
Abstract
Cholesterol dyshomeostasis has been linked to the pathogenesis of sporadic Alzheimer’s disease (AD). In furthering the understanding of mechanisms by which increased levels of circulating cholesterol augments the risk of developing sporadic AD, others and we have reported that low-density lipoprotein (LDL) enters brain parenchyma by disrupting the blood-brain barrier and that endolysosome de-acidification plays a role in LDL-induced amyloidogenesis in neurons. Here, we tested the hypothesis that endolysosome de-acidification was central to amyloid-β (Aβ) generation and that acidifying endolysosomes protects against LDL-induced increases in Aβ levels in neurons. We demonstrated that LDL, but not HDL, de-acidified endolysosomes and increased intraneuronal and secreted levels of Aβ. ML-SA1, an agonist of endolysosome-resident TRPML1 channels, acidified endolysosomes, and TRPML1 knockdown attenuated ML-SA1-induced endolysosome acidification. ML-SA1 blocked LDL-induced increases in intraneuronal and secreted levels of Aβ as well as Aβ accumulation in endolysosomes, prevented BACE1 accumulation in endolysosomes, and decreased BACE1 activity levels. LDL downregulated TRPML1 protein levels, and TRPML1 knockdown worsens LDL-induced increases in Aβ. Our findings suggest that endolysosome acidification by activating TRPML1 may represent a protective strategy against sporadic AD.
INTRODUCTION
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder that is characterized clinically by progressive impairments in memory and cognition, and neuropathologically by loss of synaptic integrity, accumulations of amyloid-β (Aβ) and phosphorylated tau proteins, and increased levels of oxidative stress [1–4]. Aβ continues to be considered an important pathogenic factor of AD [5, 6], and mutations in amyloid-β protein precursor (AβPP) and presenilin proteins result in early-onset familial AD [5]. However, the vast majority of AD cases are sporadic in nature.
Currently, pathogenic mechanisms responsible for the accumulation of Aβ in sporadic AD remain unclear, but are believed to result from complex interactions between aging, nutritional, environmental, and genetic factors [7]. Among these factors, altered levels of circulating cholesterol have been implicated in the pathogenesis of sporadic AD [8–12]. More specifically, as the single strongest genetic risk factor for sporadic AD [7, 13], ApoE4 is clearly associated with elevated plasma levels of low-density lipoprotein (LDL) and decreased levels of high-density lipoprotein (HDL) [14, 15]. High levels of plasma LDL, independent of APOE genotypes, increase the risk of developing sporadic AD [8, 16–20], and low levels of plasma HDL, independent of the APOE genotype, are associated with decreased risk of developing AD [11, 22].
How altered circulating cholesterol contributes to the pathogenesis of sporadic AD in brain remains unclear, especially when essentially all brain cholesterol is locally synthesized and there is no exchange of LDL particles across an intact blood-brain barrier (BBB) [23]. As such, apoB-100, the exclusive LDL carrier protein in circulating blood, is not present in normal brain [24]. However, under conditions when the BBB is compromised, as occurs early in sporadic AD [25–30], plasma LDL can enter brain parenchyma where it may contribute to the pathogenesis of sporadic AD. In support, impairment of the BBB can increase cholesterol flux from the peripheral circulation into brain [31], and apoB-100 is present in AD brain, where it has been shown to co-localize with Aβ [9, 32–34]. In addition, others and we have shown that high levels of plasma cholesterol disrupts the BBB [35–37] and increases brain levels of apoB-100 [9, 38]. Thus, circulating LDL, when entering brain parenchyma through a compromised BBB, could have a direct effect on the development of AD in brain.
In brain, LDL can be internalized into endolysosomes via receptor-mediated endocytosis, and thus LDL could directly disturb endolysosome function. Consistent with findings that endolysosome dysfunction is an early and prominent pathological feature in sporadic AD [39, 40], we have shown that LDL de-acidifies neuronal endolysosomes, induces endolysosome dysfunction, and enhances amyloidogenic processing of AβPP in endolysosomes [10]. Because endolysosome de-acidification in neurons appears to play an important role in LDL-induced amyloidogenesis, it is likely that endolysosome acidification may decrease the pathogenesis of AD [41].
Here, we compared the effects of LDL and HDL on endolysosome function and Aβ generation in neurons. We also determined the extent to which and mechanisms by which ML-SA1, an agonist of endolysosome TRPML1 channels, affects endolysosome pH and LDL-induced increases in Aβ generation in neurons. We demonstrated that LDL, but not HDL, de-acidified endolysosomes and increased Aβ. Furthermore, we found that ML-SA1 acidified endolysosomes and prevented LDL-induced endolysosome de-acidification and increases in Aβ levels. Mechanistically, we demonstrated that TRPML1 channels were specifically involved in the endolysosome acidifying effects of ML-SA1 and its protective effects on LDL-induced amyloidogenesis.
MATERIAL AND METHODS
Cells
Primary cultures of cortical neurons were prepared from embryonic day 18 Sprague-Dawley rats using a protocol approved by the University of North Dakota Animal Care and Use Committee adherent with the Guide for the Care and Use of Laboratory Animals (NIH publication number 80–23). Pregnant dams (embryonic day 18) were sacrificed by asphyxiation with CO2. The fetuses were removed, decapitated, and meninges-free cerebral cortices were isolated, trypsinized, and plated onto 35-mm poly-D-lysine-coated glass-bottom tissue culture dishes. Neurons were grown in NeurobasalTM medium with L-glutamine, antibiotic/antimycotic, and B27 supplement, and were maintained at 37°C and 5% CO2. Typically, the purity of the neuronal cultures was greater than 95% as determined by immunostaining with neuronal (mouse anti-NeuN, Millipore MAB377; or Chicken anti-MAP2 antibodies, Millipore AB15452) and astrocyte (mouse anti-GFAP antibody, Sigma G3893) markers. Neuron were kept in culture for 10–14 days, at which time they were treated with LDL (Kalen Biomedical) or HDL (Kalen Biomedical) for another 3 days. Human neuroblastoma cells (SH-SY5Y) expressing wild type AβPP were kindly supplied by Dr. Norman Haughey (John Hopkins University). SH-SY5Y cells were cultured in Eagle’s minimum essential medium (MEM) supplemented with 10% FCS, penicillin/streptomycin, nonessential amino acids, and sodium pyruvate (1 mM) at 37°C in 5% CO2. For our experiments, 4×106 SH-SY5Y cells were seeded on 60 mm2 dishes and cultured for 48 h at which time the media was changed to serum-free MEM and treated with LDL (Kalen Biomedical) or HDL (Kalen Biomedical) for 3 days. ApoB from human plasma (Sigma, A5353) and recombinant human apoE3 (Sigma, SRP4696) were used to prepare the mixture of HDL/apoB and HDL/apoE3.
