
Abstract
Depression is a common co-morbidity seen in people with Alzheimer’s disease (AD). However, the successful treatment of depressive symptoms in people with AD is rarely seen. In fact, multiple randomized controlled trials have shown selective serotonin reuptake inhibitors (SSRIs), the current best recommended treatment for depression, to be ineffective in treating depressive symptoms in people with AD. One explanation for this lack of treatment effect may be that depressive symptoms can reflect the progression of AD, rather than clinical depression and are a consequence of more severe neurodegeneration. This raises several questions regarding not only the efficacy of SSRIs in the treatment of depression in people with AD but also regarding the accuracy of diagnosis of depression in AD. However, there may be a rationale for the prescription of SSRIs in early AD. Even in the absence of depression, SSRIs have been shown to slow the conversion from mild cognitive impairment to AD. This may be attributed to the effect of SSRIs on the processing of amyloid-β precursor protein, which may cause a reduction in the accumulation of amyloid-β. Thus, although SSRIs may lack efficacy in treating depression in people with AD, they may hold therapeutic potential for treating and delaying the progression of AD especially if treatment begins in the early stages of AD. This article reviews the current consensus for SSRI treatment of depression in people with AD and highlights the possibility of SSRIs being a treatment option for delaying the progression of AD.
INTRODUCTION
The average lifespan for men and women is increasing in both high income and developing countries [1]. As life expectancy increases, so too does the predicted incidence of dementia, with age being the biggest risk factor. It is estimated that more than 50 million people are living with dementia worldwide. This number is predicted to rapidly rise with over 9.9 million new cases every year. Perhaps most concerning is that even the most effective treatment options currently available will not be able to prevent dramatic increases in the prevalence of dementia. One approach to improving the effectiveness of current treatments is to develop better diagnostic tools enabling earlier identification of dementia [2]. This will allow both pharmacological and lifestyle interventions to be prescribed to limit neurodegeneration, preserve cognitive function and maintain a higher quality of life before the severity of the disease progresses.
Alzheimer’s disease (AD) is the most common form of dementia accounting for over 60% of dementia diagnoses [3] and is characterized by the accumulation of amyloid-β (Aβ) and the formation of neurofibrillary tangles resulting from abnormal tau protein phosphorylation [4, 5]. In addition, loss of synaptic function and neuronal loss are widely reported [6, 7]. The generation of highly toxic, soluble oligomeric forms of Aβ in addition to elevated oxidative stress and impaired immune functioning have been proposed as key players in the progression of AD. The subsequent accumulation of these neuropathological lesions leads to neurodegeneration, reduced cognitive ability and impaired everyday functioning which is ultimately fatal [8 –10].
Neuropsychiatric symptoms (NPS) refer to a heterogenous grouping of non-cognitive symptoms that are common in people with AD, such as changes in mood, perception, and behavior. The presentation of NPS is related to a poorer quality of life and an increase in progression from mild cognitive impairment (MCI) to severe AD. Worsening of NPS is also related to an increased risk of cognitive decline and onset of AD [11, 12]. In people with AD, apathy and depression are the most commonly diagnosed NPS [13]. Apathy is characterized by the loss of motivation that is not attributable to cognitive decline or emotional distress. Often apathy is considered as a symptom of depression. Although they can present together, the key distinction of apathy from depression is the involvement of emotional distress [14] and therefore effective treatment for both of these NPS may be different. It has also been found that depression and apathy are associated with different measures of cognitive dysfunction [15]. Current pharmacological treatment for apathy in AD, including the use of selective serotonin reuptake inhibitors (SSRIs) has shown mixed results for alleviating symptoms [16, 17]. Despite limitations of the research outcomes by the difficulty in measuring clinically significant apathy and inconsistent methodologies, treatment with escitalopram reduced apathy and positively impacted measures of disability [18].
