
Abstract
The amyloid hypothesis (AH) is still the most accepted model to explain the pathogenesis of inherited Alzheimer’s disease (IAD). However, despite the neuropathological overlapping with the non-inherited form (NIAD), AH waver in explaining NIAD. Thus, 30 years after its first statement several questions are still open, mainly regarding the role of amyloid plaques (AP) and apolipoprotein E (APOE). Accordingly, a pathogenetic model including the role of AP and APOE unifying IAD and NIAD pathogenesis is still missing. In the present understanding of the AH, we suggested that amyloid-β (Aβ) peptides production and AP formation is a physiological aging process resulting from a systemic age-related decrease in the efficiency of the proteins catabolism/clearance machinery. In this pathogenetic model Aβ peptides act as neurotoxic molecules, but only above a critical concentration [Aβ]c. A threshold mechanism triggers IAD/NIAD onset only when [Aβ]≥[Aβ]c. In this process, APOE modifies [Aβ]c threshold in an isoform-specific way. Consequently, all factors influencing Aβ anabolism, such as amyloid beta precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) gene mutations, and/or Aβ catabolism/clearance could contribute to exceed the threshold [Aβ]c, being characteristic of each individual. In this model, AP formation does not depend on [Aβ]c. The present interpretation of the AH, unifying the pathogenetic theories for IAD and NIAD, will explain why AP and APOE4 may be observed in healthy aging and why they are not the cause of AD. It is clear that further studies are needed to confirm our pathogenetic model. Nevertheless, our suggestion may be useful to better understand the pathogenesis of AD.
Keywords
INTRODUCTION
The non-inherited form of Alzheimer’s disease (NIAD) accounts for 50–70% of the 47 million people with dementia worldwide [1, 2] and for over 99% of all AD cases [2]. With a global aging estimation of 30 million people, as aging is a primary risk factor for NIAD, the ratio of NIAD to all AD cases continues to increase. According to current statistics, the global prevalence of NIAD is expected to double in a decade, making NIAD a critical economic concern for society [2, 3]. However, despite a number of studies investigating NIAD pathogenesis, no cause has been identified and accepted so far. Thus, as underlined by the therapeutic failure of over 400 candidate drugs in the last two decades [4, 5], effective treatments are also missing.
For over 100 years, scientists have recognized a strong correlation between the clinical signs of dementia and the abnormal deposition of β-sheet protein in central nervous system. This evidence has led in subsequent years to the amyloid school of thought, i.e., to the concept that amyloid protein deposition in the brain is the cause of dementia. Accordingly, it is currently widely accepted that both the inherited form of AD (IAD), and NIAD are caused by the same pathologically-defined condition due to the accumulation of amyloid-β (Aβ) peptides in the brain cortex. However, whereas the triggering cause of Aβ accumulation in IAD is well known [6–9], in NIAD, it is still missing [1, 10]. Moreover, despite in vitro and in vivo evidence supporting Aβ deposition as the cause of IAD/NIAD, recent data have shown that amyloid plaques (AP) resulting from the Aβ deposition are also present in brain cortex of healthy elderly [11–18], suggesting that Aβ overproduction and AP formation is a physiological process of aging. Therefore, the main question still is: Is Aβ deposition responsible for IAD/NIAD onset? The search of an answer to this question has stimulated the formulation of a number of alternative pathogenetic hypotheses. However, despite the profusion of mechanisms that have been proposed in the recent years [19], the amyloid hypothesis (AH) [20] remain the most widely accepted pathogenetic mechanism for IAD, still leaving the role of Aβ deposition in NIAD pathogenesis an open question.
More questions arise from the genetic role of the APOE. The missing genetic causes of Aβ deposition in NIAD have stimulated the scientific community to search for alternative genetic components underlying NIAD pathogenesis. The gap was filled in the course of 20 years when the allelic variants ɛ4 and ɛ2 were discovered to be the main genetic factor increasing [21–25] or decreasing [21, 26–28] the incidence of NIAD. However, despite the amount of experimental data, no relevant conclusions have been formulated [29–32]. Therefore, their role remains undefined, with several questions still waiting for answers: why is the ɛ4 allelic variant the major genetic risk factor of NIAD? Is the allelic variant ɛ4 responsible for NIAD onset? Why is the allelic variant ɛ2 a genetic protective factor for NIAD? And also, in the hypothesis of a common pathogenetic pathway underlying IAD and NIAD, what is the role of the ɛ4 and ɛ2 allelic variants in IAD?