Measurement of endolysosome pH
Endolysosome pH was measured using a ratio-metric lysosome pH indicator dye (LysoSensor Yellow/Blue DND-160, Invitrogen); a dual excitation dye that permits pH measurements in acidic organelles independently of dye concentration. Neurons were loaded with LysoSensor (2μM) for 5 min at 37°C. Light emitted at 520 nm in response to excitation at 340 nm and 380 nm was measured for 20 ms every 5 s using a filter-based imaging system (Zeiss and 3i). The ratios of light excited at 340/380 nm and emitted at 520 nm were converted to pH using a calibration curve established using 10μM of the H +/Na+ ionophore monensin, and 20μM of the H +/K+ ionophore nigericin dissolved in 20 mM 2-(N-morpholino) ethane sulfonic acid (MES), 110 mM KCl, and 20 mM NaCl adjusted to pH 3.0 to 7.0 with HCl/NaOH.
Lysosome fraction preparation
Crude lysosome fractions were prepared using a lysosome isolation kit (Sigma). Cells were homogenized in extraction buffer, and homogenates were centrifuged at 1,000×g for 10 min at 4°C. Supernatants were collected and pellets were homogenized in 2 volumes of 1x extraction buffer and centrifuged at 1,000×g for 10 min at 4°C. The two supernatants were pooled and then centrifuged at 20,000×g for 20 min at 4°C. Pellets were resuspended in 0.5 ml of 1x extraction buffer and this fraction contained a mixture of light mitochondria, endosomes, lysosomes, peroxisomes, and endoplasmic reticulum.
Measurement of BACE1 activities
BACE1 activity was determined using 50 mM synthetic peptide substrates containing the BACE1 cleavage site (MCA-Glu-Val-Lys-Met-Asp-Ala-Glu-Phe-(Lys-DNP)-OH) (Calbiochem). Equal amounts of protein (50μg) were used from each sample lysate. The fluorescence was measured using a fluorescence microplate reader with an excitation wavelength set at 320 nm and an emission wavelength set at 383 nm. As a control for specificity, BACE1 activity was tested in the absence and presence of the BACE-1 inhibitor, H-Lys-Thr-Glu-Glu-Ile-Ser-Glu-Val-Asn-Stat-Val-Ala-Glu-Phe-OH (Calbiochem).
Immunoblotting
Cells were lysed with RIPA buffer (Pierce) plus 10 mM NaF, 1 mM Na3VO4 and Protease Inhibitor Cocktail (Sigma). After centrifugation (14,000×g for 10 min at 4°C), supernatants were collected, and protein concentrations were determined with a DC protein assay (Bio-Rad). Proteins (10μg) were separated by SDS-PAGE (12% gel) and transfer to polyvinylidene difluoride membranes (Millipore). The membranes were incubated overnight at 4°C with antibodies against acid phosphatase (Abcam, ab58688), TRPML1 (Sigma, HPA031763), N-terminal AβPP (Thermofisher, 14-9749-80), BACE-1 (Thermofisher, MA1-177), LAMP-1 (Sigma, L1418), v-ATPase V1A1 (Santa Cruz, sc-374475), v-ATPase V0a1(Thermofisher, PA5-69418). GAPDH (Abcam, ab8245) was used as a gel loading control. The blots were developed with enhanced chemiluminescence, and bands were visualized and analyzed using our OdysseyFc multi-modal imager system (Li-Cor). Quantification of results was performed by densitometry and the results were analyzed as total integrated densitometric volume values (arbitrary units).
Immunostaining
Cells were fixed with cold methanol (– 20°C) for 10 min, washed with PBS, blocked with 5% goat serum, and incubated overnight at 4°C with primary antibodies including amyloid beta mouse antibody MOAB-2 (Biosensis, M-1586), BACE1 mouse antibody (Thermofisher, MA1-177) and lysosome-associated membrane protein-1 (LAMP1) rabbit antibody (Sigma, L1418). After washing with PBS, cells were incubated with fluorescence-conjugated secondary antibodies including Alexa 488-conjugated goat anti-mouse antibody (Invitrogen) and Alexa 546-conjugated goat anti-rabbit antibody (Invitrogen). Images were taken using our confocal microscope (Zeiss 800). Controls for specificity included staining with primary antibodies without fluorescence-conjugated secondary antibodies (background controls) and staining with only secondary antibodies; these controls eliminated auto-fluorescence in each channel and bleed-through (crossover) between channels. Averaged fluorescent intensities per cell was quantified using Image J software (NIH). Percentage of colocalization were quantified using Image J software (NIH) and data were normalized to control.
Quantification of Aβ levels
Secreted and intracellular Aβ levels were measured using human/rat Aβ 1-40 and Aβ 1-42 ELISA kits (Wako). For secreted Aβ measurements, media from cultured cells was collected, diluted 1:4 with standard diluent buffer, and each sample was analyzed in duplicate. Total cellular protein levels were determined by a DC protein assay (Bio-Rad). Aβ levels were normalized to total protein content in each sample. For intracellular Aβ measurements, cells were diluted 8-times (w/v) with ice-cold 5 M guanidine-HCl/50 mM Tris-HCl cells and were homogenized. The homogenates were diluted with ice-cold reaction buffer (Dulbecco’s phosphate-buffered saline containing 5% BSA and 0.03% Tween-20 supplemented with 1x protease inhibitor cocktail). Following centrifugation at 16,000×g for 20 min at 4°C, supernatants were collected, diluted 1:1 with standard diluent buffer, and quantified by colorimetric sandwich Aβ ELISA kits (Wako). Intracellular Aβ levels were normalized to total protein content in the samples.
RNA interference
TRPML1 (mucolipin1) protein levels were knocked down with specific rat siRNAs (RSS305451, RSS305452, RSS305453 from Thermofisher) at a final concentration of 20 nM; negative siRNAs (4404020 from ThermoFisher) were used as controls. Before siRNA transfection, fresh Neurobasal media was added to cultured neurons plated for 10 days. The transfection cocktail containing transfection buffer (SignaGen), TRPML1 siRNA stock, and GenMuteTM reagent was added carefully to each dish along with fresh Neurobasal media. After incubation (37°C, 5% CO2) for 5 h, the transfection media was replaced with fresh Neurobasal media or treated with LDL for another 48 h. Knockdown efficiency was measured by immunoblotting.
Statistical analysis
All data were expressed as means and SEM. Statistical significance for multiple comparisons was determined by one-way ANOVA plus a Tukey post-hoc test or by Student’s t-test. p < 0.05 was considered to be statistically significant.