Depression precedes 10–15% of dementia cases and the risk of developing late-life depression is significantly higher in people with MCI (21.7%) and dementia (24.7%) compared to cognitively healthy populations (10.5%) [19, 20]. Depressive symptoms can also manifest concurrently in people with MCI [21]. In fact, the association between depression and a decline in cognitive impairment may be more pronounced in MCI compared to those who already have a diagnosis of AD [22]. Research has shown that depression is associated with an increased risk of AD [23] and recurring depressive episodes are particularly pernicious [24]. In addition, episodes of depression in mid and late life, not only increase the risk of AD but are also thought to speed up the progression of AD manifestation and degeneration, especially in people with MCI [25 –29]. The prodromal nature of AD may be a causative factor in the development of late-life depression. For example, the presentation of executive dysfunction, which contributes to the development of clinically significant cognitive impairment, is common in both AD and late-life depression [30]. However, not all people with ‘depression-executive dysfunction syndrome’ progress to AD [31, 32]. This provides a challenge when determining onset of depressive symptoms independent of cognitive decline and other closely related syndromes [33].
‘DEPRESSION’ IN AD
The overlapping symptoms of AD and depression provide clinicians and researchers with a challenge. Are symptoms that are normally associated with depression in late life present as a result of the neurodegenerative course of AD or are they symptoms of depression? This distinction is of course vital to the successful treatment of late-life depression and may help to elucidate why treatment resistance is greater in older age populations [34 –36]. The symptoms of depression can manifest as a combination of prolonged sadness, agitation, feelings of worthlessness and hopelessness, and reoccurring suicidal thoughts. Depression in AD is typically of a milder presentation than major depression, yet it can still significantly effect quality of life [37].
A worsening of depressive symptoms in older adults has been shown to be a significant predictor of those who may develop AD [38]. The risk of depression in AD has been linked to several factors such as, perturbed neurotransmitter signaling, elevated inflammation, tau pathology, and Aβ accumulation [39]. In addition, AD and late-life depression are associated with overlapping neuroanatomical changes. A reduction in hippocampal volume and increased neuronal death in areas related to emotional regulation and cognition are associated with both conditions [40]. There are, however, some subtle differences that may be a powerful tool to distinguish depression due to AD and late-life depression. People with AD demonstrate greater total brain volume deficits and asymmetrical degeneration of the hippocampus and amygdala. These asymmetries may also be associated with the progression of AD [41].
THE RELEVANCE OF AMYLOID-β FOR DEPRESSION IN AD
The accumulation of Aβ appears to have some association with depression as people with a lifetime history of major depression were seen to have increased brain Aβ deposition [42, 43] and interestingly people with AD, who have a history of major depression show even greater Aβ plaques and neurofibrillary tangles in the hippocampal region, compared to people with AD without depression [44]. However, without a clear cut-off for the relationship between Aβ in the brain and the development of clinical symptoms it is difficult to draw causative conclusions. Nevertheless, the validity of plasma Aβ as a marker of depression in AD has been of significant interest, as collection of a plasma sample is minimally invasive and easily accessible. Higher levels of plasma Aβ have been associated with an increased risk of clinically relevant depressive symptoms, especially in people who go on to develop AD within 11 years [45, 46]. The longer amyloid peptide, Aβ42 is prone to aggregation and has been shown to be the most cytotoxic peptide, due to its interaction and uptake into cells [47]. Aβ42 has been found to be a positive predictor of both late-onset depression and AD [48]. In addition, people with a higher ratio of Aβ40 :42 were found to have the greatest impairment in cognitive function and represented a prodromal manifestation of depression in AD, termed ‘Aβ associated’ depression. An important consideration when determining if Aβ associated depression reflects the prodromal onset of AD is that not all people with an elevated Aβ load develop AD. The lack of relationship between Aβ and AD in some people is a major challenge in AD research.