Until today no conclusive interpretation has been formulated, and a pathogenetic model including the role of Aβ deposition and APOE unifying IAD and NIAD pathogenesis is still missing. Based on the experimental evidence, in the present paper we propose our interpretation of the AH in an attempt to answer these pending issues. It is important to note that communication in the field of AD clinics and genetics is sometimes unclear because even though widely accepted definitions are used, they still lend themselves to misinterpretstion. Accordingly, an attempt to clarify terms, definitions, and misunderstandings is reported in Table 1.
Definitions, acronyms, and symbols used in the text
THE AMYLOID HYPOTHESIS
The concept that both IAD and NIAD are a pathologically-defined condition due to the accumulation of Aβ was first proposed in 1984, when Glenner and Wong isolated Aβ from AD brain cortex. They suggested for the first time that the accumulation of Aβ in cerebral cortex could be the cause of AD. This is probable the first statement of the AH [33]. In the same year, they also found that the amino acid sequence of Aβ was the same as that isolated from the brain cortex of individuals with Down’s syndrome [34], always developing AD [35]. In 1985, the gene encoding the amyloid beta precursor protein (APP) was located on chromosome 21 [36]. Two years later, in 1987, the gene APP was cloned at locus 21q21.3 [37–39].
Since the official statement in 1992 [20], AH was improved in several aspects [40–43] up to a more recent reinterpretation [44–46], in which Aβ deposition resulted from an imbalance between Aβ peptides production and/or clearance. This imbalance “ ... leads to gradual accumulation and aggregation of the peptide in the brain, initiating a neurodegenerative cascade ... ” [47], giving a central role to the protein homeostasis as a result of a balance between anabolism and catabolism/clearance. This and other statements of AH, however, did not change the basis of the initial hypothesis: a linear pathway that begins with Aβ peptides production, continues with Aβ deposition and AP formation, and ends with IAD/NIAD onset. Therefore, the AH has become one of the most accepted models for IAD/NIAD pathogenesis, so that Aβ deposits are recognized as a histological hallmark of postmortem brain tissues [40, 48], and considered a fundamental feature of the histopathological diagnosis [49]. In fact, until today, a clinical diagnosis of IAD/NIAD can only be “possible” or “probable” [50–52] and confirmed only after death with the identification of Aβ deposits in the brain cortex [53, 54].
Thus, since both IAD and NIAD share common clinical and neuropathological characteristics and assuming the “lex parsimoniae” stated by the Franciscan friar William of Ockam (1287–1347) that “frustra fit per plura quod fieri potest per pauciora”, the simplest explanation of the causes suggests that both IAD and NIAD also share the same pathogenetic mechanism proposed by the AH, that implies a common genetic background. Thus, the “lex parsimoniae” becomes “the genetic approximation” of the AH. In fact, whereas mutations in the genes APP, PSEN1, or PSEN2 are recognized as causal factors in the Aβ deposition in IAD, in NIAD, if any, are still missing [1, 10]. Moreover, the recent data indicating a similar amyloid burden in cognitively healthy elderly brain [11–18] also suggested that not even Aβ deposits are the cause of AD. Finally, in recent years, it has been well-recognized that the major genetic risk factor for NIAD, the ɛ4 allelic variant of the APOE gene, is not a cause of the disease [55–57].
Aβ ANABOLISM
It is well known that proteolytic processing of the amyloid beta protein precursor (AβPP) leading to Aβ production follows two parallel biochemical pathways that are in equilibrium with each other [58]. These pathways are commonly known as the “non-amyloidogenic” and the “amyloidogenic” pathway, since the latter produces the family of Aβ, that are non-soluble and sticky, prone to aggregation to form oligomers, while the first produces a family of peptides, the amyloid α-peptides (Aα) that are soluble, and correctly metabolized by the neurons. In fact, despite its historical name, the AβPP is the precursor of both Aα and Aβ peptides (Table 1).
In detail, the AβPP processing pathway is a two-step mechanism in which the first step is catalyzed by the α-secretase or the β-secretase enzymes, in the non-amyloidogenic or the amyloidogenic pathway respectively, followed by a second common step catalyzed by the γ-secretase, producing Aα first and Aβ second. Notably, α - and β -secretases have opposite effects in generating Aα and Aβ due to their competitive cleavages of AβPP. In fact, increased α-secretase activity through protein kinase C stimulation decreases Aβ generation [59–61], while BACE1 knockout mice lack cerebral Aβ [62, 63,]. Therefore, the equilibrium between the two biochemical pathways is fundamental for Aβ anabolism. However, the most important difference is that Aβ peptides, and most of its oligomers, seem to be neurotoxic [64–69], while Aα seems to be neuroprotective [70–73]. Thus, a physiological age-related unbalance of this mechanism may be the first responsible for an age-related overproduction of Aβ, such as those observed in aging, and for a first addressing of the organism towards neurodegeneration or neuroprotection.
