
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder, where neuroinflammation and immune cells are key pathological factors. Recently, it was suggested a possible association between AD and human herpesvirus 6 (HHV-6) infection. Since we recently observed that multiple sclerosis patients with KIR2DL2 expression on natural killer (NK) cells are more susceptible to herpesvirus infection, we tested the possible implication of KIR/HLA genetic for HHV-6A infection. We identified, for the first time, a possible implication of a specific KIR/HLA subset in AD. The combination KIR2DS2/KIR2DL2/C1 correlated with a lower MMSEDi score, representative of a severe AD status and an increased susceptibility to HHV-6A infection. Therefore, the results seem to converge on the hypothesis that herpesvirus infection might play a role in AD. If this hypothesis finds experimental confirmation, a new therapeutic strategy, modulating KIR2DL2 expression on NK cells, for AD might be envisaged.
INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disease worldwide, representing 60–70% of all dementia cases [1]. Neuroinflammation is a key factor in AD pathology, and immune cells play a functional role in modulating soluble molecules including neuroendocrines, neurotransmitters, and cytokines [2, 3]. Several studies have reported that the presence of proinflammatory cytokines and lymphocyte subset distribution can be disturbed in AD [2, 3]. Natural killer (NK) cells are key effectors of the immune system, due to their human leukocyte antigen (HLA)-restricted cytotoxic activity. NK inhibitory receptors prevent NK cell-mediated attack against normal cells whereas cells with compromised HLA class I expression (e.g., by tumor transformation or viral infection) are susceptible to NK cell-mediated killing. There are two main types of NK inhibitory receptors: 1) killer Ig-like receptors (KIRs), specific for determinants shared by groups of HLA-A, HLA-B, or HLA-C allotypes [4] and 2) CD94/NKG2A, recognizing HLA-E [5]. Inhibitory KIRs have long cytoplasmic tails (KIR-L) containing two immunoreceptor tyrosine-based inhibition motifs (ITIMs) that, upon phosphorylation, shut down NK cell activation. Each KIR displays two (KIR2DL) or three (KIR3DL) extracellular Ig-domains conferring specificity for HLA allotypes. The inhibitory KIR2DL1, KIR2DL2, and KIR2DL3 receptors recognize distinct HLA-C allotypes, KIR3DL1 ligands are HLA-B molecules with the Bw4 epitope, and KIR3DL2 binds HLA-A3 and HLA-A11.
Although the immunobiology of NK cells in neurodegenerative diseases such as multiple sclerosis (MS) is well studied, little is known regarding their role in AD [3]. NK cells have two main functions: killing of target cells and cytokine production. NK cells killing takes place by secretion of perforin and granzymes and mediators of apoptosis (TNF-related apoptosis-inducing ligand and FasL). NK cells may secrete both immunostimulatory and immunomodulatory cytokines such as interferon (IFN)-γ, TNF-α, granulocyte monocyte colony-stimulating factor, IL-5, IL-13, IL-10, and transforming growth factor (TGF)-β. The immune system in AD patients is severely affected and several studies reported that proinflammatory circulating cytokines and lymphocyte subset distribution can be disturbed in AD [3]. The implication of NK cells in AD immunopathogenesis was observed by Krishnaraj [6] who showed that cholinesterase inhibitors could suppress expansion and cytotoxic function of NK cells in AD patients, with an improvement of clinical status. Araga et al. [7] showed that the functional potential of NK cells in AD patients was significantly lower than in normal controls. Also, other researchers reported decreased NK cytotoxic function [3]. Thus, evaluating the role of NK cells in the immunopathogenesis of AD may open new horizons for designing novel therapeutic methods.
In our recent studies on MS, we observed that patients with KIR2DL2 expression are more susceptible to herpesvirus infection [8, 9]. In particular, MS patients positive for KIR2DL2 expression on NK cells failed to control herpes simplex virus 1 (HSV-1) infection and secreted high levels of Th17 cytokines, while MS patients negative for KIR2DL2 on NK cells had an efficient killing of virus-infected cells and released Th1 cytokines. Furthermore, we have evidence that the lack of activation of KIR2DL2 positive NK cell takes place in the presence of other human herpesviruses such as EBV and HHV-6 (Rizzo et al., manuscript in preparation). A lower NK cells intracerebral surveillance triggered by herpesvirus infection could have an important role in MS pathogenesis.