RESULTS
LDL, but not HDL, increases Aβ
Given epidemiological findings that LDL increases, whereas HDL decreases, the risk of developing AD, we first determined the extent to which LDL and HDL affects intraneuronal and secreted levels of Aβ. It should be noted that there is a species difference in the composition of lipoproteins. Human liver produces only apoB100, whereas rodent liver produces a mixture of ApoB100 and ApoB48 [42]. Thus, human LDL particles contain only ApoB100, whereas rodent LDL particle contains both apoB100 (slightly dominating) and apoB48. Because apoB48 lacks the canonical LDLR binding site, human LDL particles and rodent LDL particles exhibit different receptor binding property [43]. Rodent HDL particles is also different from that of humans. ApoE is the major protein component of rodent HDL, whereas human HDL contain only small amounts of ApoE. Thus, in rodents, HDL particles are the predominant class of lipoproteins that transport plasma cholesterol; but in humans, LDL particles are the major transporter of plasma cholesterol [43]. Due to these differences in lipoproteins and lipid profiles, rodents are relatively resistant to the development of atherosclerosis. For these reasons, LDL and HDL derived from human plasma are used in the present study. In SH-SY5Y cells, LDL treatment (50μg/ml for 3 days) significantly increased secreted levels of Aβ 1-40 and Aβ 1-42 (Fig. 1A), and intraneuronal levels of Aβ 1-40 and Aβ 1-42 (Fig. 1B). However, HDL treatment (50μg/ml for 3 days) did not significantly increase secreted or intraneuronal levels of Aβ. In contrast to LDL, HDL decreased significantly secreted levels of Aβ 1-40 (Fig. 1A). Neither LDL nor HDL affected protein levels of full-length AβPP (Fig. 1C), but LDL (not HDL) increased protein levels of BACE1 (Fig. 1D). At the concentrations used, neither LDL nor HDL induced significant levels of cell death as indicated by LDH releasing assay (data not shown). To determine whether LDL-induced increases in Aβ is due to increased cell proliferation, we measured total protein levels in cells treated with LDL and HDL for 3 days as compared with cells without any treatment for 3 days (control). We found that total cellular protein concentrations (μg/μl) following treatment with LDL (0.37±0.13), HDL (0.31±0.03), or control (0.34±0.04) were not different (n = 5, p > 0.05). Given that cells were seeded at the same density at the beginning of treatment and total cellular proteins were collected in the same way, our finding suggests that LDL does not affect cell proliferation when compared with HDL.
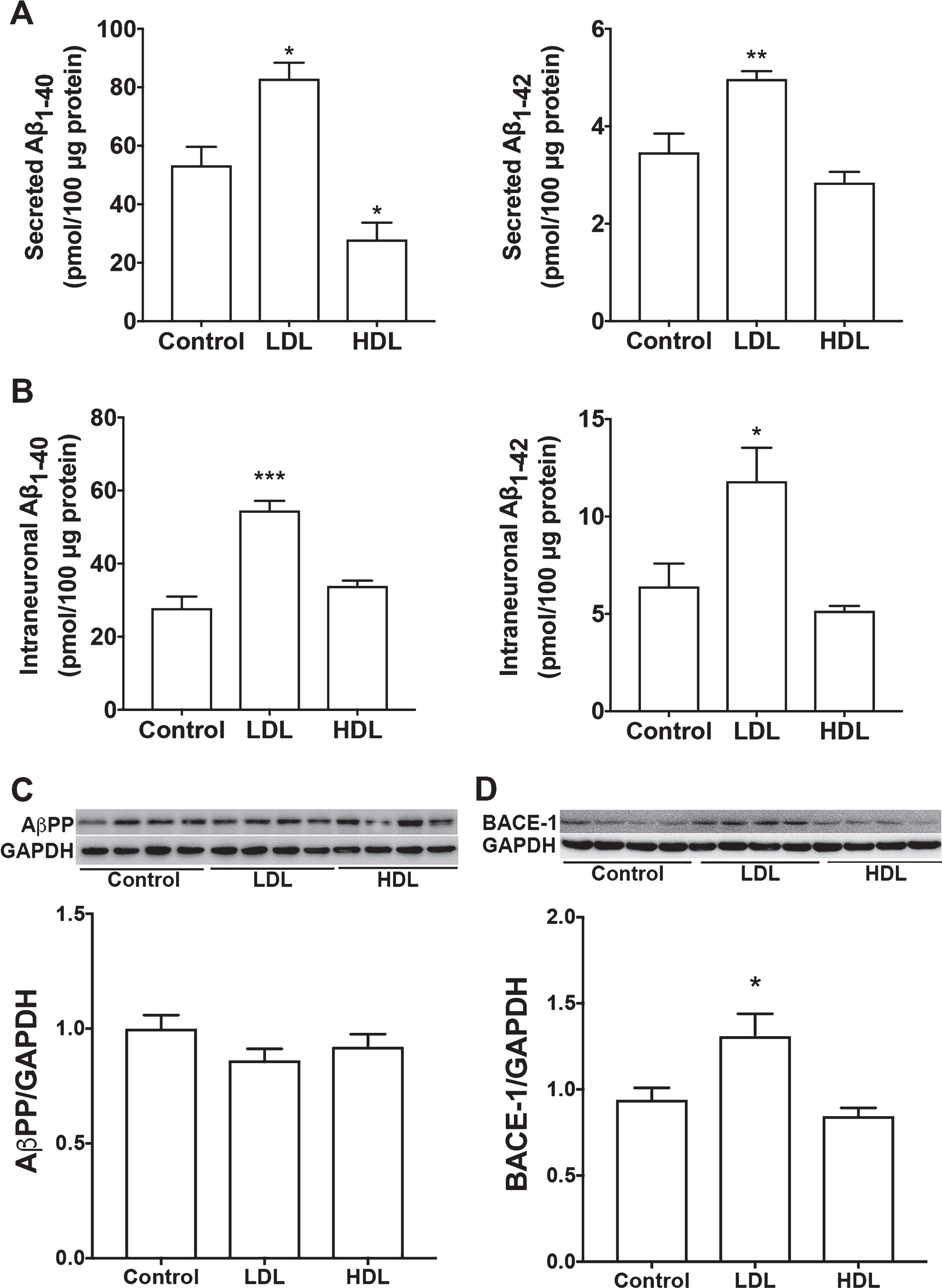
LDL, but not HDL, increased Aβ. A, B) In SH-SY5Y cells expressing wild-type AβPP, LDL (50μg/ml for 3 days) treatment significantly increased secreted levels Aβ 1-40 and Aβ 1-42, as well as, intraneuronal levels of Aβ 1-40 and Aβ 1-42 (*p < 0.05; **p < 0.01; ***p < 0.001 versus control; n = 5). HDL (50μg/ml for 3 days) treatment did not significantly increase secreted or intraneuronal levels of Aβ (n = 5). C) Neither LDL nor HDL affected protein levels of full-length AβPP. D) LDL treatment, but not HDL, increased significantly protein levels of BACE1 (*p < 0.05; n = 4).