To schematize this process, assuming that Aβ overproduction and deposition is a physiological process of aging, in the weight scale of the anabolism of amyloid peptides, given as
However, since the balance between protein anabolism and catabolism/clearance determines the quantity of a protein in a biological system [44, 46], the final [Aβ] in brain cortex is determined by the balance between Aβ anabolism and catabolism/clearance [74].
Aβ CATABOLISM/CLEARANCE
The concept that the balance between protein anabolism and catabolism/clearance determines the quantity of a protein in a biological system is well known [44, 46]. Thus, the final [Aβ] in the brain cortex is determined by the balance between Aβ production and Aβ degradation/clearance [74]. This means that all factors influencing Aβ anabolism and/or Aβ catabolism/clearance decide the final [Aβ] in the brain cortex.
A summary of the possible alterations in Aβ homeostasis is summarized in Fig. 1. Assuming a physiological (Phy) age-related unbalance of this mechanism as the first cause of an age-related overproduction and deposition of Aβ [11–17], during aging,

The relationship between Aβ anabolism and β catabolism/ clearance in the pathogenesis of IAD/NIAD. A) The overproduction of Aβ is a physiological process of aging resulting from an age-related reduction of protein catabolism/clearance in the organism, increasing Aβ concentration [Aβ]. This is a very slow process leading to Aβ deposition and amyloid plaque (AP) formation. B) In the non-inherited AD form (NIAD), a series of environmental and physiological factors further decrease Aβ catabolism/clearance, increasing [Aβ]. This is a slow process leading to Aβ deposition and AP formation. C) In the inherited AD form (IAD), mutations in the amyloid-precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) causes the production of at least more 50% (in the heterozygotes) of Aβ, increasing [Aβ]. This is a very fast process leading to Aβ deposition and AP formation.
and [Aβ] slowly increases (Fig. 1A). However, during aging, Aβ catabolism/clearance may decrease in several ways, such as a decrease in the enzymatic activity of the major Aβ-degrading enzymes, neprilysin [45, 76], leading to an age-related reduced clearance [77], or due to the series of AD risk factors such as immune system dysregulation [78–80], mitochondrial dysfunction and oxidative stress [81, 82], metabolism abnormalities and diabetes [83–86], cholesterol homeostasis failure [87, 88], and deregulation of cell cycle pacing mechanisms [89, 90], all affecting Aβ catabolism. Thus, during aging
As a consequence,
and [Aβ] increases faster than the normal physiological conditions, leading the organism toward a slow neurodegeneration (Fig. 1B). This may be the condition in which NIAD onset. Conversely, mutations of APP, PSEN1, and PSEN2 genes may instead affect Aβ anabolism. Thus, in presence of APP, PSEN1, and PSEN2 gene mutations
As a consequence,
and [Aβ] rapidly increases, leading the organism toward a fast neurodegeneration (Fig. 1C). This may be the condition in which IAD onset.
THE APOLIPOPROTEIN E
The pivotal role of APOE in the neuropathology of the central nervous system is widely documented [91], and the genetics of the APOE polymorphism has been well investigated [92–94]. Briefly, the three common genetic variants known as ɛ2, ɛ3, and ɛ4 of the APOE gene are determined by three of the four haplotypes resulting from the combination of the alleles of the T-to-C and C-to-T single-nucleotide polymorphisms (SNPs) rs429358 and rs7412 at the APOE gene locus. Among the three genetic variants, ɛ3 (haplotype T-C) has been observed as the most common in the population worldwide, and therefore considered as the “normal variant”, followed by ɛ4 (haplotype C-C), and ɛ2 (haplotype T-T), being the fourth haplotype C-T (variant ɛ3r) the rarest [94, 96]. Interestingly, both SNPs change the first base of two CGC codons encoding arginine (Arg) resulting in TGC codons encoding cysteine (Cys). Thus, at codons 112 and 158, the APOE encoded by the three genetic variants carries the different amino acid combinations Cys-Arg (APOE3), Arg-Arg (APOE4), and Cys/Cys (APOE2), with the Arg-Cys (APOE3r) the rarest [94, 96].
Since 1991, involvement of the E4 isoform in the clinic and neuropathology of AD has been well documented as the main genetic risk factor for NIAD [21–24] compared with the normal E3 isoform. Until now, APOE is still confirmed as major risk factor for NIAD, namely the main gene with a semi-dominant inheritance [25]. Indeed, a protective role of the E2 isoform in NIAD has also been proposed [21, 26–28]. However, despite this evidence, it has been well demonstrated that, from a genetic point of view, the presence of the ɛ4 allele is neither necessary nor sufficient to cause AD [55–57]. Accordingly, the clinicopathologic series in which the role of APOE in the diagnosis of NIAD was estimated [55, 96] did not support a rationale for its genotyping in the clinical setting [97]. Conversely, it has been well documented that in the presence of a clinical diagnosis of possible/probable NIAD, APOE genotyping may be a useful diagnostic tool [98–102].
