
Abstract
Epidemiological studies suggest that individuals with diabetes mellitus are at greater risk of developing Alzheimer’s disease. A well-known insulin-sensitizing drug and the most widely prescribed oral medication for diabetes is metformin. There is evidence that metformin acts in a neuroprotective manner via the AMPK/mTOR pathway by inhibiting the tau phosphorylation. In addition, it is known that metformin upregulates Fgf21, which in turn activates the AMPK/mTOR pathway and mediates neuroprotection. Thus, metformin-induced Fgf21 release may be involved in AMPK/mTOR activation. However, some studies reported that metformin causes cognition impairment. Due to the controversial data on the neuroprotective properties of metformin, we treated Apolipoprotein E deficient (ApoE– /–) mice, a mouse model of tauopathy, with metformin for 18 weeks. Metformin-treated mice revealed increased expression of lipogenic genes, i.e., lxrα and srebp1c. In line with this, metformin caused an increase in plasma triglyceride leading to enhanced gliosis as indicated by an increase of GFAP-positive cells. Although the systemic Fgf21 concentration was increased, metformin did not activate the FgfR1c/AMPK/mTOR pathway suggesting a Fgf21-resistant state. Further, metformin-treated mice showed increased tau phosphorylation and reduced numbers of NeuN-and PSD95-positive cells. Thus, metformin-associated lipogenesis as well as inflammation aggravated neurodegenerative processes in ApoE– /– mice. Consequently, this study supports previous observations showing that metformin causes impairment of cognition.
INTRODUCTION
Epidemiological studies identified a strong correlation between type 2 diabetes mellitus (T2DM) and Alzheimer’s disease (AD) [1–3]. Furthermore, Morris et al. [4] showed an insulin resistance in the central nervous system (CNS) in AD patients. Likewise, Craft and co-workers described insulin abnormalities in AD [5, 6]. In addition, they reported that a reduced hippocampal insulin-degrading enzyme in late-onset AD is associated with the Apolipoprotein E (ApoE) status, which in turn influences the risk to develop AD [5]. A well-known insulin-sensitizing drug is metformin, the most widely prescribed oral medication for T2DM in the world. Metformin is also described to have neuroprotective properties [7]. In this context, a longitudinal aging study showed that long-term treatment (>6 years) with metformin among T2DM patients was significantly associated with lowest risk of cognitive impairment [8]. It is thought that metformin activates the AMP-activated protein kinase (AMPK) [9]. Activated AMPK leads to decrease of mammalian target of rapamycin (mTOR) signaling activity by inactivation of the mTOR/p70S6K-mediated negative feedback loop to insulin receptor substrate-1 [10], which enhances the insulin signaling. Beside this, repressed mTOR signal inhibits tau hyperphosphorylation and reduces formation of neurofibrillary tangles as a neuropathological hallmark of AD [11].
Further, it is described that metformin increases circulating fibroblastic growth factor 21, a starvation hormone (Fgf21) [12]. Fgf21 activity occurs when Fgf21 binds to fibroblast growth factors receptor (Fgfr) and β-klotho, a single transmembrane protein [13]. Fgfrs consist of seven major isoforms (1b, 1c, 2b, 2c, 3b, 3c, and 4), whereby the isoform Fgfr1c is the primary receptor of Fgf21 in the mediation of its activity in in vivo studies [14]. Upon receptor binding of Fgf21, AMPK becomes activated [15]. Beside beneficial effects on metabolism through increasing insulin sensitivity and energy expenditure [16], there is growing evidence that Fgf21 exerts neuroprotective effects and improves cognition [17]. Accordingly, we recently showed in ApoE-deficient mice (ApoE– /–) that caloric restriction-associated neuroprotection seems to be Fgf21/AMPK/mTOR-dependent [18]. ApoE– /– mice represent a well-established mouse model of tauopathy with memory deficits [19, 20]. ApoE binds to tau protein and prevents hyperphosphorylation of tau protein [10], leading to a slowdown of formation of neurofibrillary tangles. In turn, ApoE deficiency results in tau hyperphosphorylation.
There are controversial data on the cerebral effect of metformin. Barini et al. [21] and Li et al. [22] reported that cognitive function of metformin-treated mice with tauopathy was restricted despite reduced tau phosphorylation. Furthermore, a clinical study in diabetic patients showed that metformin impairs cognition [23], whereby Ng et al. [8] described the opposite. To clarify this discrepancy, the present study examined to what extent metformin has neuroprotective properties in ApoE– /– mice and focused on the Fgf21/AMPK/mTOR pathway (Fig. 1A).