AD has been associated with a possible infective etiology [10], and almost three decades ago was suggested for the first time a possible involvement of herpes simplex virus type 1 (HSV-1) [11]. The ever-increasing number of these studies warrants re-evaluation of a possible association between HSV infection and AD. In humans, herpesvirus brain infection has been associated with AD-like pathology [12–14]. In mice and in cell culture, amyloid-β (Aβ) deposition and tau abnormalities typical of AD are observed after infection with HSV-1 [15–18], and a direct interaction between AβPP and HSV-1 has been reported [19]. Antivirals, including acyclovir, block in vitro HSV-1-induced Aβ and tau pathology [20]. HSV-1 reactivation and seropositivity correlate with incident AD [12, 13] and the use of antiherpetic drugs reduces the relative risk of senile dementia by a factor of 10 [14]. Apolipoprotein E (APOE), a risk factor for AD, increases HSV-1-induced brain damage [21]. Inside the brain, ApoE, characterized by three allelic forms: APOE2, APOE3, APOE4, helps to clear Aβ, a plaque component. ApoE ɛ2 appears to perform this function more effectively than ApoE ɛ4, with ApoE ɛ3 in the middle. Interestingly, ApoE ɛ4 is a risk factor for vascular causes of cognitive impairment and dementia [22, 23], can increase the risk of atherosclerosis and stroke [24], may induce the response to stress or injury [24], increases the risk for AD, and lowers the age of onset [25].
Furthermore, it has been suggested that there is a cumulative correlation between multiple herpesvirus infections and cognitive decline in patients with vascular disease [26]. Also, a connection between cytomegalovirus (CMV) and AD was made, showing that CMV IgG and IFN-γ levels in cerebrospinal fluid correlate with neuropathologic characteristics of AD [27] and with an increased rate of cognitive decline [28].
Recently it was suggested a possible association between AD and human herpesvirus 6 (HHV-6) infection [29]. The authors, constructing multiscale networks of late-onset AD, observed increased presence of HHV-6A in AD patients than in controls, suggesting viral activity as a general feature of AD. HHV-6 is classified in two distinct species, HHV-6A and HHV-6B [30], but the term HHV-6 remains in usage for both species. The two viral species are quite similar, sharing many similarities, but differ for cell receptors, specific tropism, and disease association. HHV-6 preferentially replicates in activated CD4+ T lymphocytes [31, 32], but can infect also NK cells, endothelial cells, astrocytes, oligodendrocytes, and microglial cells [31]. The in vivo host tissue range of HHV-6 includes brain, tonsils, salivary glands, kidneys, liver, lymph nodes, endothelial cells, and monocytes/macrophages [31, 33–35].
Interestingly, similarly to MS, AD patients also show decreased NK cell activity [2]. These two observations (i.e., HHV-6A presence and NK cell decreased activation) might support the hypothesis that NK cells and herpesvirus infection might contribute to AD immune-pathogenesis.
Our aim was to test the possible implication of KIR/HLA genetic background in AD, and its association with HHV-6A infection.
MATERIALS AND METHODS
Study subjects
We recruited 98 patients (55–87 years old) with an AD clinical diagnosis in accordance with the criteria of the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association (NINCDS–ADRDA) [36]. These criteria include a mild-to-moderate severity, neuroimaging analysis, and a Mini-Mental State Examination (MMSEi) [37] score of 11.0 to 29.0 at diagnosis (Table 1). MMSE values were corrected by age and educational level as proposed by Magni et al. [38]. After correction, the MMSEi scores ranged from 12.3 to 24.7. We recruited 1 severe, 42 moderate, and 55 mild patients. All patients were recruited at the Alzheimer Center of the University Hospital, Arcispedale S. Anna, during the 2004-2006 period. Disease severity was re-evaluated after one year of follow up. The study was approved by the Ethics Committee of the University of Ferrara, and the subjects and the caregivers consented to the study.
Baseline characteristics
MMSEDI, Mini-Mental State Examination at diagnosis, corrected by age and education; ADLDI, Activities of Daily Living at diagnosis; IADLDI, Instrumental Activities of Daily Living at diagnosis. Higher score indicates better performances in normal daily activities (ADL, IADL) or better cognitive functions.
Healthy members of societies for retired people were recruited for the cognitively-normal control group. Only ethnic Italians without known dementia in first degree relatives were included.
Genotyping KIR and HLA
Genomic DNA was isolated from whole blood using the QIAamp DNA Blood Mini kit (QIAGEN, Hilden, Germany). Genotyping of KIR and HLA alleles was performed by Olerup typing kit (West Chester, PA, USA) following manufacturer’s protocols.