Because ApoB, the exclusive apolipoprotein of LDL, is not present in plasma HDL or in brain in situ synthesized ApoE-rich lipoprotein, we then determined the effect of HDL mixed with ApoB or ApoE3 on the production of Aβ in SH-SY5Y cells. ApoE-rich lipoproteins synthesized in situ in brain is thought to be HDL-like particles composed of phospholipids and un-esterified cholesterol [44–46]. To model brain in situ apoE-rich HDL-like lipoproteins, we pre-incubated apoE3 with HDL with a fixed ratio of 0.4 for apoE protein/HDL protein. This ratio was based on findings that the phospholipid content of human plasma HDL is about 29% and the protein content of human plasma HDL is about 40%; thus, the ratio of phospholipids/protein content of HDL is about 0.7 [47]. Furthermore, the apoE/phospholipid ratio in CSF is about 0.6 [48]. Therefore, if one assumes that the phospholipid (HDL) content is similar in plasma and CSF, the calculated ratio of apoE protein/HDL protein content in CSF is 0.42. Similarly, we pre-incubated apoB with HDL with a fixed ratio of 0.4 for apoB protein/HDL protein. We demonstrated that treatment with HDL (50μg/ml) mixed with apoB (20μg/ml) for 3 days significantly increased secreted levels of Aβ 1-40 and Aβ 1-42 (Fig. 2A) and intraneuronal levels of Aβ 1-40 and Aβ 1-42 (Fig. 2B). However, treatment with HDL (50μg/ml) mixed with apoE3 (20μg/ml) for 3 days did not significantly increase secreted or intraneuronal levels of Aβ (Fig. 2).
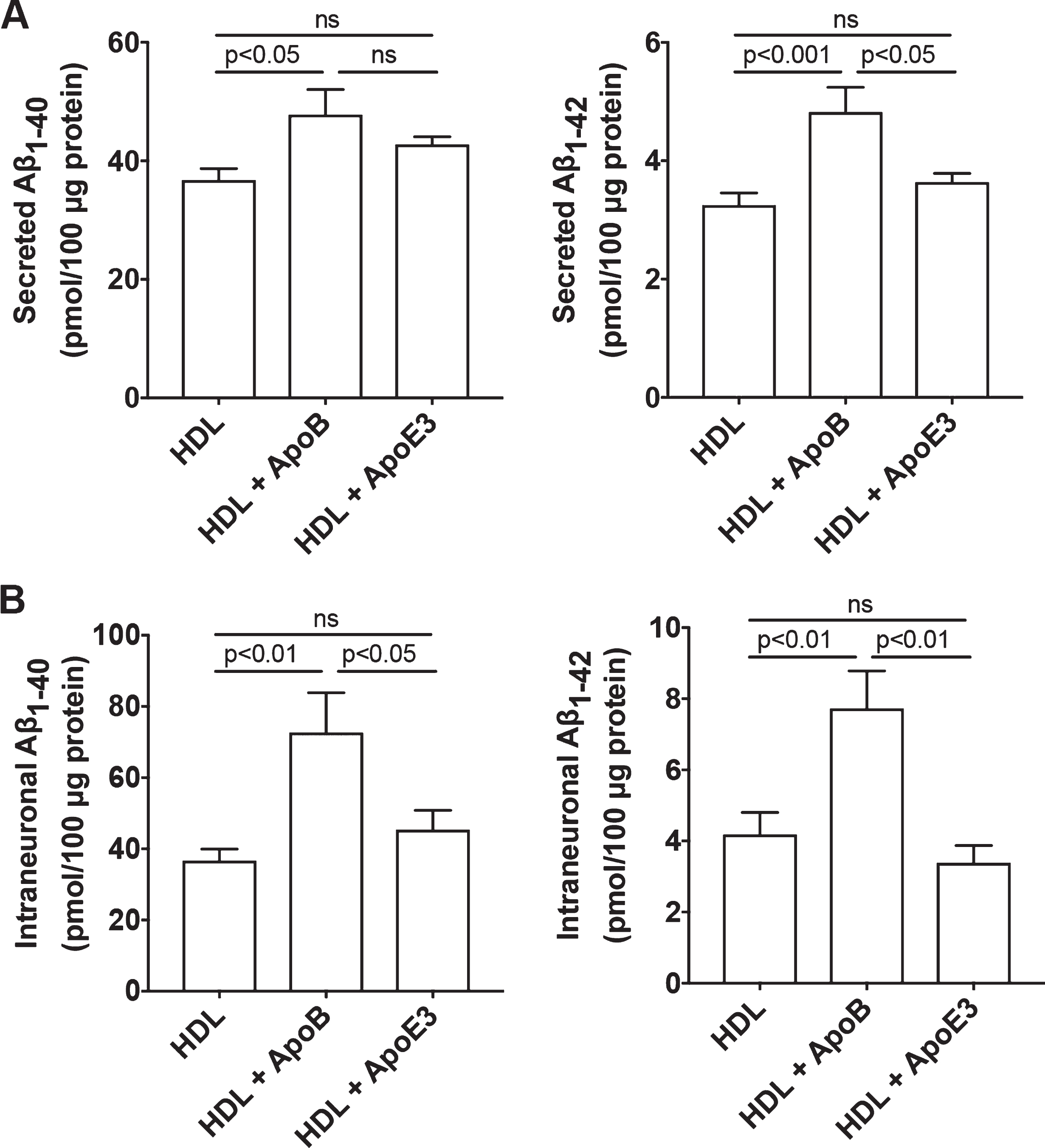
HDL mixed with apoB, but not HDL mixed with apoE3, increased Aβ. A) In SH-SY5Y cells expressing wild-type AβPP, treatment with HDL (50μg/ml) mixed with apoB (20μg/ml) for 3 days significantly increased secreted levels Aβ 1-40 and Aβ 1-42. Treatment with HDL (50μg/ml) mixed with apoE3 (20μg/ml) for 3 days did not significantly increase secreted levels of Aβ (n = 5). B) In SH-SY5Y cells expressing wild-type AβPP, treatment with HDL (50μg/ml) mixed with apoB (20μg/ml) for 3 days significantly increased intraneuronal levels of Aβ 1-40 and Aβ 1-42. Treatment with HDL (50μg/ml) mixed with apoE3 (20μg/ml) for 3 days did not significantly increase intraneuronal levels of Aβ (n = 5).