MATERIAL AND METHODS
Animals
For the experiments, female ApoE deficient (ApoE– /–) mice were used at 10 weeks of age. Female mice were chosen because of the comparability to previous studies [18]. The mice received either drinking water without additives (H2O group, n = 8) or drinking water with metformin ∼300 mg/kg/day in accordance to Barini et al. [21] (metformin group, n = 8) for 18 weeks (Fig. 1B). All mice were housed individually in standard cages in a temperature-controlled room (22°C±2°C) on a 12 h light/dark cycle with free access to food and water under specific pathogen free (SPF) conditions. The drinking quantity was measured weekly as well as the body weight. The experimental protocol was approved by the local Animal Research Committee (Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei (LALLF) of the state Mecklenburg-Western Pomerania (LALLF M-V/TSD/7221.3-1.1-002/14) and all animals received humane care according to the German legislation on protection of animals and the Guide for the Care and Use of Laboratory Animals (NIH publication 86–23 revised 1985).

Schematic illustration of the working hypothesis (A) and of the animal experiment (B). (A) The present study hypothesized whether metformin caused indirectly via hepatic Fgf21-induced release or directly the Fgfr1c-activated AMPK/mTOR pathway. An activation of AMPK/mTOR pathway may lead to a reduction of tau phosphorylation and in consequence to a neuroprotection. (B) Ten-week-old ApoE– /– mice received either drinking water without additives (H2O group, n = 8) or drinking water with metformin ∼300 mg/kg/day (metformin group, n = 8) for 18 weeks. The drinking quantity was measured weekly as well as the body weight. At the end of the experiment, all mice at the age of 28 weeks were exsanguinated by puncture of the vena cava inferior for immediate measurement of blood sugar and separation of plasma, followed by harvest of brain tissue.
Sampling and assays
At the end of the experiment, all mice at the age of 28 weeks were exsanguinated by puncture of the vena cava inferior for immediate measurement of blood sugar (AccuCeck Active, Roche Diagnostics) and separation of plasma, followed by harvest of brain tissues. Measurement of plasma malondialdehyde (MDA), serving as an indicator of lipid peroxidation and oxidative stress, was performed using the MDA-586 method according to the manufacturer’s instructions (OxisResearchTM, Portland, USA). Measurements of triglycerides, Fgf21, β-hydroxybutyrate, and insulin in plasma were performed using the assay kit methods according to the manufacturer’s instructions (Fgf21: R&D System, MI, USA, triglycerides and β-hydroxybutyrate: Cayman Chemical Company, MI, USA, ultra sensitive insulin: Crystal Chem, IL, USA).
For measurement of hepatic cholesterol concentrations, lipids were extracted by means of the Bligh-Dyer method [24]. Briefly, livers were incubated with a mixture of chloroform and methanol (1:2). After vortexing, one part of chloroform and one part of H2O was added and spun down by 3000×g to separate phases. The organic phase (lower layer) was collected and concentrated by vacuum pump. The cholesterol content was analyzed by using the cholesterol/cholesteryl ester quantitation kit method according to the manufacturer’s instructions (Calbiochem, Merck KGaA, Darmstadt, Germany).
RNA analysis of brain tissue
Total RNA was isolated using the RNeasy® Mini Kit (Qiagen) in accordance with the manufacturer’s instructions. 2μg of total RNA was reverse-transcribed with SuperScript™ First Strand Synthese System (Invitrogen) as described by the manufacturer. Real-time quantitative PCR assays were performed by using Lightcycler 1.5 using the Lightcycler® FastStart DNA MasterPlus SYBR Green I kit (Roche Diagnostics). Each amplification mixture (20μl) contained 5μM primer, 19μl of universal PCR Mastermix, and 1μl 1:2 diluted cDNA solution. PCR thermocycling parameters were 95°C for 10 min and 40 cycles of 95°C for 10 s, 55°C for 5 s, and 72°C for 10 s. All samples were analyzed for ribosomal protein S18 (rps18) expression. For analysis of the relative change in gene expression we used the 2–ΔΔCt method. A cDNA pool of livers of untreated C57BL/6J mice served as the control and therefore as the first Δ. The second Δ is represented by Ct-values of rps18 amplification. Specificity of the amplification was verified by melt-curve analysis and evaluation of efficiency of PCR amplification. The primers are listed in Table 1.