Haplotype reconstruction
Maximum-likelihood KIR haplotype frequencies were estimated using an expectation–maximization (EM) algorithm implemented in Arlequin 3.1 [39]. KIR-inferred haplotypes were compared with those available on the web site of the official allele nomenclature (https://www.ebi.ac.uk/ipd/kir/nomenclature.html).
Genotyping APOE
APOE alleles were determined by genotyping at two single nucleotide polymorphisms (SNPs), rs7412 and rs429358. Genotyping was performed using predesigned Taqman Assays (C_904973_10 and C_3084793_20, Applied Biosystems) on the Applied Biosystems 7900 HT Fast Real Time PCR. Deviation from Hardy Weinberg Equilibrium (HWE) was calculated for the APOE locus, and both cases (p = 0.86) and controls (p = 0.36) were found to be in HWE.
NK92 cell nucleofection
NK92 (ATCC CRL-2407) cell line was grown in MEM-Alpha medium (Gibco BRL, Invitrogen Corporation, Carlsbad, CA, USA) supplemented with 20% of FCS (fetal calf serum), 0.1 mM 2-Mercaptoethanol, 100 U/mL of penicillin and streptomycin, and 150 U/mL of IL-2. Cell cultures were maintained at 37°C in humidified atmosphere of 5% CO2.
KIR2DL2-GFP-tagged was inserted in the plasmid pCMV6-AC-GFP (OriGene Technologies, Rockville, MD USA). The recombinant plasmid was amplified in One Shot® TOP 10F’ E. Coli (Invitrogren). Plasmid DNA was extracted with plasmid MAXI kit (QIAGEN, Hilden, Germany) and nucleofected in NK92 cells (Ingenio®Electroporation Kit, Mirus Bio LLC, WI USA). Stable cell colonies were selected in the presence of 1000μg/mL of Geneticin® (G418 Sulfate, Gibco®). KIR2DL2 NK92 cells and NK92 control cells were co-cultured with infected cells in a 1:5 ratio and collected for subsequent analysis [8, 9]. Each experiment was performed in triplicate.
Images were acquired with Nikon Eclipse TE2000S fluorescence microscope equipped with a digital camera.
KIR2 e beta-actin mRNA analysis
Total RNA was extracted from PBMC by TRIzol (Invitrogen). RNA samples were digested with DNase (1 U/ug RNA) and stored at –85°C in ethanol until cDNA synthesis. Two micrograms of total RNA were reverse transcribed using SuperScripttrademark First-Strand Synthesis System (Invitrogen). cDNA was amplified by specific oligonucleotide primers for KIR2DL2 [40] and house-keeping control gene beta-actin [41, 42]. Amplification products were analyzed by ethidium bromide–stained agarose gel electrophoresis.
HHV-6 infection
The human T cell line J-Jhan was cultured in RPMI-1640 (Gibco BRL, Invitrogen Corporation, Carlsbad, CA, USA) with 10% FCS, supplemented with 100 U/ml each of penicillin and streptomycin and maintained at 37°C in humidified atmosphere of 5% CO2.
J-Jhan cells were infected with an HHV-6A (U1102) cell free inoculum as previously described [42]. The day after infection, J-Jhan cells were harvested for DNA and RNA quantification and for APOE mRNA analysis or NK92 cells were added at a 1:5 ratio. Each experiment was performed in triplicate.
HHV-6 analysis
HHV-6 DNA viral load was analyzed by real time quantitative (qPCR) in duplicate, as described [43]. RNA cell extraction was performed using the RNeasy kit (Qiagen, Hilden, Germany). No DNA contaminated the RNA samples, as shown by control β-actin PCR without retrotranscription [41, 42]. Reverse transcription was performed by the RT2 First strand kit (Qiagen, Hilden, Germany). cDNA aliquots corresponding to 200 ng RNA were used for virus transcription analysis, performed by qPCR detecting the expression of U42 gene, as previously reported [44].
Sp1 phosphorylation status analysis
Sp1 phosphorylation was evaluated by western blot. Proteins were extracted by RIPA buffer and ready-to-use cocktails of protease and phosphatase inhibitors (Abcam, UK). Protein concentration was quantified by Bradford assay (Bio-Rad Laboratories). Similar amounts of protein were loaded in 12% TGX-Pre-cast gel (Biorad, Segrate, MI, Italy), with subsequent electroblotting. The membrane was incubated with 1:200 Sp1 (ProSci Incorporated; Poway, CA, USA) or anti-phospho Sp1 (pThr) (Sigma-Aldrich; Saint Louis, MO, USA) monoclonal antibodies. A horseradish peroxidase (HRP)-conjugated antimouse antibody (1:5000; Amersham Biosciences, NJ, USA) was added and developed with the ECL kit (Amersham Biosciences, NJ, USA). The images were acquired by GelDoc EZ System (BioRad; Milano; Italy).