Aβ42 accumulation is also one of a number of factors affected by the genetic mutation of Apolipoprotein E (APOE). People who carry the one or two copies of the APOE ɛ4 allele are at an increased risk of developing AD [49] and carriers of APOE ɛ4 are more likely to display increased accumulation of Aβ42 in the brain at a younger age [50, 51]. APOE ɛ4 carriers can be at a 50% greater risk for developing AD when compared to people carrying one copy of the protective APOE ɛ2 gene. This translates into an 18–23 year difference in age of onset [49]. Interestingly, APOE ɛ4 carriers are also at an increased risk of developing depression, further implying a shared mechanism between depression and AD pathology [52].
Amyloid-β protein precursor (AβPP) processing is crucial to Aβ generation, and gaining a better understanding of altered AβPP processing may provide insight into the role of Aβ in AD and late-life depression. Amyloid-β is formed via enzymatic cleavage of AβPP by a β-secretase enzyme. This cleavage liberates the secreted amino-terminal fragment-β (AβPPβ) and an Aβ intracellular C-terminal fragment (CTF) of 99 amino acids. Alternatively, AβPP cleavage can be initiated by an α-secretase. This pathway precludes Aβ formation by liberating the neuroprotective secreted amyloid-β protein precursor α (sAβPPα) amino terminal fragment and an 83-amino acid CTF. The cleavage of these CTFs by γ-secretase results in the formation of p3 or Aβ and the amino-terminal AβPP intracellular domain (for reviews, see [53, 54]).
Members of the ‘a disintegrin and metalloproteinase’ (ADAM) family are known to have α-secretase activity and are thought to act with redundancy; however, compelling evidence suggests that ADAM10 is the major physiologically relevant α-secretase involved in the processing of AβPP [55, 56]. Beta-site cleaving enzyme (BACE-1) is the major enzyme in β-secretase AβPP cleavage [57]. Research into the effects of late-life depression on AβPP processing is very limited, due to the difficulty in accessing suitable tissue and difficulty in producing a depression-like state in cellular models. However, platelets represent an excellent model for studying AβPP processing as they express all of the required machinery to generate Aβ peptides. In addition, platelets can secrete neurotransmitters, inflammatory markers and growth factors, such as brain-derived neurotrophic factor, which are altered in depression and AD patients [58, 59]. In platelets AβPP is expressed either as the 751 kDa or 770 kDa isoforms in contrast to the 695 kDa isoform in neurons. Despite this difference in sequence/peptide length, all isoforms are processed in the same way by the secretase enzymes as described earlier at both the cell membrane and inside the cell in the secretory pathway [59]. Not only this, platelet expression and activity of ADAM10, BACE-1, and the ratio of secreted AβPP have all been found to be significantly altered in people with AD [60 –65]. Crucially, these platelet biomarkers are thought to reflect the changes in AβPP processing seen in the AD brain that, combined with a reduced ability to clear Aβ peptides, may be a causative factor of AD [8, 66].
THE EFFICACY OF SSRIs IN THE TREATMENT OF DEPRESSION IN AD
Antipsychotics, acetylcholinesterase inhibitors, anti-convulsant, and hormone-replacement therapy are all typical treatments that are commonly prescribed to alleviate depression in AD. However, many of these drugs have been found to be inadequate [67] in treating depression in AD and as a result, SSRIs are more widely prescribed. SSRIs are often associated with a reduced risk of an adverse event [68, 69], and are therefore considered safer.
SSRIs target the serotonin receptor transporters (SERT), preventing serotonin re-uptake from the synapse and therefore, increase signaling and ligand-receptor binding. Initially, SSRIs were assumed to possess therapeutic effects based on the ‘monoamine hypothesis’ of depression which postulates that defective neurotransmitter signaling is a central feature [70]. However, multiple downstream effects have been suggested. Elevated neuro-inflammation has been described as a prominent feature of both AD and depression. Treatment with SSRIs has been shown to reduce circulating cytokines and interact with inflammatory signaling pathways that are typically elevated in chronic inflammatory conditions [71]. In addition to inflammation, hyperactivation of the hypothalamic-pituitary axis and lowered neurotrophin production are key neurobiological links between depression and AD that may be modulated by SSRIs [72]. Thus, plausible mechanisms of action for SSRIs do exist in both depression and AD and in the interaction of these conditions.