To place APOE in the AH frame, its interaction with Aβ must be considered. It is well known that APOE binds and transports Aβ [103–106]. Interestingly, the different APOE isoform shows a different binding affinity for Aβ, according to the following order:
In the present interpretation of the AH (Fig. 2), we assume that the neurotoxic action of Aβ follows a threshold model in which the AD onset is a consequence of a critical Aβ concentration ([Aβ]c), i.e., when
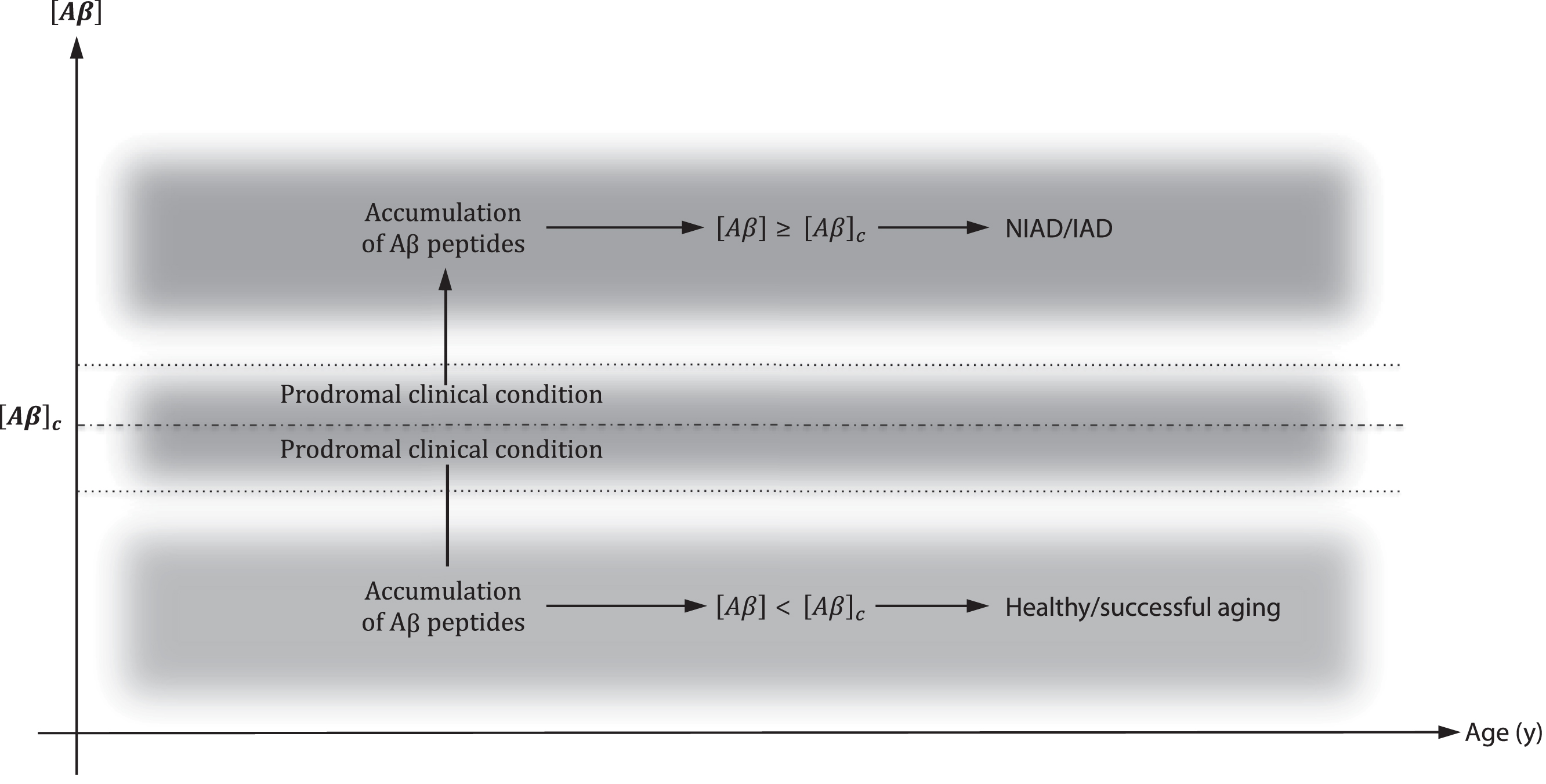
A critical concentration of Aβ ([Aβ]c) determined the onset of IAD/NIAD. Until [Aβ] < [Aβ]c, healthy aging progress. When [Aβ]≥[Aβ]c, IAD/NIAD onset. Around [Aβ]c probably exists a shaded area in which a clinical prodromal condition may be identified.