PKA analysis
PKA was extracted from 10∧7 cells with PepTag® Non-Radioactive Protein Kinase Assays (Promega, Milano, Italy). The inhibition of PKA activity was obtained with increasing concentrations (0.25, 0.5, 1.0, 2.0, 2.5μM) of KT 5720 (Sigma-Aldrich, St. Louis, MO, USA), a specific inhibitor of protein kinase A.
Carboxyfluorescein diacetate succinimidyl ester (CFSE) analysis
Infected cells were assessed for cell-mediated cytoxicity, using 7AAD/CFSE Cell-mediated cytoxicity assay kit (Cayman Chemicals, Ann Arbor, MI, USA), according to the manufacturer’s protocol. Labelled cells were analyzed with FACSCantoII flow cytometer (Becton Dickinson, Milano, Italy) and FlowJo software (FlowJo LLC, OR, USA).
APOE expression
J-Jhan cells, infected with HHV-6A (U1102) were harvested after 1, 3, 7, 14, and 21 days post infection. Total RNA was extracted (RNeasy kit, Qiagen, Hilden, Germany) and checked for the absence of contaminating DNA [41]. RNA reverse transcription was performed as described above. APOE expression analysis was performed on DNA aliquots corresponding to 200 ng RNA using Applied Biosystems Gene expression analysis (Hs00171168_m1).
Statistical analyses
To detect significant departure from Hardy–Weinberg equilibrium, we used the extended version of Fisher’s exact test implemented in Arlequin 3.01 [39]. Tests of association between genotypes and cognitive changes were performed with a logistic regression adjusted for age, sex, baseline MMSEDi, to test the possible association between disease status and KIR. Biological variables were evaluated by Student t test by Graph pad software. p-value <0.05 was considered to be statistically significant. p values were corrected (pc) for multiple comparisons, using Bonferroni correction.
RESULTS
KIR/HLA combinations
Patient characteristics are shown in (Table 1). Frequencies and distributions of KIR alleles were in Hardy Weinberg equilibrium and are summarized in (Table 2). Patients with AD showed a significantly increased frequency of the activating receptor KIR2DS2 (62% versus 37.5%; pc: 2.2×10–3) and a reduced frequency of the activating receptor KIR3DS1 (16.6% versus 32.7%; pc:2.9×10–3). A higher number of AD patients had increased inhibitory receptor KIR2DL2 (62.2% versus 36.4%; pc: 2.2×10–3). No significant differences were found in the distribution of KIR genotypes (AA or Bx) but there was an increase in HLA-C1/C2 genotype in AD patients (45.9% versus 22.7%; pc: 2×10–3). When analyzing KIRs and their respective ligands, the combination KIR2DS2/KIR2DL2 present/C1 present, was significantly increased in AD patients (48% versus 28.2%; pc:0.037) while the combination KIR2DS2/KIR2DL2 absent/C1 present was significantly decreased in AD patients (6.1% versus 30.9%; pc:5.1×10–5). After including all variables in logistic regression, the increased frequency of KIR2DL2 and increased frequency of the combination KIR2DS2/KIR2DL2 present/C1 present maintained a strict correlation with AD (p = 0.02; p = 0.01, respectively).
KIR/HLA frequency
p, p value obtained by Fisher’s exact test. pc, corrected p value for multiple comparisons (Bonferroni correction).
There was a significant lower MMSEDI score at diagnosis in patients with the KIR2DS2/KIR2DL2/C1 combination (p = 0.0002) (Fig. 1a). Disease severity at diagnosis, ADLI, and IADLI did not present any correlation with KIR/HLA combinations (Fig. 1b, c). Interestingly, the worsening of the disease status after one-year follow-up was more evident in patients with the KIR2DS2/KIR2DL2/C1 combination. In fact, 14 of 20 patients with a worsened disease status had KIR2DS2/KIR2DL2/C1 (p = 0.0045).

J-Jhan T cell lines were infected with HHV-6A at a multiplicity of infection of 100 genome equivalent/cell. Virus presence (DNA) and transcription (RNA) were evaluated respectively by qPCR and RT-qPCR performed on U94 and U42 virus genes respectively, at 1, 3 and 7, 14, and 21 d.p.i., as already detailed. mRNA ApoE ɛ4 expression was evaluated in J-Jhan cells at 1, 3 and 7, 14, and 21 d.p.i. RQ, relative quantity of ApoE ɛ4 mRNA.
