38 in the hippocampus but not in cortex [250]. Tau is postulated to get phosphorylated at Ser202/thr205 sites following p38 MAPK interaction as shown in neural cells in vitro [251]. TXNIP co-localization with thioredoxin leading to dissociation of TRX from ASK1 and the consequent p38 MAPK and JNK activation might be considered as the main way TXNIP may prompt p38 MAPK phosphorylation activity [252, 253]. This phenomena also leads to chromatin modification and is known to take part in TXNIP-induced apoptosis [180, 254] as well as inflammation in retinal endothelial cells under diabetic conditions in vitro [255]. The presumptive contribution of TXNIP to tau phosphorylation has been confirmed by a recent report indicating Aβ1 - 42-induced tau phosphorylation is ameliorated by TXNIP inhibition which abrogates p38 MAPK phosphorylation and subsequent tau phosphorylation at Ser202/Thr205 in SH-SY5Y cells or in the hippocampus of the 5xFAD mice [211]. Furthermore, the intriguing connection might be indirectly affirmed with an earlier study demonstrating high tau phosphorylation in brains with high levels of IL-1β, as the immediate product of TXNIP/NLRP3/Caspase-1 activation. The evidence has been produced by observing higher tau phosphorylation and 4–6 fold increase in plaque formation as well as microglial activation at the site of neuroinflammation in transgenic mouse model of AD, with sustained IL-1β overexpression (IL-1βXAT). The changes paralleled with increased p38MAPK and GSK3β activity contributing to tau phosphorylation [256].
TXNIP and autophagy deregulation
Cortical biopsy from AD brains demonstrates abundant autophagic vacuoles within neurite processes in AD brains. In fact, the profound accumulation of immature autophagosomes in dystrophic neurites is suggestive of the impaired authophagy probably in the transportation system required to deliver un-functional proteins (e.g., NFTs) to lysosomes to maintain cellular homeostasis [257, 258]. Further studies unraveled TXNIP contributing effects on reducing autophagic clearance in hyperglycemic conditions. Both in mice with diabetic nephropathy and in cultured human renal proximal tubular cells exposed to HG, the concurrent increase in TXNIP and markers of (LC3/LC3-II and p62) is associated with a rise in autophagic vacuoles where TXNIP siRNA transfection reversed the due observations [259]. This was corroborated with later evidence indicating reduced autophagic clearance in tubular cells of human diabetic kidneys is reversed by TXNIP DNAzyme. With more mechanistic in vitro experiments, TXNIP was suggested to control autophagy through activation of the mTOR signaling pathway [260]. However, more precise examinations showed TXNIP may augment autophagy through formation of a complex between TXNIP and regulated in development and DNA damage responses 1 (REDD-1). That is REDD-1, which rapidly inactivate mTORC1 in a TSC1/2-dependent manner, may form a complex with TXNIP and suppresses redox-sensitive ATG4B cysteine endopeptidase activity. ATG4B repression then reduces LC3B delipidation and improves autophagy. In support of the hypothesis REDD1–/–mice show deregulated ATG4B activity and enhanced stress-induced autophagic flux in rats brain [261]. REDD1 suppression and reduced REDD1/TXNIP formation have been also shown to augment autophagy in human and mouse articular cartilage [262]. However apparently controversial to the impaired autophagy by TXNIP, these findings underline how TXNIP overexpression may correlate with immature autophagosome accumulations. In support of this, recent experiments indicate impaired autophagic flux and α-synuclein accumulation in HEK293 cells transfected with TXNIP plasmid. Furthermore, TXNIP overexpression has been shown to associate with loss of dopaminergic neuron in substantia nigra in animal model of Parkinson’s disease [263].
TXNIP AS A REASONABLE TARGET IN AD
TXNIP expression in the brain is rather confined to astrocytes and its constitutive presence in hypothalamic neurons sensing nutrient excess has no direct relevance to memory performance [168, 264]. As a delicate sensor for damage signals, TXNIP expression shows dramatic alteration in several malaise including those contributing to AD, namely diabetes [265] and cerebrovascular disorders [266]. In fact, part of hypothesized TXNIP role in AD pathology stems from data on their coincidence. As described in details, the intracellular and systemic feature of diabetes and metabolic syndrome; are associated with robust TXNIP activation, which is also an early event in AD. That is in 5xFAD mice as an early AD phenotype, TXNIP is overexpressed in the hippocampus and in the enthorinal cortex well before cortical neural apoptosis [202, 268]. TXNIP overexpression has been also detected in hippocampal capillary endothelial cells in these AD mice [267]. D-galactose-induced rat model of AD has also shown augmented expression of TXNIP [269].
The significance of TXNIP contribution to the progress of AD is being more demonstrated with the emerging information from recent investigations. With the recent findings on NLRP3 inflammasomes as a strong TXNIP effector, neuroinflammation and apoptosis were sought as main TXNIP effectors to abolish cognitive dysfunction associated with aging and ischemia. For instance, in an age-related model of AD, D-galactose receiving rats, the therapeutic advantages of the phenylpropanoid glycoside, solidroside, was shown to associate with repressed TXNIP activity [269]. A recent precise investigation into the role of TXNIP in AD pathology has shown chronic accumulation of inflammatory mediator in human neuronal cells facilitates TXNIP-NLRP3 and NLRP3-ASC, interaction leading to increased Aβ secretion. The study delineated antioxidant Dl-3-n-butylphthalide suppresses TXNIP and mitigate neuronal apoptosis in the mouse AD brains [270]. The neuroprotective effect of acupuncture in vascular dementia associated with global ischemia induced by occlusion of both common carotid arteries, has been also shown to concur with remarkable TXNIP, NLRP3, caspase-1, and IL-1β repression. Importantly, treatment with TXNIP siRNA had a therapeutic advantage on cognition and hippocampal neurons comparable to acupuncture in vascular dementia rats [271].