List of used primer pairs
Western blot analysis of brain tissue
Harvested brain tissue was further processed for protein isolation. For this purpose, brain tissue was homogenized in lysis buffer (10 mM Tris pH 7.5, 10 mM NaCl, 0.1 mM EDTA, 0.5% Triton-X 100, 0.02% NaN3, and 0.2 mM PMSF (a protease inhibitor cocktail), incubated for 30 min on ice and centrifuged for 10 min at 4°C and 10.000×g. Protein contents were assayed by the bicinchoninic acid method (Pierce Biotechnology) with 2.5% BSA (Pierce Biotechnology) as standard. On 12% SDS gels, 20μg (pTau, Tau, β-Klotho, Synapsin I a/b, VAMP2, GAPDH) and 40μg (pAMPK and AMPK) protein from brain tissue was separated and transferred to a polyvinyldifluoride membrane (Immobilon-P; Millipore). After blockade with 2.5% BSA (Pierce Biotechnology), membranes were incubated overnight at 4°C with the primary antibodies (Table 2). Afterwards, secondary peroxidase-linked anti-mouse or anti-rabbit antibodies were applied (Table 2). Protein expression was visualized by means of luminol-enhanced chemiluminescence (ECL plus; Amersham Pharmacia Biotech) and digitalized with ChemiDoc™ XRS System (Bio-Rad Laboratories). Signals were densitometrically assessed (Quantity One; Bio-Rad Laboratories) and densities normalized either to GAPDH or/and to non-phosphorylated proteins (AMPK, Tau).
List of used antibodies for western blot analyses
Immunhistology
For immunohistochemical analysis, brain tissue was fixed in 4% phosphate-buffered formalin and embedded in paraffin. From the paraffin-embedded tissue blocks, 4μm thin sections were put on X-tra Adhesive Precleaned Micro Slides (Leica) and exposed to the antibodies (Table 3). For the development of the primary antibodies with DAB chromogen Universal LSAB® kits (System-HRP; DakoCytomation, Dako) were used according to the manufacturer’s instructions. The sections were counterstained with hemalaun and analyzed with a light microscope (Olympus BX51). Images were acquired with a Color View II FW camera (Color View).
List of used antibodies for immunohistochemistry
Statistical analysis
All data are expressed as means±SEM. Statistical differences were determined using unpaired student t-test and if there was no normality Mann-Whitney Rank Sum test was used. For repeated measurements, Two-Way ANOVA test was used. Data were considered significant if p < 0.05. Statistical analysis was performed using the SigmaStat software package (Jandel Scientific).
RESULTS
Metformin caused a slight body weight reduction
The blood sugar measurement over the time of 18 weeks showed neither hypoglycemic nor hyperglycemic events and revealed no differences with 6.3±0.1 mmol/L upon metformin or with 6.1±0.2 mmol/L upon H2O application at the end of treatment period. However, the plasma insulin concentration was almost 38% less in the metformin-treated mice. The drinking behavior differed slightly as indicated by an overall 4 mL less liquid intake upon metformin exposure (Fig. 2A). This was taken into account in dose determination of metformin. Remarkably, the body weight of metformin-treated mice was significantly reduced over the observation period of 18 weeks when compared to H2O group (Fig. 2B, p < 0.001).

The drinking quantity (A) and the body weights (B). Ten-week-old ApoE– /– mice received either drinking water without additives (H2O group, n = 8) or drinking water with metformin ∼300 mg/kg/day (metformin group, n = 8) for 18 weeks. The drinking quantity was measured weekly as well as the body weight. Values are given as means±SEM. Two-Way ANOVA for repeated measurements: *p < 0.001 versus H2O.
Metformin increased systemic concentration of Fgf21
Upon metformin exposure the systemic Fgf21 concentration significantly raised and reached approx. 2-fold higher levels versus H2O-treated mice (Fig. 3A, p = 0.013). Interestingly, the phosphorylated form of cerebral FgfR1c (Fig. 3B and lower panel) and its co-receptor β-Klotho (Fig. 3C) was found 2-fold downregulated upon metformin administration.