KIR/HLA combinations and APOE alleles and expression
Frequencies and distributions of APOE alleles were in Hardy Weinberg equilibrium. The frequency of APOE ɛ4 allele was highest in AD patients (pc: 0.00018) (Table 3). APOE ɛ4 allele was significantly more frequent in AD patients with the KIR2DS2/KIR2DL2/C1 combination (p = 3.9×10–5) (Table 4).
APOE allele frequency
KIR/HLA frequency in combination with APOE allele frequency
We evaluated the possible effect of HHV-6A infection on APOE ɛ4 expression. The results, shown in (Fig. 1), evidence that APOE ɛ4 mRNA synthesis is enhanced by HHV-6A infection, in a time dependent correlation. In particular, there is a clear correlation between the enhancement in HHV-6A RNA production and the increase in APOE ɛ4 mRNA at day 3 p.i. with a peak at day 14 p.i. (Fig. 1).
KIR2DL2 receptor and HHV-6 infection
Since the KIR2DS2/KIR2DL2/C1 combination has been shown to increase the susceptibility to HSV-1 infections [9], we evaluated if this might also be the case for HHV-6A. NK92 cell line, negative for KIR expression, was nucleofected with KIR2DL2 or control plasmid (GFP) (Fig. 2d). Efficiency of transfection was 89% (Fig. 2e). NK92-GFP and NK92-KIR2DL2 cells were co-cultured with HHV-6A infected T cells, expressing HLA-C1 allele. NK92-GFP cells were able to control HHV-6A infection, reducing viral titer, while NK92-KIR2DL2 cells did not (Fig. 2f) (p < 0.001). The blocking of KIR2DL2 receptor with CD158b moAb, specific for KIR2DL2 receptor, reverted the effect of KIR2DL2 expression on NK92 cells, reducing HHV-6A titer to values similar to those obtained in NK92-GFP cells (Fig. 2f). To ensure that NK92-GFP cells killed HHV-6A infected cells and not only to prevent viral replication, cells were analyzed by flow cytometry. We observed no modification in HHV-6A infected cell percentage when co-cultured with NK92-GFP cells (Fig. 2g). On the contrary, NK92-KIR2DL2 cells reduced CFSE+ cell percentage, indicating a higher proliferation of the cells and suggesting the inability of NK92-KIR2DL2 cells to kill infected cells.
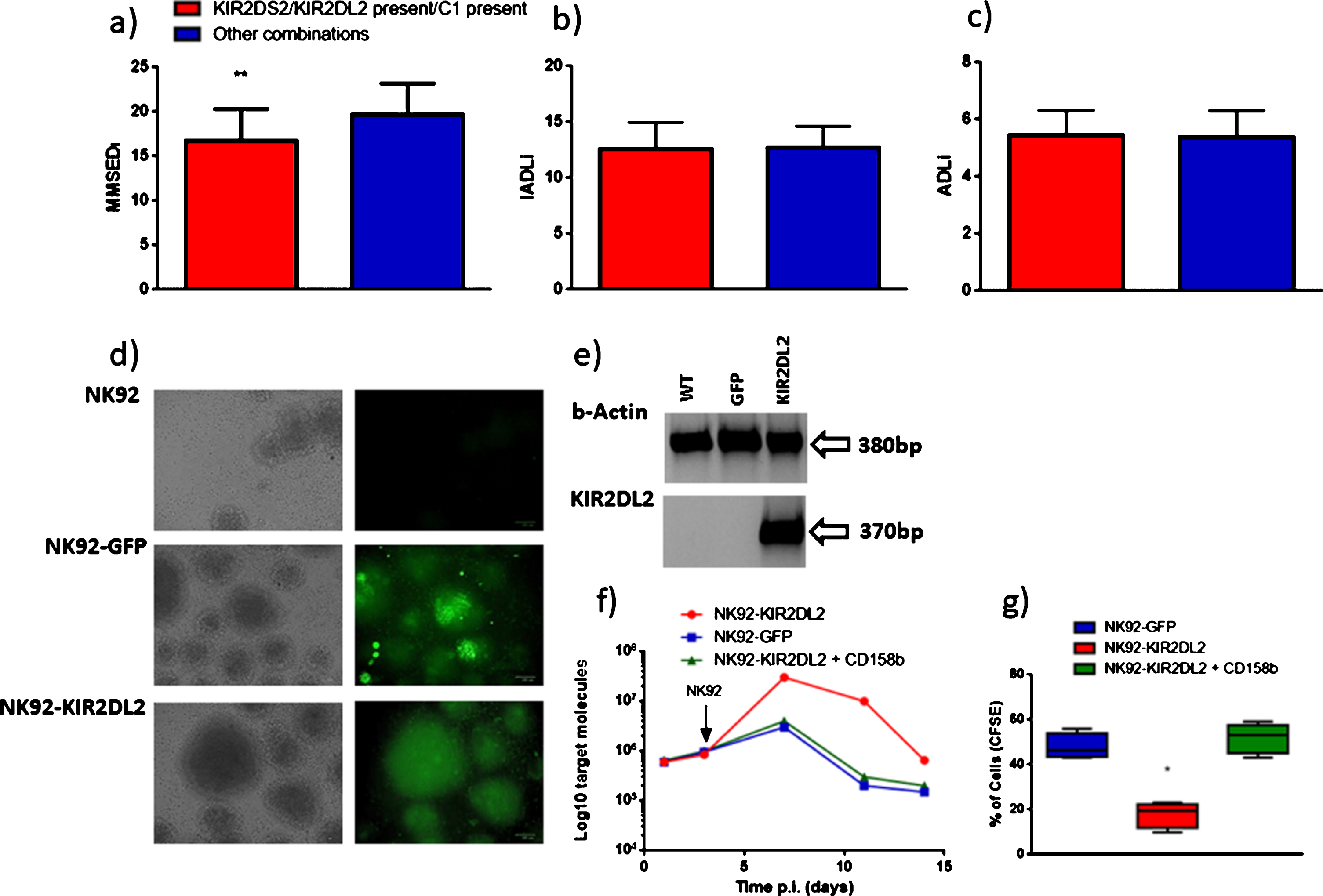