This is the necessary and sufficient condition to IAD/NIAD onset. Moreover according to the reported different affinities of the APOE isoforms for Aβ, [Aβ]c increases in the following order:
thus, predisposing one to AD onset at a lower [Aβ]c in ɛ4 and at higher [Aβ]c in ɛ2 carriers (Fig. 3). Thus, a higher amyloid burden is expected in the presence of the ɛ4 allele in a dose-dependent fashion [113, 118]. In this context, all factors affecting Aβ metabolism, and therefore [Aβ]c, may be involved in IAD and NIAD onset. Indeed, in a study investigating erythrocyte associated Aβ and its potential role as a biomarker to diagnose dementia [119], we observed increased concentrations of erythrocyte associated Aβ in the presence of APOE4 and a decreased concentration in APOE2 as compared with APOE3 carriers (data not showed).
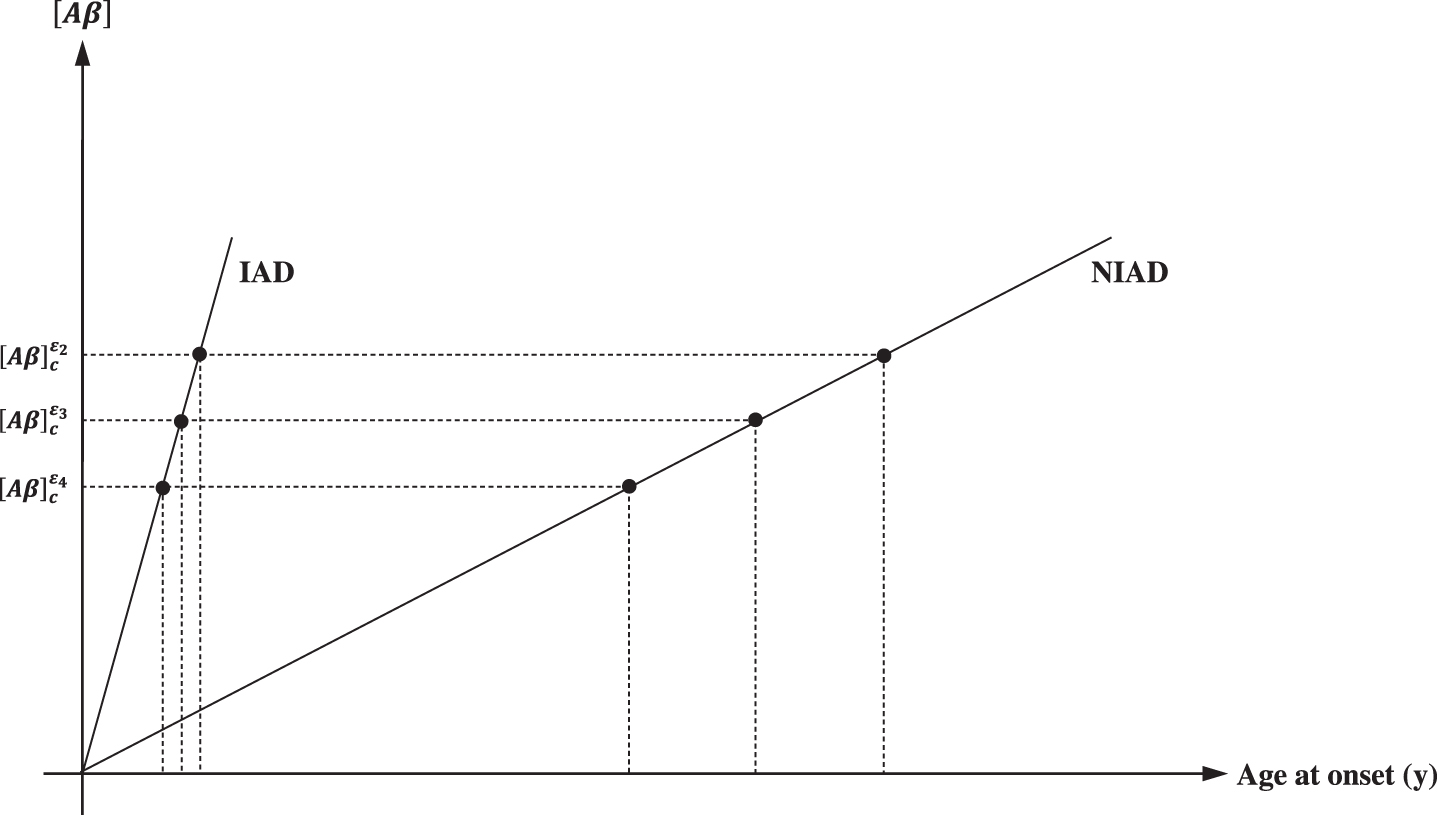
Apolipoprotein E (APOE) titrate [Aβ]. In respect to E3, which is the most common isoform, E4 have an increased Aβ affinity, lowering [Aβ]c threshold and the age at onset. Conversely, E2 have a decreased affinity for Aβ, increasing [Aβ]c threshold and age at onset. Notably in IAD, since the rapid increase in [Aβ], the effect of the APOE isoform on [Aβ]c and age at onset is negligible.. It is, conversely, significant in NIAD. The first consequence of this model is the increased amyloid burden associated with APOE4 and the decreased amyloid burden associated with APOE2.