The identification of potential mechanisms of action for SSRIs fitted well with the outcomes of early research studies, which suggested that SSRIs could improve depressive symptoms in people with AD when compared to treatment with placebo [4 , 74]. However, these studies were significantly underpowered and the need for larger trials, to prove efficacy of SSRI treatment for depression in AD, was identified. More recently, larger studies have shown that SSRIs were no better than placebo for the treatment of depressive symptoms in people with AD [75, 76]. Several randomized control trials [77 –80] have also suggested that SSRIs lack the required efficacy for treating depressive symptoms in people with AD and this was supported in a recent meta-analysis [81]. However, the lack of effect may partly be due to our inability to predict individual response to treatment and provide personalized optimal dosage strategies [36]. It has also been shown that the placebo effects in older age adult populations are much larger than in younger cohorts, and it may be more appropriate to offer psychotherapies before prescribing SSRIs as a last resort [82]. In addition, although SSRIs can have delayed effects in older populations, long term use of SSRIs was not associated with alleviated symptoms of depression in people with AD [83]. However, the prescription of SSRIs for the treatment of late-life depression in older age adults without AD (or other dementias) is largely successful [84], as up to 70% of people will recover [85]. That said, older age adult populations tend to show a poorer response to SSRIs compared to younger people. In studies comparing old and young, up to 50% of older age adults can be treatment resistant [86, 87].
There is an urgent need to understand why people with AD and depression appear not to respond to treatment. With advancing age, an individual is likely to experience (cognitive and physical) functional decline, which can diminish quality of life. As these changes can occur at a rapid rate, they will no doubt place a psychological burden on the individual and increase the likelihood of recurring depressive episodes [86]. In addition, the neurodegeneration [88] and global physiological changes associated with advancing AD, such as increased inflammation and oxidative stress [89], are likely to enhance the severity of depressive episodes. This may lead to significant cognitive impairment and reduce the likelihood of both treatment response and adherence to medication [90, 91]. In fact, people with late-life depression and AD, or both, are known to have significantly lower treatment adherence rates possibly linked to reduced cognitive functioning and a greater number of co-morbidities [92, 93]. Although adherence to SSRIs in people with AD is greater than adherence to Tricyclic antidepressants, over 50% of people with AD discontinue all antidepressant medication within 6 months [94]. This is most often due to a general lack of ability to maintain routine, related to reduced cognitive function. Poor adherence to medication can also increase the severity of comorbidities in AD, lead to a greater risk of adverse events and often a lower quality of life [95].
In over 30% of late-life depression cases, cognitive deficiencies are present independent of AD pathology. This cognitive impairment is thought to be reversible with the successful treatment of depression [96]. However, for those who do not recover from depressive episodes with treatment, the possibility of such symptoms reflecting a prodromal period of AD development cannot be ruled out [97]. In support of this view it has been shown that older individuals that respond poorest to antidepressant treatment have a greater brain Aβ load and lower cognitive function [98]. This suggests that Aβ deposition, poorer cognitive function, or greater neurodegeneration reduce the therapeutic action of antidepressant treatment or, more likely, that the individual’s depression-related symptoms are reflecting a prodromal AD state and therefore typical treatment options for depression are unsuitable. In order to determine this, robust biomarkers that predict the onset of AD are required, to avoid unnecessary prescription of SSRIs to older adults that are actually not depressed but that are developing symptomatic AD.