a) MMSEDI (Mini-Mental State Examination at diagnosis), b) ADLDI (Activities of Daily Living at diagnosis), and c) IADLDI (Instrumental Activities of Daily Living at diagnosis) distribution according with the KIR2DS2/KIR2DL2 present/C1 present combination. **p value <0.05, obtained by logistic regression. d) NK92 nucleofected cells with pCMV6-AC-GFP (RG224797, OriGene Technologies, Inc., Rockville, MD, USA) (NK92-GFP) or pCMV6-AC-KIR2DL2 (GFP- tagged- Human killer cell immunoglobulin-like receptor, two domains, long cytoplasmic tail, 2) (NK92-KIR2DL2) plasmid. GFP expression was evaluated after 12 days of selection. e) Beta actin and KIR2DL2 mRNA expression in NK92 (WT), NK92 nucleofected cells with pCMV6-AC-GFP (GFP) or pCMV6-AC-KIR2DL2 (KIR2DL2) plasmid. f) HHV-6A infection of J-Jhan cells and co-culture with NK92 nucleofected cells with pCMV6-AC-GFP (NK92-GFP) or pCMV6-AC-KIR2DL2 plasmid with (NK92-KIR2DL2 + CD158b) or without (NK92-KIR2DL2) the addition of anti-KIR2DL2 moAb (CD158b). Virus DNA presence was evaluated using an amount of 100 ng of DNA. Results are expressed as mean Log10 target molecules. g) Mean results obtained for CFSE Fluorescent cell staining of HHV-6A infection of J-Jhan cells co-cultured with NK92 nucleofected cells with pCMV6-AC-GFP (NK92-GFP) or pCMV6-AC-KIR2DL2 plasmid with (NK92-KIR2DL2 + CD158b) with (NK92-KIR2DL2 + CD158b) or without (NK92-KIR2DL2) the addition of anti-KIR2DL2 moAb (CD158b). *p value <0.05, obtained by Student t-test. Each experiment was performed in triplicate.
Involvement of PKA/Sp1 pathway
Since KIR2DL2 expression is controlled by specific transcription factors [45], we evaluated the modulation of Sp1 phosphorylation (pThr), highly represented in NK92 cell line [46, 47]. We observed an increase in Sp1 phosphorylation in NK92 cells co-cultured with HHV-6A infected cells (Fig. 3a). Since Sp1 DNA binding properties are stimulated by cAMP-dependent protein kinase (PKA) [48], we looked at possible differences in PKA activity. PKA activity increased in NK92 cells co-cultured with HHV-6A infected cells (Fig. 3b). Interestingly, Sp1 phosphorylation was decreased in the presence of KT 5720, a PKA inhibitor, even in the presence of HHV-6A infected cells (Fig. 3c). These data suggest that the PKA/Sp1 pathway might be involved in KIR2DL2 expression on NK cells, inhibiting their activation during HHV-6A infection.