LDL, but not HDL, de-acidified endolysosomes
Amyloidogenic processing of AβPP occurs predominantly in endosomes where the acidic pH is compatible with high levels of BACE1 expression and activity [49, 50]. Thus, we determined the extent to which LDL and HDL affect endolysosome pH in SH-SY5Y cells. We found that LDL (50μg/ml for 3 days), but not HDL (50μg/ml for 3 days), increased significantly endolysosome pH (Fig. 3A). Because v-ATPase is the main proton pump that maintains the acidic environment of endolysosomes, we then determined the extent to which LDL and HDL affected total levels and endolysosomal protein levels of v-ATPase. We found that LDL, but not HDL, significantly decreased total protein levels of v-ATPase V1A1 and V0a1 subunits in whole cell lysates (Fig. 3B). Furthermore, LDL, but not HDL, significantly decreased protein levels of v-ATPase V1A1 and V0a1 subunits in enriched lysosome fractions (Fig. 3C).

LDL, but not HDL, de-acidified endolysosomes. A) In SH-SY5Y cells expressing wild-type AβPP, LDL (50μg/ml for 3 days), but not HDL (50μg/ml for 3 days), significantly elevated endolysosome pH (***p < 0.001, n = 16–20 cells from 4 different plates). B) LDL, but not HDL, significantly decreased protein levels of V1A1 and V0a1 subunits of v-ATPase in total cell lysates of SH-SY5Y cells (*p < 0.05, **p < 0.01, n = 4). C) LDL, but not HDL, significantly decreased protein levels of v-ATPase V1A1 and V0a1 subunits in enriched lysosome fractions of SH-SY5Y cells (**p < 0.01, n = 4).
ML-SA1 acidified endolysosomes
The above findings suggest that endolysosome deacidification plays a role in LDL-induced increases in Aβ. To further determine a cause role of endolysosome deacidification in LDL-induced increases in Aβ, we need an agent that could acidifies endolysosomes. Given that activating TRPML1 channels with ML-SA1 has been shown to reduce endolysosome cholesterol accumulation in Niemann– Pick’s disease type C [51], we determine the extent to which ML-SA1 affects endolysosome pH in rat primary cultured neurons. We found that ML-SA1 (20μM) decreased acutely endolysosome pH in neurons (Fig. 4A). Given that ML-SA1 is an agonist of TRPML1, we determined if TRPML1 was involved in the endolysosome acidifying effects of ML-SA. Thus, we knockdown the expression of TRPML1 using siRNA in primary cultured neurons (Fig. 4B). We found that TRPML1 knockdown did not affect basal levels of endolysosome pH; however, TRPML1 knockdown blocked significantly the magnitude of ML-SA1-induced acidification of endolysosomes (Fig. 4C). These findings indicate that ML-SA1 induced endolysosome acidification is, at least, in part by activating TRPML1.

TRMPL1 is involved in ML-SA-induced endolysosome acidification. A) Ratiometric endolysosome pH trace shows the effect of ML-SA1 (20μM) on endolysosome pH in primary cultured rat cortical neurons. Quantified data shows that ML-SA1 (20μM) decreased significantly endolysosome pH (2 min following ML-SA1 treatment versus baseline, n = 19 neurons of 4 different plates from two different animals). B) Immunoblotting shows that TRPML1 siRNA decreased significantly protein levels of TRPML1 in primary cultured neurons (n = 4). C) Basal levels of endolysosome pH did not change following treatment withTRPML1 siRNA; however, the magnitude of ML-SA1-induced acidification of endolysosomes (2 min following ML-SA1 treatment) was decreased in TRPML1 siRNA treated neurons when compared with negative siRNA treated neurons (*p < 0.05, n = 15–20 neurons of 4 different plates from two different animals).
ML-SA1 prevented LDL-induced increases in Aβ
Consistent with findings described above using SH-SY5Y cells, we demonstrated in primary cultured rat cortical neurons that LDL (50μg/ml for 3 days) increased secreted levels of Aβ 1-40 (Fig. 5A) and Aβ 1-42 (Fig. 5B). Importantly, ML-SA1 (20μM) co-treatment decreased significantly basal level of secreted Aβ and blocked significantly LDL-induced increases in secreted levels of Aβ 1-40 (Fig. 5A) and Aβ 1-42 (Fig. 5B). Using an antibody (MOAB-2) specific to Aβ (residues 1–4) that is capable of differentiating Aβ from AβPP [52] and an antibody targeting lysosome-associated membrane protein 1 (LAMP1), we then determined the effects of LDL and ML-SA1 on endolysosome accumulation of Aβ. We demonstrated that LDL increased significantly immunopositive signals of Aβ (Fig. 5C, D) and increased the co-localization of Aβ with LAMP1 positive lysosome (Fig. 5E), and that such effects were prevented by ML-SA1 (Fig. 5C-E). In addition, we found that LDL treatment has changed the distribution of LAMP1-positive vesicles. In control neurons, LAMP1-postive endolysosomes were relatively small and homogeneous in size and were distributed primarily in axon hillock region. However, in neurons treated with LDL, LAMP1-postive signals were increased, LAMP1-postive endolysosomes were markedly enlarged, often clumped together, and distributed throughout neuron soma (Fig. 5C).