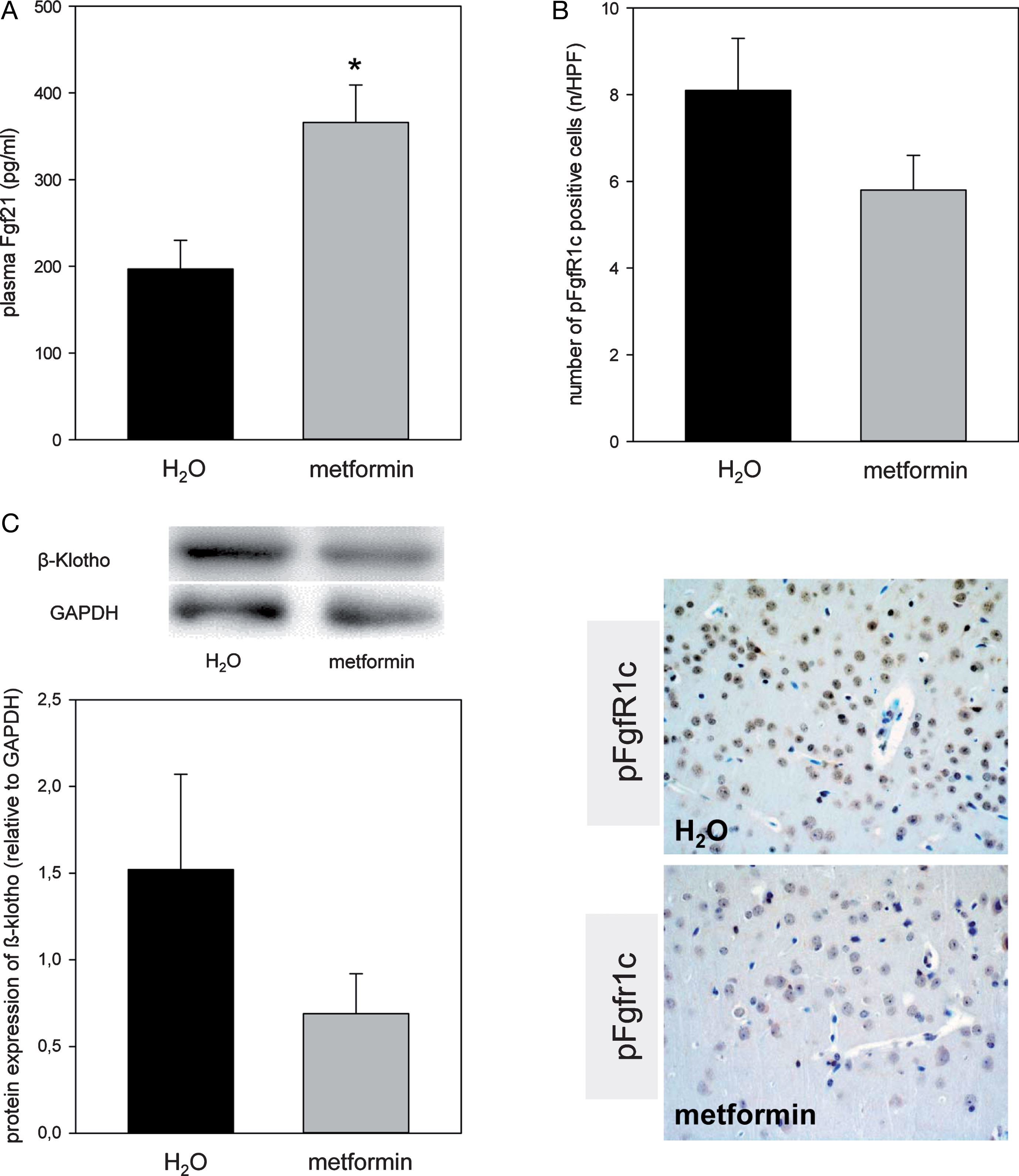
Quantitative analysis of plasma Fgf21 (A), pFgf1c positive neuronal cells given as number per high power field (HPF, B, n = 4) and the cerebral protein expression of β-Klotho (C) in the ApoE– /– mice. Representative immunohistochemical images (B), lower panel, original magnification ×400) of pFGFr1c positive neuronal cells in the cortex. Mice received either drinking water without additives (H2O group, n = 8) or drinking water with metformin ×300 mg/kg/day (metformin group, n = 8) for 18 weeks. Signals were normalized to GAPDH. Values are given as means±SEM. Unpaired Student-t-Test followed by Man-Whitney Rank sum test: *p < 0.05 versus H2O.
Metformin increased tau phosphorylation
Metformin did neither influence phosphorylation of AMPK nor of mTOR when compared to the H2O group (Fig. 4A–C). However, metformin caused an almost 2-fold increase of cerebral tau phosphorylation (Fig. 4D).