DISCUSSION
The amyloid hypothesis
In the present study, we proposed an interpretation of the AH aimed to address some missing answers, mainly regarding the pathogenetic roles of Aβ deposition and the APOE polymorphism. An overall summary of the proposed model, starting from AβPP processing and ending with IAD/NIAD onset or healthy aging is summarized in Fig. 4.

A summary of the present interpretation of the AH. The original formulation suggested the amyloid plaques (AP) as the cause of the onset of inherited and non-inherited Alzheimer’s disease (IAD, NIAD). However, it did not explain why AP were observed in healthy brain, and the role of the apolipoprotein E (APOE) isoforms APOE2 and APOE4. (1) Aβ anabolism. The competition between α- and β-secretase to cleave the amyloid-beta protein precursor (AβPP) is the first determinant of the physiological Aβ concentration ([Aβ]). (2) Aβ catabolism/clearance. The equilibrium between Aβ anabolism and Aβ catabolism/clearance is the second determinant of the physiological [Aβ]. (3) Mutations in the amyloid-beta precursor protein (APP), presenilin-1 (PSEN1), or presenilin-2 (PSEN2) genes strongly affect Aβ anabolism, increasing [Aβ] and unbalancing
In the present understanding we assume that exists a critical concentration of Aβ peptides and oligomers that is determined by an unbalance of the Aβ homeostasis, the APOE genotype, and individual physiological conditions do exist, and that AD arises only when the concentration of Aβ peptides and oligomers exceed this threshold. This means that the neurotoxic effect of Aβ is manifested only beyond this threshold. In this context, AP formation is a direct consequence of Aβ production, but independent of the neurotoxic action, thus it becomes a necessary but not sufficient condition for AD onset. In fact, it is well established that AP are observed both in AD and in healthy brain, since Aβ deposition is a normal physiological process of aging [11–18]. Therefore, considering that the AβPP non-amyloidogenic pathway did not form AP, we can observe healthy brains with and without AP. Conversely, we can observe AD brains only with AP. This mechanism of action also explains why Aβ deposition begins years before the AD clinical onset and why AP are observed years before AD clinical onset. Notably, since the topography of Aβ deposits in AD brain follows a well-defined geographic progression [120, 121], it may be possible that AP distribution may be different between IAD, NIAD, and a normal aged brain.
The second question concerns the role of the APOE polymorphism, and in particular of the ɛ4 allele variant in NIAD, because despite a well-documented role as a major genetic risk factor increasing NIAD risk 4-fold in ɛ3/ɛ4 heterozygous and 8-fold in ɛ4/ɛ4 homozygous, its presence is neither necessary nor sufficient to cause the disease. This means that a genetic test identifying the ɛ4 allele variant in asymptomatic patients might be prognostic, since it increases the risk of NIAD, but not diagnostic because it is not the cause of the disease. In the present model, we suggest that APOE4 is associated to a higher amyloid burden, lowering the age at onset. This mechanism identifies the ɛ4 allele variant as a genetic risk factor directly involved in the NIAD pathogenesis but not responsible for its onset. This hypothesis also answers the other two questions. Is the ɛ2 allelic variant a genetic protective factor for AD? Is Aβ deposition prodromal to AD? The first answer is yes, since APOE2, with a lower affinity for Aβ, is associated with a lower amyloid burden increasing the age at onset. This explains the role of APOE2 as a protective factor regardless of NIAD causes of onset. Conversely, the second answer is no, because when [Aβ] < [Aβ]c, the disease does not occur. It is clear that a prodromal role of [Aβ] near [Aβ]c cannot be excluded. However similarly to APOE4, Aβ deposition is a necessary but not sufficient condition for disease onset. Notably this model implies that the higher/lower amyloid burden associated with APOE4 the first, and APOE2 the second observed in the AP, are similar in AD and healthy brain. Therefore, AD onset is an event resulting from a physiological process of Aβ production that can be accelerated by a series of environmental and non-environmental factors and much more rarely by mutations in AD genes. In this context, both AP and APOE4 are necessary but not sufficient to cause AD. It is notewhorthy that, the AβO hypothesis [64–66, 69] (see also Table 2), that places the Aβ peptides and oligomers, and not the Aβ fibrils, first for their neurotoxic role [68] may well fit with our AH interpretation. Indeed, the importance of the AβO hypothesis has grown so much that recently, it has been reported that “ ... it has all but supplanted the amyloid cascade ... ” [67].