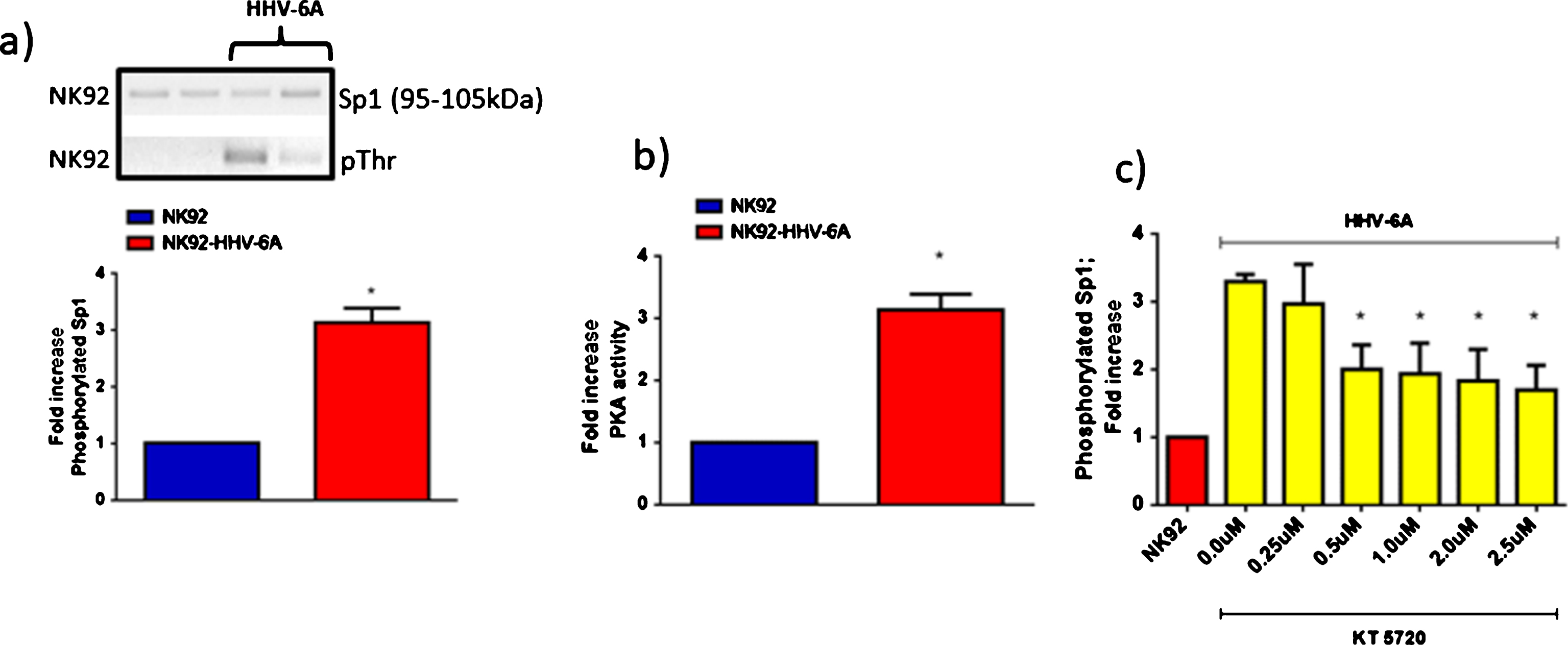
a) Western blot analysis for the evaluation of Sp1 and pThrSp1 expression in NK92 cells in co-culture with HHV-6A infection of J-Jhan cells (HHV-6A). b) PKA activity in NK92 cells in co-culture with HHV-6A infection of J-Jhan cells (HHV-6A). c) Fold increase of pThrSp1 in NK92 cells in co-culture with HHV-6A infection of J-Jhan cells (HHV-6A) in the presence of increasing concentrations (0.25, 0.5, 1.0, 2.0, 2.5μM) of KT 5720, a specific, cell-permeable inhibitor of protein kinase A. *p value <0.05, obtained by Student t-test. Each experiment was performed in triplicate.