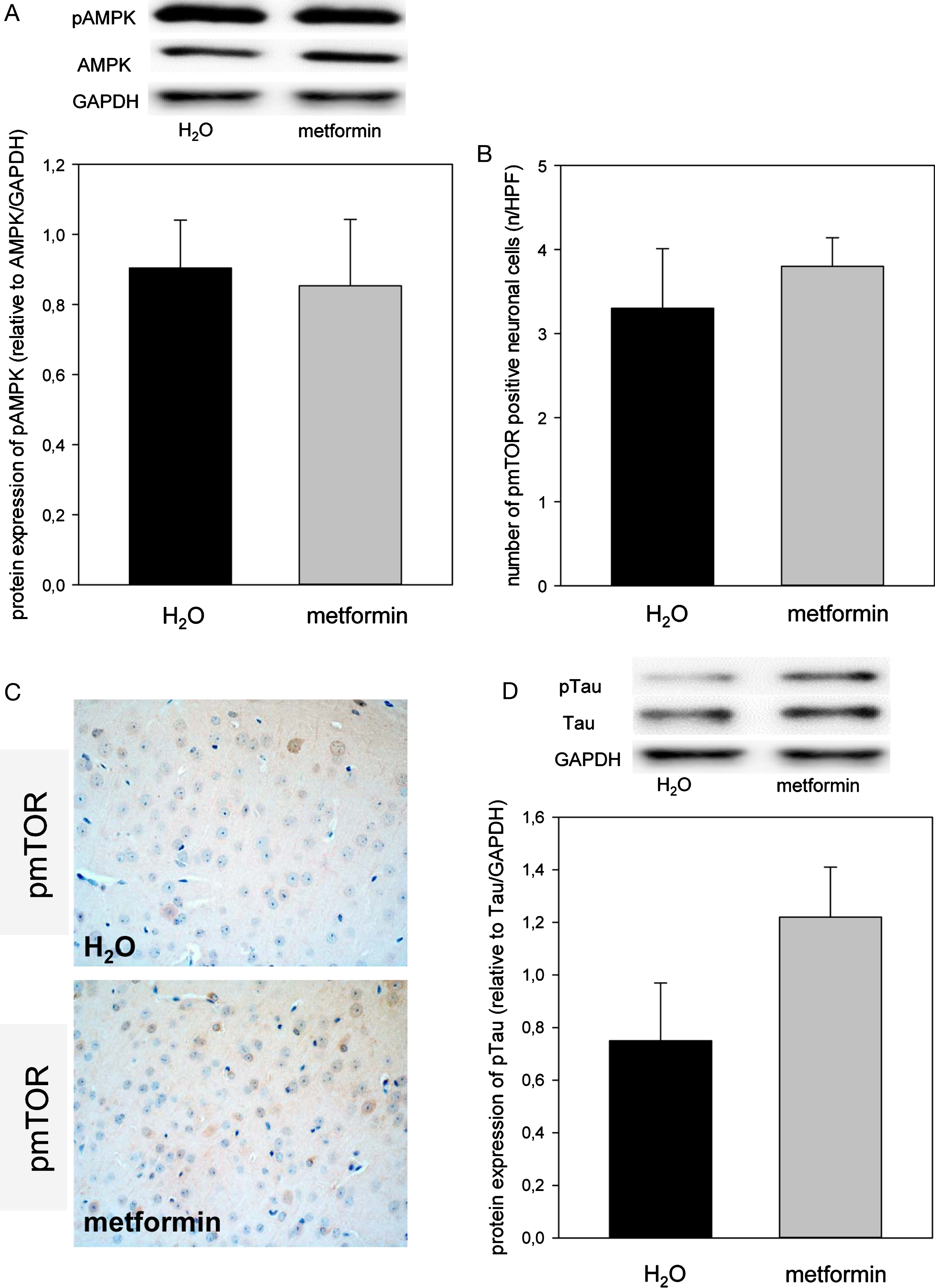
Representative western blot and densitometric analysis of cerebral pAMPK (A). Quantitative analysis of pmTOR positive neuronal cells, given as number per high power field (HPF; B) and representative immunohistochemical images (original magnification ×400; C) of cortical pmTOR expression in the brain of ApoE– /– mice. Representative Western blot and densitometric analysis of pTau in the brain of ApoE– /– mice. Mice received either drinking water without additives (H2O group, n = 8) or drinking water with metformin ∼300 mg/kg/day (metformin group, n = 8) for 18 weeks. Signals were normalized to GAPDH and to AMPK or tau. Values are given as means±SEM.