AD pathogenesis: The AH and the first event schools of thought
*Articles in which the hypothesis was proposed for the first time. 1Pathogenetic hypothesis valid for both IAD and NIAD. 2Pathogenetic hypothesis valid only for NIAD. 3Mitochondrial dysfunction resulting by an age-related accumulation of mtDNA mutations. 4Aβ production as a protective adaptation to the disease. 5Soluble Aβ acting as neuroprotective factor. 6Pathogenetic hypothesis valid only for IAD. 7Causing presenilin loss of function. 8The initial Aβ production as antimicrobial peptide will be beneficial on first microbial challenge but will become progressively detrimental as the infection becomes chronic and reactivates from time to time. 9Age related Aβ-deposition. 10A vicious cycle of inflammation has been formed between Aβ accumulation, activated microglia, and microglial inflammatory mediators, which enhance Aβ deposition and neuroinflammation. 11Factors associated with AD can be seen as partial stressors within the matrix of signaling pathways that underlie cell survival and function. Senescence pathways are triggered when stressors exceed the cells ability to compensate for them. 12Causing impaired AβPP metabolism.
In conclusion, assuming a threshold model in which [Aβ] is titrated by APOE and AD occurs only beyond [Aβ]c, the most common questions about the role of APOE in the AH can be considered answered.
Other hypotheses
In the present paper, by means of an interpretation of the AH including the role of aging and APOE, the major risk factors for AD, we try to offer a complete overview of IAD and NIAD pathogenesis [122]. Clearly, this interpretation takes for granted Aβ deposition as a pivotal event in the pathogenetic process. However, since the AH is still criticized [47, 123–133], all the hypotheses considering Aβ deposition as a secondary event, if any, cannot be excluded (Table 2). These hypotheses include: 1) the mitochondrial hypothesis suggesting an age-related increase of oxidative stress [82, 134–138], also resulting from mitochondrial mutations [139–141], sometimes in synergistic effect with protein glycation [142] or abnormalities in mitotic signaling [90, 144], either individually [147] or in a synergistic manner [90, 148]; 2) the infection hypothesis suggesting a bacterial/virus infection as the first event [145–148], also considering inflammation as a secondary event [149–152]; and 3) the vascular hypothesis suggesting vascular deficits as first event [153–155]. The final hypothesis in which AD is the result of an interaction among these causes suggesting that it may be a complex disease [126, 157] must be also considered.
Notably, a protective role of Aβ deposition which acts as protective adaptation to the neuronal stress resulting from the disease, have been also suggested [128, 159]. It is interesting to note that the confirmation or confutation of the amyloid hypothesis is crucial for the treatment to be followed, i.e., the anti-amyloid or the non-anti-amyloid treatment [160–167]. Paradoxically, in the acceptance of a protective role of Aβ in AD, all therapeutic efforts aimed at lowering the Aβ production or removing Aβ deposit will only serve to exacerbate the disease process [128].
Aβ and τ
Of interest is the integration of the role of τ in the AH, asking a further question. What is the connection between AH and τ pathology? Indeed, some studies consider τ deposition as the first event in AD pathogenesis, thus formulating a further non-amyloid hypothesis, the “τ hypothesis” [172]. However, from the “amyloid point of view”, currently the AH has no answer to this question [135]. It is clear that if τ deposition is considered the first pathological event in AD, the question is up-side down [168], i.e., what is the connection between the τ hypothesis and Aβ deposition? In fact, it has recently been reported that τ lesions occurred before Aβ accumulation [169, 170], suggesting that AD progression is strongly associated with τ pathology, rather than Aβ pathology. Although the transmission mechanism of τ cell-to-cell aggregates is still not clear, τ pathology spreads in the brain in a well-defined way, and it has been demonstrated that its distribution can be correlated with the clinical stages of the disease [171]. In fact, it is reported that τ pathology correlates better than Aβ pathology with clinical features of AD [172]. As a result, it has been recently reported that the increase of AβPP with or without familial AD mutations, not Aβ, can function as an abnormal τ fibrils receptor and promote the intracellular τ aggregation [172], suggesting that AβPP, rather than Aβ, can accelerate τ accumulation and propagation [172]. In animal models, it has already been reported that the suppression or deletion of τ has a profound protective effect against brain damage and neurological deficit [173–178]. Therefore, it may be possible that AD is a disorder triggered by impairment of AβPP metabolism and progresses through τ pathology. However, despite these suggestions, a complete connection to add the role of τ in the AH is still missing. Accordingly, the AT(N) marker grouping [179] of the National Institute on Aging and Alzheimer’s Association (NIAA) Research Framework [180] place amyloid biomarkers at the apex of the preclinical biomarker hierarchy, with τ biomarkers as second.