THE EFFICACY OF SSRIs IN THE TREATMENT OF AD
The possibility of late-life depression reflecting a prodromal period of AD is supported by several key observations: Both depression and AD share similar neurobiological and peripheral biochemical changes that reflect a disease state, for example elevated inflammatory and oxidative stress markers and increased cerebrovascular damage or degeneration [42 , 100] are common in depression and AD. Depression and AD may be linked bi-directionally as depression is a risk factor for AD and AD is a risk factor for depression. Alterations in brain Aβ accumulation are often seen in people with a life-time history of depression [101]. Although SSRI treatment may not always be efficacious for alleviating depressive symptoms in AD, interest in the effectiveness of SSRIs for treatment of cognitive decline has been gaining attention. Current research suggests that long-term treatment with SSRIs may delay the progression from MCI to AD [102, 103]. In fact SSRI treatment has been associated with a reduced mortality rate at two-year follow up, although this effect dissipates after four years [104]. However, those receiving long term treatment may have had an earlier onset of depression-related symptoms. Those who only receive SSRI treatment later in life may instead have depressive symptoms reflecting an early manifestation of AD and therefore, are more likely to develop to clinically significant AD. This raises an important consideration. Effective treatment of major depression with SSRIs can induce neurogenesis and reverse hippocampal atrophy [105, 106]. However, once the clinical symptoms of AD become apparent the associated neurodegeneration is irreversible. Therefore, the treatment of AD with SSRIs is not curative or necessarily able restore a loss of function, but may delay the disease process and crucially, prolong cognitive function into older age. In order to achieve this therapeutic effect, SSRIs must act through mechanisms that interfere with aberrant pathological processes seen in people with AD and there is emerging evidence to support this effect.
One mechanism that may contribute to this effect may be a regulation of Aβ accumulation; however, neither sAβPPβ or cerebrospinal fluid Aβ42 levels changed in response to SSRIs in these studies. It is possible that SSRI treatment had a positive effect on the α-cleavage of AβPP via ADAM10, which would lead to production of the neuroprotective soluble AβPPα. Interestingly, platelet ADAM10 expression can increase with use of SSRIs in people with AD [107]. If this effect is confirmed and is mirrored in the brain, then SSRI use in AD could have implications for the progression of neurodegeneration seen in AD. If the accumulation of Aβ could be slowed by SSRIs increasing the non-amyloidogenic cleavage of AβPP, then further Aβ accumulation could be prevented.
Further to this, reduced neuroinflammation and elevated brain-derived neurotrophic factor are all potential mechanisms by which SSRI treatment may contribute to the delayed progression of AD [103]. AD is a multifactorial disease. Both increased oxidative stress and a chronic low-grade inflammation are associated with AD [108, 109]. Neuroinflammation has been implicated in the progression of AD by increasing amyloidogenic processing of AβPP, resulting in elevated Aβ and increasing the susceptibility of cells to neurodegenerative processes [110]. SSRIs can reduce peripheral inflammatory markers such as Interleukin-6, C-reactive protein, and tumor necrosis factor-α and prevent microglia activation in the brain [111]. This is of particular relevance to AD as the blood-brain barrier is more susceptible to leakage of cytokines into the brain. In addition, the anti-inflammatory effect of SSRIs is reflected in the brain by preventing elevated serotonin reuptake from the synapse as a result of elevated cytokine signaling or by direct action on reducing cytokine production [112, 113]. This highlights a possible therapeutic mechanism of action in slowing the global inflammatory response seen as a result of AD progression [114]. Further, SSRIs may also be effective in lowering oxidative stress. This may be due to either increased endogenous antioxidant capacity or activity, or through possible antioxidant properties of the drugs itself suggesting alternative protective action [115, 116]. Higher levels of the oxidative stress marker F2-Isoprostane at baseline have also been associated with a poorer treatment response for depression [100, 117]. All the current research assessing the ability of SSRIs to alter inflammation and oxidative stress has been conducted in animal models or in participants with depression alone, and therefore, given the potential interactions in pathologies between depression and AD, studies are needed to explore the effects of SSRIs in people with depression and AD.