Metformin decreased neuronal integrity
Metformin led to a 20% reduction of NeuN positive cells (Fig. 5A, C) and to a significant reduction of PSD95 (post-synaptic marker) positive neurons in the cortex (p = 0.015, Fig. 5B, D). In line with this, the protein expression of VAMP2 (synaptic integrity marker; Fig. 6A; p = 0.054) and of Synapsin I a/b (pre-synaptic marker; Fig. 6B) was markedly decreased in metformin-treated mice.

Representative immunohistochemical images (original magnification ∼400) of NeuN (A) and PSD95 (B) of brain sections of ApoE– /– mice as wells as the quantitative analysis of cortical NeuN- (C) and PSD95- (D) positive cells, given as number per high power field (HPF). Mice received either drinking water without additives (H2O group, n = 8) or drinking water with metformin ∼300 mg/kg/day (metformin group, n = 8) for 18 weeks. Values are given as means±SEM. Unpaired Student-t-Test followed by Man-Whitney Rank sum test: *p < 0.05 versus H2O.

Representative western blot and densitometric analysis of cerebral protein expression of VAMP2 (A) and of Synapsin I a/b (B) of ApoE– /– mice. Mice received either drinking water without additives (H2O group, n = 8) or drinking water with metformin ∼300 mg/kg/day (metformin group, n = 8) for 18 weeks. Signals were normalized to that of GAPDH. Values are given as means±SEM.
Metformin induced fat accumulation
Metformin slightly increased plasma cholesterol content (Fig. 7A) and led to significant rise of plasma triglycerides (Fig. 7B; p = 0.008) while the plasma concentration of ß-hydroxybutyrate and of MDA as signs of ketogenesis and oxidative stress remained unchanged (Fig. 7C, D). The mRNA expression of genes, indicative for lipogenesis, i.e., lxrα and srebp1c were almost 2-fold higher in metformin-than in H2O-treated mice (Fig. 8A, B) whereas the mRNA expression of abca1, a reverse cholesterol transporter, was almost unchanged (Fig. 8C).

Analysis of plasma concentrations of cholesterol (A), triglycerides (B), β-hydroxybutyrate (C) and malondialdehyde (MDA; D). Mice received either drinking water without additives (H2O group, n = 8) or drinking water with metformin ∼300 mg/kg/day (metformin group, n = 8) for 18 weeks. Of note, the significant increase of plasma triglyceride upon metformin administration. Values are given as means±SEM. Unpaired Student-t-Test followed by Man-Whitney Rank sum test: *p < 0.05 versus H2O.

Quantitative real-time PCR of the nuclear receptors lxrα (A), srebp1c (B), and abca1 (C) and representative immunohistochemical images (D) of cortical GFAP positive cells in the brain of ApoE– /– mice. Signals were corrected to that of rps18. Notable is the increased number of GFAP positive cells indicating a rise of inflammatory processes upon metformin treatment. Mice received either drinking water without additives (H2O group, n = 8) or drinking water with metformin ∼300 mg/kg/day (metformin group, n = 8) for 18 weeks. Values are given as means±SEM. Unpaired Student-t-Test followed by Man-Whitney Rank sum test: *p < 0.05 versus H2O.
Metformin increased neuro-inflammation
Metformin caused gliosis as indicated by an increase of GFAP positive cells in the cortex (Fig. 8D).
DISCUSSION
Although it is described that metformin treatment may cause hypoglycemic events [25], the current study showed no serious changes of chronic blood glucose concentrations in metformin-treated mice. However, the concentration of insulin was almost 38% decreased which might indicate increased insulin sensitivity. Accordingly, metformin is characterized as an insulin-sensitizing drug. Furthermore, metformin has been suggested to be neuroprotective. Therefore, it is supposed that especially the tau phosphorylation is reduced by metformin [22, 26], although this does not necessarily increase neuronal integrity [21]. Due to these findings and the controversial results relating metformin-associated neuroprotection in diabetic patients [8, 23], we used ApoE– /– mice, a well-known AD mouse model of tauopathy, and treated them with metformin.