Aβ and biomarkers
In addition to treatment, to accept or not of Aβ deposition as the first pathological event in AD also defines its role as early biomarker of the disease. In particular, although according to the present interpretation of the AH Aβ deposition alone is not sufficient to cause AD, we cannot exclude that, it may be sufficient to cause downstream pathologic changes (i.e., tauopathy and neurodegeneration) that ultimately lead to AD [52]. This suggests doubts that Aβ alone could serve as an early biomarker or as the defining signature of AD. However, both Aβ and τ deposits are necessary to fulfill neuropathologic criteria for AD diagnosis, which suggests that evidence of abnormalities in both Aβ and pathologic τ biomarkers should be present to apply the label “Alzheimer’s disease” in a living person [54, 181]. Thus, although it is possible that AP and neurofibrillary τ deposits are not causal in AD pathogenesis, these abnormal protein deposits define AD as a unique neurodegenerative disease among several disorders that can lead to dementia [185]. In this context, the definition of AD as a biological construct will allow a more accurate characterization and understanding of the sequence of events leading to cognitive impairment associated with AD, as well as the multifactorial etiology of dementia. This approach will also allow a more precise approach to interventional trials in which specific pathways can be targeted in the disease process and in the appropriate patients [185, 182].
CONCLUSIONS
In the present paper, we propose an interpretation of the AH that unifies the pathogenetic models for IAD/NIAD and explains why AP and APOE4 are also observed in healthy aging and why they are not the cause of AD.
Of interest may be the implications of the proposed AH interpretation in influencing future clinical and research directions. The key point that we propose as central in the model, namely the neurotoxic action of Aβ only above a critical concentration, suggests that raising [Aβ]c may be a correct action to delay IAD/NIAD onset, and therefore the way to setup a pharmacological treatment against IAD/NIAD. In this context, an action mechanism that mimics those of the APOE interaction with Aβ, in which the first titrates the second, may be the correct model to follow to develop new drugs for AD. It is clear that the APOE cannot be a therapeutic target itself, just as non-physiological Aβ deposition as those observed in the use of antibodies against Aβ must be avoided. Moreover, since Aβ is needed for a certain number of physiological functions, in addition to its role in the pathogenesis of IAD/NIAD, a failure or adverse effect resulting from actions against Aβ production may be hypothesized. It is also clear that is not possible to exclude a role toward IAD/NIAD of the treatment that modulates the actions of the risk factors that revolve around IAD/NIAD pathogenesis. Thus, the correct understanding of the IAD/NIAD pathogenesis can open new previously missing therapeutic pathways.
Clearly, the proposed interpretation of the AH did not exclude the validity of other model considering Aβ deposition as a secondary event. Undoubtedly, among these non-amyloid hypotheses, the most reliable is to put mitochondria and oxidative stress as the first event. This hypothesis clearly includes age as a primary risk factor for AD. Aging is the accumulation of changes that increases the risk of death. Accordingly, neurons involved in the lesions associated with NIAD are apparently aging at a faster rate than normal since the same lesions are observed at later ages in normal individuals. There is a growing consensus that these deleterious changes are produced by free radical reactions, initiated largely by mitochondria during normal metabolism at a rate that progressively increases with age, while the life span of an individual is determined by the rate of such damage to the mitochondria. Accordingly, Aβ deposition will follow as a consequence of an increase in reactive oxygen species concentration as a secondary event. This hypothesis has been well investigated and confirmed by a series of papers [82, 138–143]. However, despite this pathogenetic hypothesis including age as a major risk factor for AD, the role of APOE as a main genetic risk factor for AD is far from being explained. Similarly, while this hypothesis fits well with NIAD onset, there are some difficulties in justifying it as the cause of the mutation responsible for IAD onset.
In conclusion, more studies are needed to better understand the molecular mechanisms underlying the behavior of Aβ and APOE in the AH context to verify the correctness of our understanding.
Footnotes
ACKNOWLEDGMENTS
This work was fully supported by “Ministero della Salute”, I.R.C.C.S. Research Program, Ricerca Corrente 2018–2020, Linea n. 2 “Meccanismi genetici, predizione e terapie innovative delle malattie complesse” and by the “5×1000” voluntary contribution to the Fondazione I.R.C.C.S. Ospedale “Casa Sollievo della Sofferenza”.