Conclusions
In this study, we describe a possible role for a specific KIR/HLA subset in AD. We observed an increased frequency of KIR2DS2/KIR2DL2/C1 in AD patients that correlates with a lower MMSEDi score at diagnosis and a worsening of the disease status in the one-year follow-up. Since we have previously shown that the combination KIR2DS2/KIR2DL2/C1 results in a higher susceptibility of MS patients to herpesvirus infections, this genetic background might have a role also in herpesvirus infection in AD. Interestingly, both activating KIR2DS2 and inhibiting KIR2DL2 receptors resulted increased in AD patients. KIR2DL2 and KIR2DS2 are in linkage disequilibrium, implying they are “preferentially associated” with one another. However, when KIR2DS2 and KIR2DL2 are co-expressed, NK cell inhibition via KIR2DL2 over-rides NK cell activation via KIR2DS2 [49]. We hypothesized that AD patients with KIR2DS2/KIR2DL2/C1 might be less prone to control HHV-6A infection and, in more general terms, herpesvirus infection. The in vitro experiments on NK92-KIR2DL2 cells showed an impaired control of HHV-6A infection in comparison with wild type NK92-GFP. Interestingly, NK92 cells challenged with HHV-6A infected cells increased the phosphorylation status of Sp1due to both PKA activity and the direct transactivating effect of HHV-6 on Sp1 binding sites [50]. Since KIR2DL2 promoter has a Sp1 binding site [45], the activation of Sp1 could affect the expression of KIR2DL2 on the surface of NK cells in the presence of herpesvirus infected cells [9]. Meanwhile, the activation of PKA could have a role in AD neurofibrillary pathology, directly participating in the tau pathological hyperphosphorylation [51]. We hypothesize that HHV-6A infection might induce PKA activation, tau hyperphosphorylation, and Sp1 phosphorylation, thereby increasing its binding with the KIR2DL2 promoter. Overexpression of KIR2DL2 will consequently inhibit NK cells [9], possibly with a direct role in AD. Previous studies have already described the possible involvement of NK cells in AD immune-pathogenesis [7], showing a lower NK cell functional potential in AD patients [2]. Solerte et al. suggested that the overactivity of NK cells AD patients could be related to the dysregulation of PKC (protein kinase C) expression [52], similarly to what we observed for PKA.
Taking into consideration the genetic background of AD patients, it was necessary to look at APOE allelic distribution. Inside the brain, ApoE, characterized by three allelic forms: APOE2, APOE3, APOE4, helps to clear Aβ, a plaque component. We confirmed that APOE ɛ4 allele has an increased frequency in AD patients (Tables 3 and 4), and, interestingly, this was more frequent in KIR2DS2/KIR2DL2/C1 AD patients. These results suggest that APOE ɛ4 allele and KIR2DS2/KIR2DL2/C1 could have a combined effect in a multi-gene form of AD. In particular, APOE is able to inhibit the activation of immune cells, and therefore it could enhance the NK inhibition associated to KIR2DS2/KIR2DL2/C1. We also observed that HHV-6A infection up-modulates ApoE ɛ4 expression. Moreover, ApoE increases PKA activity and, conversely, the secretion of ApoE occurs via a PKA-dependent pathway [54]. Therefor a loop could be established, with HHV-6 increasing ApoE ɛ4 expression and this would further decrease an already reduced KIR2DS2/KIR2DL2/C1 NK patrolling activity.
The involvement of NK cell in AD could include their defensive role toward HHV infections, leading to the deregulation of immune system and resulting in the AD progression [55]. Our results with NK92-KIR2DL2 cells, expressing only KIR2DL2 and not other KIR receptors that could confound the analysis, supports this possibility. Higher herpesvirus infection might lead to hyperphosphorylation and polymerization of tau proteins, induce Aβ peptide fibrilization and deposition [56], suggesting that herpesviruses might have an important role also in several features of AD pathology.
In conclusion, the possible role of herpesvirus infection in AD development/control is supported by: 1) the increase of KIR2DL2/C1 genotype in AD patients (Table 2), 2) a lower anti-herpetic activity of KIR2DL2 positive NK cells (Fig. 1f) [9], 3) a lower NK activity in AD [2], 4) an increased abundance of neurotropic herpesviruses in AD [29] that promote Aβ deposition [56], and 5) the creation of a molecular environment favorable for tau fibrilization (Fig. 2b).
To confirm this hypothesis, further studies are needed on ex vivo samples from AD patients. If this hypothesis finds experimental confirmation, a new therapeutic strategy for AD might be envisaged. In fact, modulation of KIR2DL2 expression on NK cells, as already proposed for other pathological conditions [57, 58], could restore their anti-herpetic surveillance.