To more specifically address AD brain hallmarks, empirical evidence confirms TXNIP direct contributing effects to Aβ or tau pathology. As of fundamental findings unraveled in islet cells, TXNIP was found to reduce miR-124a expression, which in turn decrease FoxA2 expression and human IAPP mRNA, contributing to cellular apoptosis [208]. Further examination then highlighted the implication of TXNIP in tau phosphorylation in AD brains. Interesting findings first established associations between IRE1α and PERK phosphorylation, TXNIP and NLRP3 expression, and tau phosphorylation in SH-SY5Y cells exposed to okadaic acid, which was further ameliorated by quercetin, a natural flavonoid [210]. This has been more recently corroborated with result showing Aβ1 - 42 leads to TXNIP overexpression, p38 MAPK activation, and tau phosphorylation at Ser202/Thr205, which were all prevented by TXNIP silencing. In this work suggesting the involvement of excitotoxicity, the calcium channel blocker Verapamil was shown to prevent TXNIP expression and tau phosphorylation at in the hippocampus of the 5xFAD mice [211].
On the contrary side to the documented evidence positing TXNIP as trigger to drive AD, TXNIP together with its joint effector NLRP3 inflammasome has been also frequently defined as downstream to Aβ and NFT pathology to amplify the consequent neuroinflammation and oxidative stress. As significant proposed mechanisms in this line, the membrane receptor for glycation end-products (RAGE) is proposed to mediate Aβ and NFT effects to induce TXNIP. In fact, remarkable TXNIP expression has been recorded in rat cerebral capillary endothelial cells lines, RBE4 cells, exposed to Aβ or hyperglycemia. Given the specific blockage of RAGE reversed TXNIP induction, RAGE was postulated to mediate the effect [267]. The significance of RAGE stimulation comes to remarkable significance when dealing with metabolic syndrome and brain diseases with the established rise in circulatory levels of AGE [272, 273]. Noteworthy, RAGE co-localization with TXNIP in brain astrocytes may further support the contribution [168, 274]. Nevertheless, the place of RAGE/TXNIP axis in AD progress still requires more verification; the importance of TXNIP/NLRP3 inflammasome to engage microglial inflammatory responses are unnegotiable in AD pathology or its risk factor diabetes mellitus [85, 204].
CONCLUDING REMARKS
The intricate cross talk between the periphery and the brain might explain the increased risk of AD with systemic disorders. Inflammation and oxidative stress as the pivotal involved pathways are strongly amplified with TXNIP function to drive the feedforward loop to connect metabolic derangements to inflammatory AD brains. Nevertheless, not all diabetic patients are affected by AD [151] indicating that such cross-linking effects are not always practically deteriorating. Accordingly, the existing evidence implies the TXNIP overexpression is likely to link AD to T2DM in the periphery or type 3 diabetes within the brain. Given that deficient brain insulin signaling, particularly in hippocampal formation, has been demonstrated as a typical feature in AD patients [275, 276], the capability of TXNIP to aid insulin resistance and metabolic dysfunction in the brain forms a strong basis for TXNIP contribution to AD. The fundamental link between brain insulin resistance and metabolism with AD has been intriguingly addressed by recent analysis on human brain imaging. Accordingly, a close spatial correlation has been detected between expression of genes implicated in insulin resistance (e.g., GLUT1) and tau or Aβ pathologies [277]. Importantly, TXNIP overexpression has been confirmed in postmortem human AD brains. That is microarray data as well as real time transcripts evaluations has specified TXNIP amongst the four significantly upregulated genes in AD hippocampus [213]. Such findings have been further confirmed by enhanced TXNIP immunostaining in human postmortem AD brains [270]. In our recent unpublished study on postmortem AD hippocampus and cortex compared with age- and gender-matched controls, we consistently detected a significant co-localization of TXNIP and inflammasome components in the vicinity of Aβ plaques, as determined by double immunostaining. Such coincidence affirming the hypothesis of TXNIP involvement in AD pathology might also implicitly support the most recent perspectives on AD etiology in which baseline differences in the metabolic reliance on glycolysis and insulin signaling determine the vulnerability of different brain regions to AD pathology [278]. To verify TXNIP involvement in AD development, rigorous investigation is required to spot the actual association between the hallmarks of insulin resistance, AD pathology and T2DM-related genes and the corresponding alterations after pharmacologic or genetic TXNIP ablation. The significant contribution of TXNIP in brain insulin resistance and AD suggests that therapeutic strategies focused on specific targeting of TXNIP and inflammasomes may work as promising therapeutic approaches in AD.
Few neuroprotective agents like butylphthalide and salidroside could modulate the expression of TXNIP and mitigate injury in animal models of AD. Nevertheless, no TXNIP inhibitor have yet been tested in clinical trials. The current lack of specific inhibitors probably has limited TXNIP direct targeting as a potential therapeutic tactic. Appropriate molecular modeling and drug development approaches may ease specific TXNIP targeting. TXNIP is established to induce cell cycle arrest and drive apoptotic events [279, 280], and pan-TXNIP inhibitors are highly concerned for neoplastic potentials. Thereby, highly specific ligands blocking particular TXNIP pharmacophores or partial agonist inhibitors retaining part of TXNIP biological activities may provide practical lead compounds to avoid potential side effects subsequent to intensive TXNIP ablation.