As SSRIs may alter the processing of AβPP, and therefore the generation of Aβ, they are a potential candidate treatment to delay the progression of AD. Elevated serotonin signaling has been associated with decreased interstitial fluid Aβ peptide and Aβ plaque load [118]. In addition, SSRI use over a 5-year period has been associated with lower Pittsburgh compound-B Aβ load in cognitively healthy participants using SSRIs [118, 119]. SSRIs have also been shown to modulate the Aβ peptide species generated and therefore can reduce the toxicity associated with oligomeric forms of Aβ [120]. This may also have downstream benefits on tau-hyperphosphorylation by minimizing the potential for Aβ-tau cross talk [121]. SSRIs may also help by reducing synergistic toxic effects of Aβ and hyperphosphorylated tau and the associated synaptic and neuronal dysfunction seen in people with AD [121, 122]. In fact, this effect has been demonstrated in primary rodent neurons [123].
Some of the possible mechanisms by which SSRIs could alter the processing of AβPP are via altered AβPP expression or increased trafficking of AβPP to the cellular membrane, increased ADAM10 activity and/or reduced BACE-1 activity. The majority of work to explore these mechanisms has taken place in vitro or in animal models; nevertheless AβPPα increased 3.4-fold after treatment with citalopram, a commonly used SSRI, which precludes the formation of Aβ [124]. This data is supported by the finding that agonists of the serotonin receptor lowered Aβ in brain interstitial fluid but had no effect on gene expression, suggesting that AβPP processing is altered at protein level in the cell cytoplasm, with ADAM10 being the most likely candidate for upregulation. In support of this, broad pharmacological inhibition of α-secretases prevented the action of citalopram and led to increased Aβ in brain interstitial fluid [125]. Elevated expression of ADAM10 has also been shown in response to fluoxetine administration in a triple-transgenic mouse model of AD. This alteration in enzyme activity was further supported by an increase in sAβPP-α and C83 protein fragments. The effects of fluoxetine were ascribed to maintaining or rescuing Wnt/ β-catenin signaling pathways through the phosphorylation of GSK3β and direct action on β-catenin expression, which are thought to