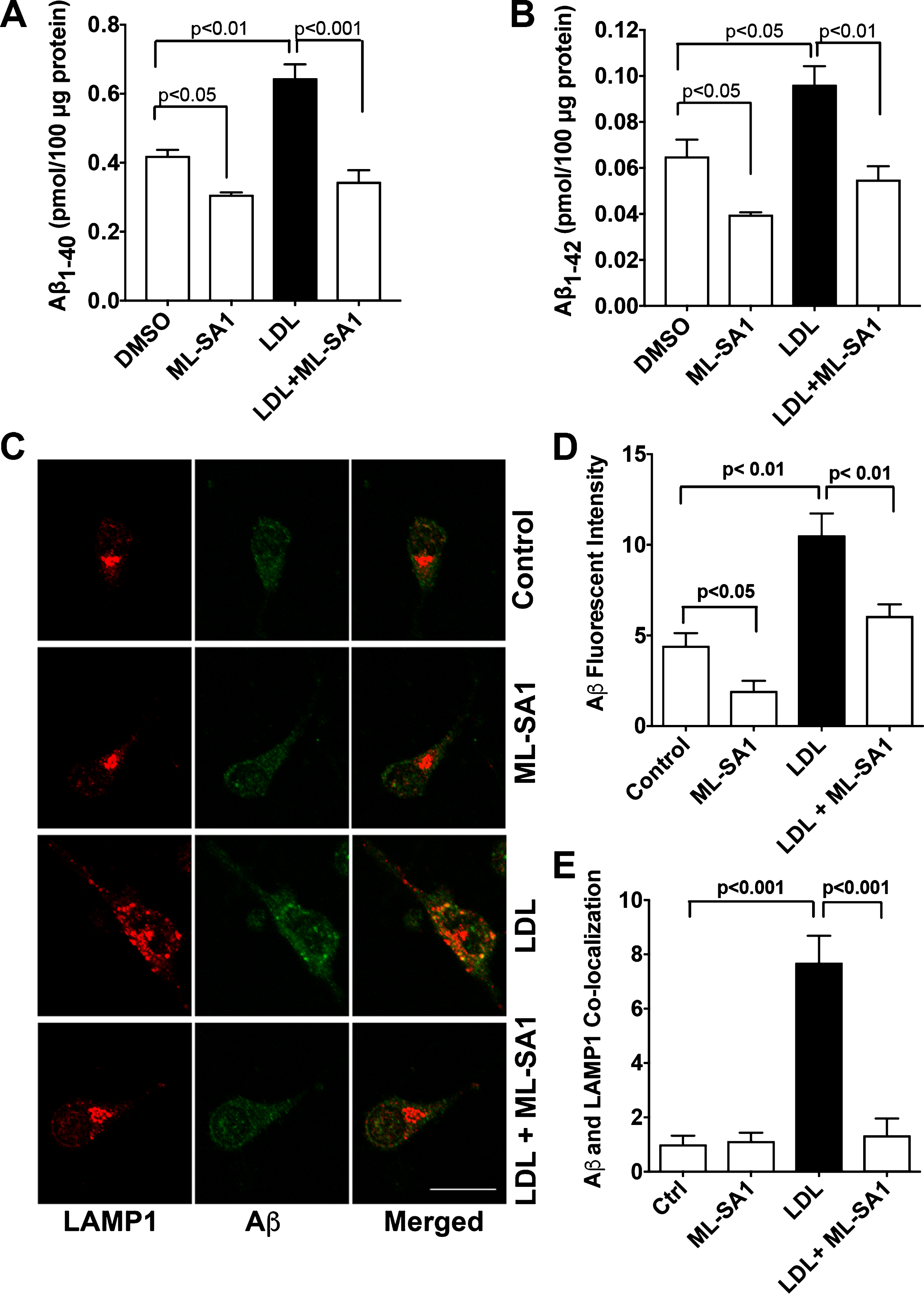
ML-SA1 prevented LDL-induced increases in Aβ. A, B) LDL (50μg/ml for 3 days) increased secreted levels of Aβ 1-40 and Aβ 1-42 in primary cultured rat cortical neurons. ML-SA1 treatment (20μM for 3 days) decreased significantly basal level of secreted Aβ and blocked significantly LDL-induced increases in secreted levels of Aβ 1-40 and Aβ 1-42 (n = 4). C, D) LDL (50μg/ml for 3 days) increased significantly immunopositive signal of Aβ in primary cultured neurons (Bar = 20μm, n = 15–20 neurons of 4 different plates from two different animals). E) ML-SA1 (20μM for 3 days) prevented significantly LDL-induced increased co-localization of Aβ with LAMP1-positive endolysosomes (n = 15–20 neurons of 4 different plates from two different animals).
Given that BACE1 is the critical enzyme for Aβ generation, we further determined the extent to which LDL and ML-SA1 affect BACE1. We found that LDL (50μg/ml for 3 days) increased immunopositive signals of BACE1 and increased co-localization of BACE1 with LAMP1 positive lysosomes, and that such effects were prevented by ML-SA1 (Fig. 6A, B). In addition, LDL treatment changed the sizes and the distribution of LAMP1-positive endolysosomes similar to those observed in Fig. 5. Furthermore, LDL (50μg/ml for 3 days) increased significantly BACE1 activity, an effect that was blocked by ML-SA1 (Fig. 6C).

ML-SA1 prevented LDL-induced increases in the colocalization of BACE1 with LAMP1 and increases in BACE1 enzyme activity. A,B) In primary cultured rat cortical neurons, LDL (50μg/ml for 3 days) increased immunopositive signals of BACE1 and the co-localization of BACE-1 with LAMP1 positive endolysosomes, and such effects were prevented by ML-SA1 (20μM) co-treatment (n = 15–20 cells of 4 different plates from two different animals). C) In primary cultured rat cortical neurons, LDL (50μg/ml for 3 days) increased significantly BACE1 activity, and ML-SA1 (20μM for 3 days) blocked significantly LDL-induced increases in BACE1 enzyme activity (n = 4).
TRPML1 knockdown potentiated LDL-induced increases in Aβ
Given that TRPML1 is involved in ML-SA1-induced endolysosome acidification and that LDL de-acidifies endolysosomes, we determine if LDL treatment could affect TRPML1 protein levels. We demonstrated that LDL (50μg/ml for 3 days) significantly decreased protein levels of TRPML1 in neurons (Fig. 7A). Next, we determined the extent to which siRNA knockdown of TRPML1 (Fig. 4B) affected LDL-induced increases in Aβ. We found that TRPML1 knockdown, per se, did not affect secreted levels of Aβ 1-40 or Aβ 1-42; however, TRPML1 knockdown significantly exacerbated LDL-induced increases in secreted levels of Aβ 1-40 and Aβ 1-42 (Fig. 7B).