It is reported that metformin stimulates the Fgf21 expression in primary hepatocytes [27] and increases circulating Fgf21 in high-fat-diet rats [12]. Further, release of Fgf21 indicates increased lipolysis and ketogenesis [28, 29]. In line with this, we could previously show that caloric restriction in ApoE– /– mice cause increased lipolysis and ketogenesis, Fgf21-release, as well as activation of the Fgfr1c/AMPK/mTOR pathway in the brain [18]. Although the present study detected a significant increase of systemic Fgf21 in metformin-treated mice, β-hydroxybutyrate was found almost unchanged and activation of the FgfR1c/AMPK/mTOR pathway failed. Moreover, the protein expression of the Fgfr1c co-receptor β-Klotho was reduced.
There are two possible explanations for these unexpected findings. One explanation for failed AMPK activation might be a different regulation of energy balance upon metformin treatment in ApoE– /– mice. It is known that AMPK is involved in the fundamental regulation of energy balance at the whole body level by responding to hormonal and nutrient signals in the central nervous system and peripheral tissues that modulate food intake and energy expenditure [30]. Thus, during energy deprivation, for instance upon caloric restriction, the AMPK pathway is activated [18]. However, metformin-treated mice revealed an energy abundance, probably making due to enhanced lipogenesis. Therefore, the activation of AMPK pathway seems to be dispensable. The present study demonstrated a cerebral increase of lipogenic genes such as srebp1c and lxrα upon metformin exposure. In addition, the mRNA expression of hepatic lxrα was increased upon metformin exposure (unpublished data) suggesting enhanced lipogenesis of the whole body. On the other hand, hepatic mRNA expression of pparα was markedly decreased (unpublished data) indicating a reduction of lipolysis, which in turn enhances the lipogenesis and may explain failed AMPK activation. A further argument for increased fat accumulation would be— besides ApoE deficiency— an almost unchanged abca1 mRNA expression implicating failure of the reverse cholesterol transport [31]. Accordingly, plasma concentrations of both triglyceride and cholesterol were found increased upon metformin treatment. Since it is discussed that fat accumulation in the periphery and therewith the increase of inflammatory processes influences the whole body including the brain [32, 33], it is conceivable to state that metformin-enhanced lipogenesis may cause cerebral gliosis, as observed in the present study. Although an increased lipogenesis was observed, the body weight was significantly reduced upon metformin treatment. Since it is known that lipogenesis is an energy-consuming step to store lipids and an energetically inefficient way [34], it is likely to state that increased lipogenesis causes weight loss although the visceral organs become fatty. A typical example of this scenario gives the NPC1 mutant mouse, also a mouse model of neurodegenerative disease [35]. This mutant mouse shows a strong adiposity of the organs due to a disturbed cholesterol transport, but loses enormously body weight [36, 37].
The second explanation for the absence of Fgfr1c activation is as follows: In line with findings in adipose tissue showing reduced β-klotho expression upon TNFα application [38], inflammatory processes in metformin-treated ApoE– /– mice may be responsible for failed FgfR1c/β-klotho activation. Thus, a Fgf21-resistante state might be present in ApoE– /– mice, similarly to the report that obesity-associated inflammation causes Fgf21-resistance [39]. Fgf21-resistance is frequently accompanied with insulin-resistance and in turn with a downregulation of glucosetransporter-1 (glut-1) [40]. Glut-1 is commonly reduced in case of AD [41]. Thus, AD might be associated with a Fgf21-resistance, particularly as AD is known to be characterized by neuro-inflammation [42]. In addition, it is described that a high fat diet as means of exogenous-induced fat accumulation aggravates amyloid and tau pathologies [43, 44]. Thus, metformin-associated fat accumulation may enhance tauopathy and may worsen AD-like symptoms as shown by increased tau phosphorylation as well as reduced numbers of NeuN-and PSD95-positive cells and reduced protein expression of VAMP2 and Synapsin I a/b. Overall, these results show that not only post-synaptic but also pre-synaptic and intra-synaptic integrity is impaired upon metformin treatment. In support of this view, Barini et al. [21] and Li et al. [22] reported that metformin impairs cognition in mice, which was also observed in a clinical study [23]. In conclusion, metformin-associated lipogenesis and inflammation may lead to enhanced neurodegeneration in ApoE– /– mice (Fig. 9). In light of our findings, the use of metformin as a neuroprotective drug shall be with caution. Finally, the application of metformin in case of ApoE risk groups of AD patients has to be proven in future clinical studies.