be altered in AD [126]. The therapeutic effects of fluoxetine on preventing neuron degeneration and synapse dysfunction and for attenuating memory loss have also been well documented in mice [127, 128]. Increased platelet ADAM10 expression has also been reported in participants with AD who were taking various SSRI medication [107] and this may be mediated by alterations in tetraspanin-5, which is known to alter the trafficking of ADAM10 in platelets [129].
Together, there is a strong case warranting further research into the use of SSRI medication as a treatment option to delay the progression of AD, targeting multiple mechanisms of action (see Fig. 1).
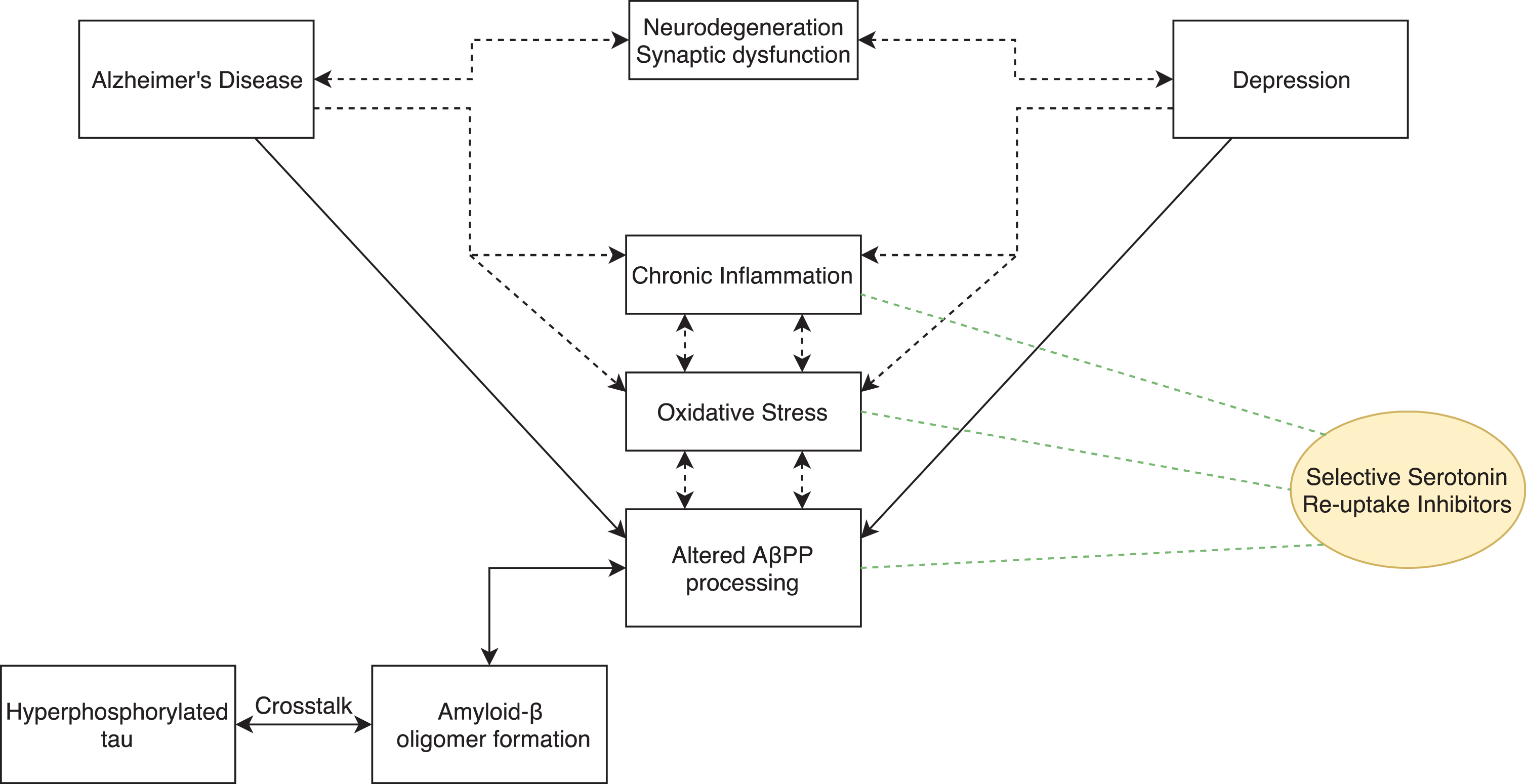
Diagrammatic representation of the therapeutic targets SSRIs may positively affect that are altered in both depression and AD.
CONCLUSION
Unfortunately, the effectiveness of SSRI treatment seems to become less obvious as AD progresses [77 , 131], yet the minimal side effects seen with SSRI use might make this a suitable option for slowing cognitive decline. The lack of benefit seen from SSRI treatment of symptoms of late-life depression, especially in people with AD, suggests that current recommendations for SSRI prescription need to be reviewed. A crucial step toward more effective treatment of AD is to identify accessible biomarkers of AD pathology in order to be able to properly monitor depressive symptoms that manifest as a characteristic of AD, rather than depression. Although both conditions share overlap in neuroanatomical features related to emotional regulation and cognitive function, asymmetric degeneration in AD may be one such way of distinguishing depression as a result of AD and comorbid late-life depression. In addition, the potential for SSRIs to promote non-amyloidogenic processing of AβPP suggest an alternative role for SSRIs in AD treatment. Research focusing on the mechanisms of action of SSRIs for both depression in AD and for AD alone are much needed and may provide an alternative approach for delaying the progression of AD. Platelets provide an easily accessible, cost-effective sample to assess AβPP processing. Further research into the effects of SSRI use for depression in AD and AD alone in platelets is warranted.
DISCLOSURES STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0780r2).