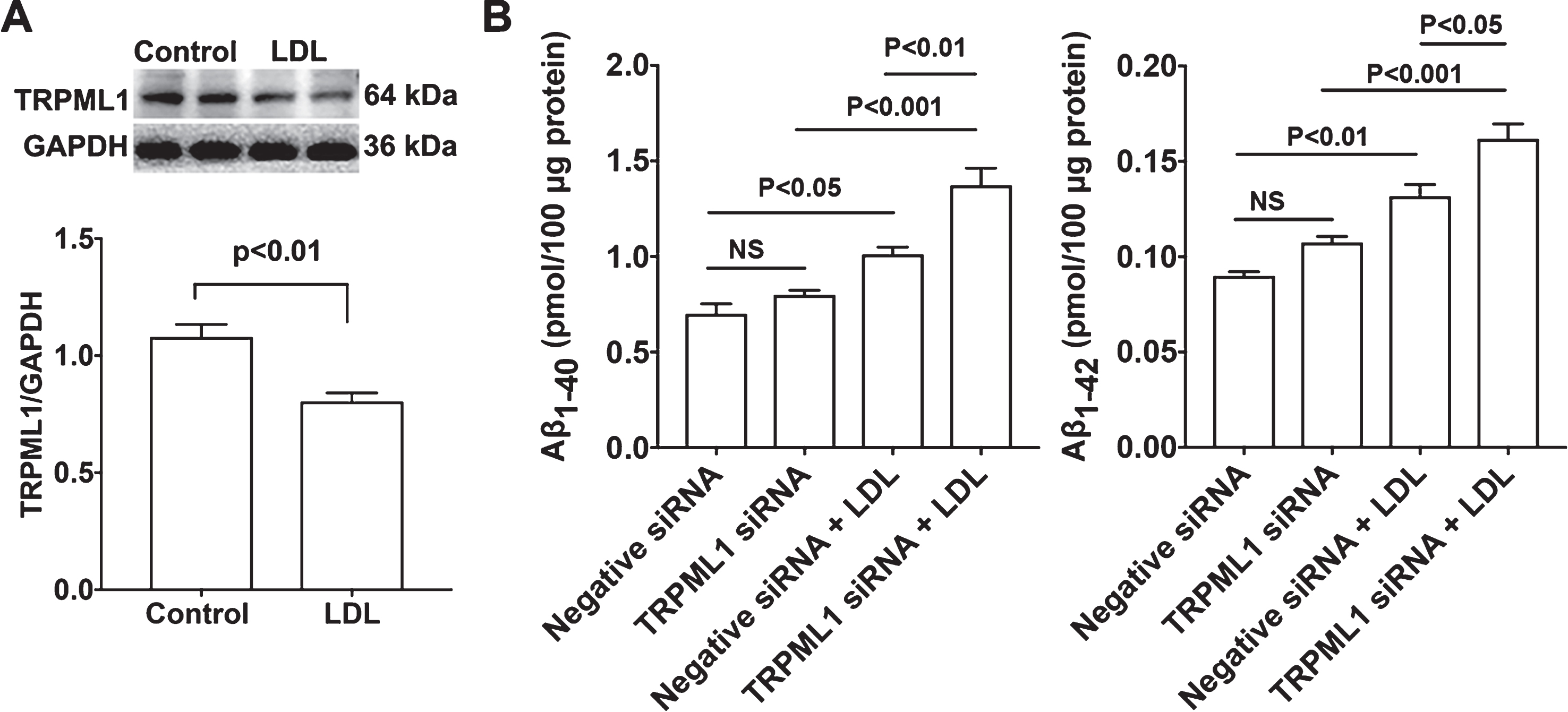
TRPML1 knockdown potentiated LDL-induced increases in Aβ. A) LDL (50μg/ml for 3 days) significantly decreased protein levels of TRPML1 in primary cultured rat cortical neurons (n = 4). B) TRPML1 knockdown, per se, did not affect secreted Aβ levels. However, TRPML1 knockdown significantly exacerbated LDL-induced increases in Aβ levels in primary cultured rat cortical neurons (n = 4).
DISCUSSION
Dyshomeostasis of circulating levels of cholesterol have been implicated in the pathogenesis of sporadic AD [8–12, 22]. Our studies here were focused to determine specifically how LDL-induced endolysosome de-acidification increased Aβ levels in neurons and to elucidate mechanisms by which endolysosome acidification protected against LDL-induced increases in Aβ levels. The main findings were 1) that LDL, but not HDL, de-acidified endolysosomes and increased intracellular and secreted levels of Aβ, 2) that TRPML1 was responsible for the endolysosome acidifying effects of ML-SA1, 3) that acidifying endolysosomes with ML-SA1 prevented LDL-induced increases in Aβ, and 4) that LDL decreased protein levels of TRPML1 and TRPML1 knockdown worsened LDL-induced increases in Aβ. Thus, acidification of endolysosomes by activating TRPML1 might be targeted for new therapeutic strategies against sporadic AD.
Altered cholesterol homeostasis, independent of APOE genotypes, has been linked to the pathogenesis of sporadic AD. However, it is not fully understood how cholesterol dyshomeostasis in circulating blood contributes to the development of AD in brain. Whether synthesized in the periphery or in brain, cholesterol is the same; however, lipoproteins associated with cholesterol differ in size, lipid composition, and mechanisms governing their transport and uptake. In plasma, LDL is the main lipoprotein particle that mediates the transport of cholesterol and lipids into periphery tissues, whereas HDL is the lipoprotein particle that mediates the reverse—cholesterol efflux from periphery tissues. LDL is a 20–25 nm sized particle that has the highest cholesterol content, and apoB-100, which is exposed at the surface of LDL allowing receptor recognition, is the exclusive apolipoprotein that mediates LDL transport and uptake. LDL particles constitute 90% of circulating apoB-containing lipoproteins, with each LDL particle containing a single apoB-100 molecule. HDL, a protein-rich disc-shaped particles, is about 8–10 nm in size, has lower cholesterol content, and primary apolipoproteins that mediate its transport and uptake are apoA-I, apoC-I, apoC-II, and apoE. In brain, apoE-rich HDL-like lipoproteins mediates that transport of cholesterol and lipids. Under normal conditions, the intact BBB restricts plasma LDL from entering brain parenchyma and brain cholesterol is almost completely dependent on in situ synthesis of apoE-containing cholesterol by astrocytes [53]. Although the intact BBB restricts prevent LDL particle from entering brain parenchyma, a small apoB peptide (receptor binding domain of apoB) has been shown to function as a BBB shuttle peptide without affecting the BBB integrity [54]. Unlike LDL, a small fraction of circulating HDL can enter the brain even with an intact BBB, and it has been shown that HDL has largely protective effects [55, 56]. Consistent with the reported protective role of HDL, we showed here that HDL could decreases secreted levels of Aβ.
Complex interactions between aging, nutritional, environmental, and genetic factors contribute to the development of sporadic AD [7]. Among these factors, vascular risk factors play a significant role in the development and progression of AD [57–59]. Increased plasma levels of LDL, which is present in many vascular risk factors, including atherosclerosis, hypertension, diabetes, and obesity, is directly implicated in the development of atherosclerotic cardiovascular disease [60]. Increased plasma levels of LDL, as well as, various vascular risk factors have been implicated to the breakdown of the BBB [57, 62]. Indeed, recent advanced neuroimaging studies have shown BBB breakdown in discrete brain regions of individual with mild cognitive impairment and early AD [63–66]. In addition, high prevalence of cerebral microbleeds are detected individuals with mild cognitive impairment and early AD [67–69], and these cerebral microbleeds correlate to the distribution of Aβ deposition [70]. Furthermore, pathological features of BBB breakdown, such as loss of tight junctions and extravasation of blood-derived proteins, have been confirmed in postmortem AD brains [71]. Although not all AD patients have uniform BBB breakdown, BBB integrity is compromised early in the pathogenesis of AD and proceeds the apparent brain deposition of Aβ [26].