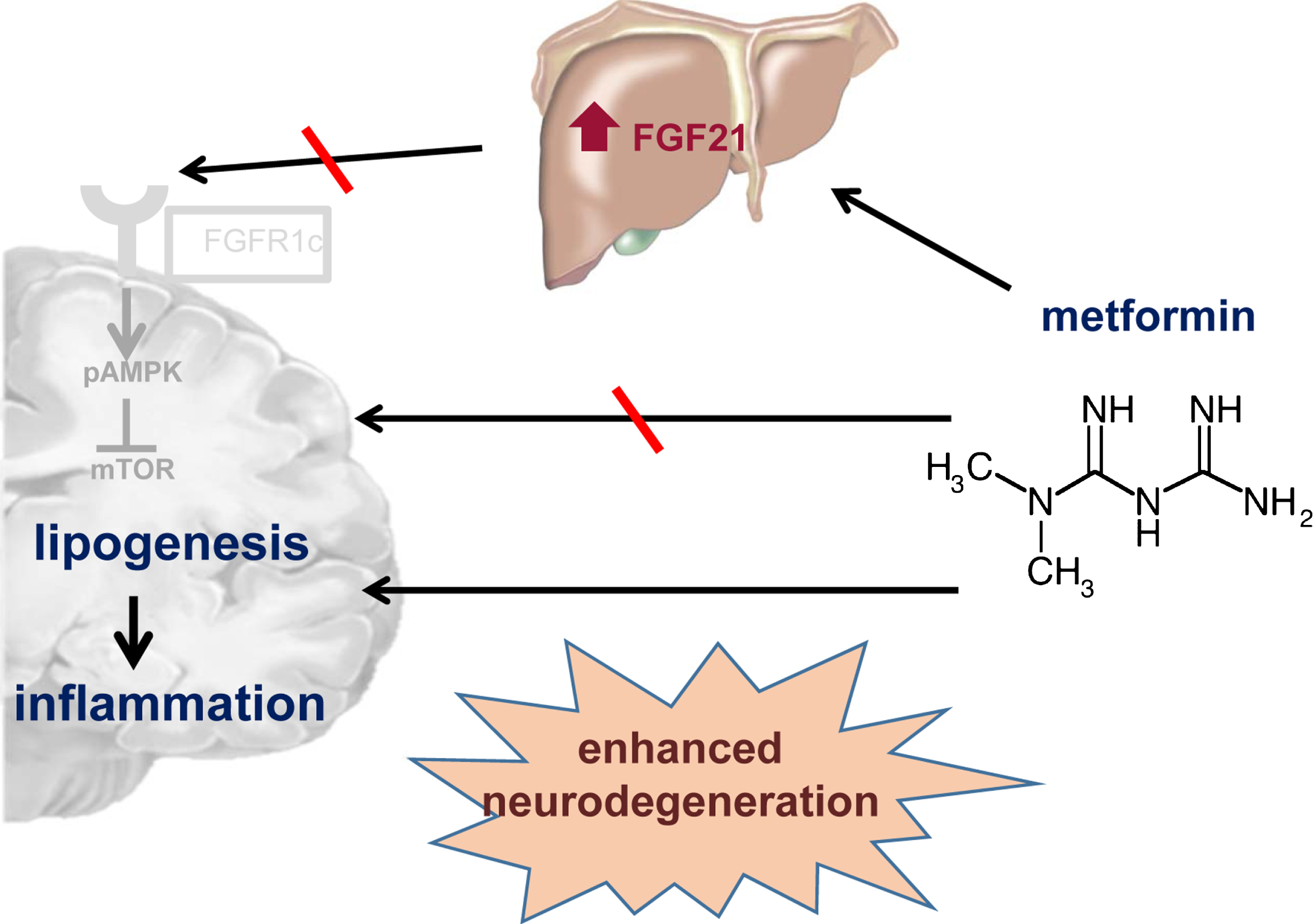
Schematic illustration summarizes the main results of the present study. In contrary to our hypothesis, the metformin-induced Fgf21 release may not be involved in the AMPK/mTOR activation but rather in stimulation of lipogenetic processes. This might lead to an increase of inflammatory processes and in consequence to enhanced neurodegenerative processes which is represented by increased tau phosphorylation.
Footnotes
ACKNOWLEDGMENTS
The authors cordially thank the technicians of the Institute for Experimental Surgery and of the Central Animal Care Facility, Rostock University Medical Center for their valuable assistance. This study was supported by a grant from the Deutsche Forschungsgemeinschaft, Bonn, Germany (KU 3280/1-2).