Under conditions when BBB integrity is compromised as occurs early in AD, a significant amount of cholesterol could flux from circulation into the brain parenchyma [31]. Indeed, as the exclusive apolipoprotein of circulating LDL, apoB100 is present in AD brain [32], where it co-localizes with Aβ in human AD brain and in animal models [19, 72]. The presence of apoB-100 in AD brain could be due to the leakage of apoB-containing LDL particles into the brain or just the leakage of ApoB into the brain. Although LDL are large particles (about 20–25 nm in size and molecular mass ranging from 2.4 to 3.9 MDa), under conditions when BBB is severely damaged as occurs with microbleeds [73], LDL particles could enter brain parenchyma. Once entering brain, apoB-containing LDL cholesterol can be internalized by neurons via receptor-mediated endocytosis with the assistance of highly expressed receptors for cholesterol uptake. Because apoB and apoE have different affinities for receptors for cholesterol uptake, neuronal uptake of apoB-containing LDL may result in drastic difference in intracellular cholesterol transport and distribution than that of apoE cholesterol. In support, it has been shown that apoB leads to cholesterol being targeted by the lysosome degradation pathway [74, 75], whereas apoE mediates cholesterol recycling [76–78]. Thus, neuronal uptake of apoB-containing LDL cholesterol could lead to cholesterol accumulation in endolysosomes [10]. Consistently, we have shown that accumulation of apoB100 and cholesterol in endolysosomes in brain of a rabbit model of sporadic AD [9, 38]. Thus, apoB-containing LDL could directly contribute to the disturbances of neuronal endolysosome structure and function - another early pathological features of sporadic AD [79, 80].
Given that highly acidic pH is critical for essential for regulating many functions of endolysosomes, the simplest explanation for the observed pathological changes of endolysosomes in post-mortem brain tissue from AD patients [79–81] is that endolysosome pH is de-acidified both in familial AD and in sporadic AD. In familial AD, presenilin 1 mutation has been shown to result in endolysosome de-acidification [82, 83], while familial AD-mutant APP overexpression may lead to a deficit in endolysosome de-acidification as implicated by endolysosomal dysfunction [84, 85]. In sporadic AD, endolysosome de-acidification may result from complex interactions between aging and environmental factors. Age-dependent lipofuscin accumulation may act as v-ATPase inhibitors and promote endolysosomal de-acidification [86], chronic oxidative stress results from aging and environmental factors may impair v-ATPase and lead to deficit in endolysosome acidification [87, 88], and lipid accumulation [89, 90] and iron overload [91–93] in endolysosome may also de-acidify endolysosomes. Consistently, we have shown that following its endocytosis into and accumulation by endolysosomes [10], apoB-containing LDL de-acidifies endolysosomes in both primary neuron and in SH-SY5Y cells. These findings provide further mechanistic insights on how cholesterol dyshomeostasis in circulating blood contributes to the development of AD in brain.
It is well known that endolysosomes play a central role in Aβ production and metabolism [80, 94]. Amyloidogenic processing of AβPP predominantly occurs in endosomes, where the pH is optimum BACE1 activity [49, 50], and such endosome-derived Aβ can be further degraded by cathepsin D in more acidic lysosomes [95–98]. Thus, simply altering endolysosome pH could have a profound effect on proteolytic AβPP processing in endolysosomes and Aβ levels. In support, chloroquine, a known weak base that neutralize the acidic pH in endolysosomes, has been shown to induce accumulation of lipids [99, 100] and AβPP by-products in endolysosomes [101] and increase Aβ levels [102, 103]. Similarly, bafilomycin, a known v-ATPase inhibitor, has also been shown to increases Aβ levels [104]. It is interesting to note that these two known alkalizing agents increase Aβ levels in wild-type AβPP expressing cells; however, they decreases Aβ levels in Swedish-mutation AβPP expressing cells [102, 105]. Currently, it is not understood what leads to such a discrepancy. In addition, U18666A, an agent that leads to cholesterol accumulation in endolysosomes, de-acidifies endolysosomes [106], and increases Aβ levels [107]. On the other hand, restoring endolysosomal acidification by GSK3 inhibition [108] or acidic nanoparticle [109–111] has been shown to improve amyloid pathology.
Because the activity of BACE-1 is optimal at a pH ≈ 5 [112, 113] and is degraded in lysosomes at a pH ≈ 4 [114] and because the activity of cathepsin D is optimal at a pH ≈ 3.5 [115], subtle de-acidification could shift lysosomes into an endosome phenotype to increase Aβ production and impair Aβ lysosome degradation, whereas subtle acidification could shift endosome into lysosome phenotype to decrease Aβ generation and enhance Aβ lysosome degradation. In the present study, LDL-induced slight increases in endolysosome pH could have resulted in decreased degradation of BACE-1 and increased accumulation and activity of BACE-1 in endolysosomes (Fig. 6), thus increasing Aβ production. On the other hand, LDL-induced increases in endolysosome pH could have resulted in inhibition of cathepsin D activity [10], thus impairing the degradation of Aβ in lysosomes and increasing Aβ accumulation in lysosomes (Fig. 5). The above scenario might have occurred when endolysosome is mildly alkalized as induced by LDL treatment. Under conditions when the acidic environment of endolysosomes is severely impaired, the drastic alkalization could block proteolytic processing AβPP, leading to accumulation of full-length AβPP in endolysosomes [116] and decreased Aβ levels. The centrality of endolysosome pH in LDL-induced amyloidogenesis in neurons is further strengthened by our findings that ML-SA1 acidifies endolysosomes and blocks LDL-induced increases in BACE-1 activity and Aβ levels. The mild acidification induced by ML-SA1 could block LDL-induced impaired degradation of BACE-1 and increased accumulation and activity of BACE-1 in endolysosomes, thus preventing LDL-induced increasing Aβ production. On the other hand, the mild acidification induced by ML-SA1 could block LDL-induced inhibition of cathepsin D activity [10], thus preventing LDL-induced impairment in the degradation of Aβ in lysosomes and increased Aβ accumulation in lysosomes.
We demonstrated that the endolysosome acidifying effects of ML-SA1 is, at least in part, dependent on the activation of endolysosome-resident TRPML1 cation channels. Although the mechanisms by which TRPML1 activation affects endolysosome pH are unclear it is possible that calcium released from TRPML1 activates calcium-activated potassium channels and increases an exchange of potassium and hydrogen ions [117–119]. Alternatively, calcium released from TRPML1 might affect the activity of v-ATPase, the key proton pump that maintains the acidic environment of endolysosomes. Finally, the endolysosome de-acidifying effects of LDL could be linked directly to TRPML1 because LDL decreases protein levels of TRPML1 and TRPML1 activation has been shown to activate v-ATPase [120]. Regardless of the mechanism, our findings suggest that endolysosome de-acidification plays a critical role in LDL-induced amyloidogenesis and that endolysosome acidification by activating TRPML1 might represent a therapeutic strategy against AD.
Footnotes
ACKNOWLEDGMENTS
We greatly acknowledge the help of Dr. Bryon Grove and Ms. Sarah Abrahamson in using the Edward C. Carlson Imaging and Image Analysis Core Facility. This work was supported by grants from the National Institutes of Health including P30GM103329, R01MH100972, R01MH105329, and R21DA040519.