
Abstract
Alzheimer’s disease (AD) is the most common dementia and a progressive neurodegenerative disorder aggravated by chronic hypoperfusion (HP). Since numerous evidence suggests that inflammation is related with AD pathology, we investigated the expression change of two anti-inflammatory markers, inter-alpha-trypsin inhibitor heavy chain H4 (ITIH4) and alpha-2-HS-glycoprotein (AHSG), in a novel AD model (APP23) with HP at 12 month of age. As compared with wild type (WT, n = 10), immunohistochemical analysis showed a higher ITIH4 and a lower AHSG expressions in the cerebral cortex, hippocampus, and thalamus of the APP23 + HP group (n = 12) than the simple APP23 (n = 10) group (*p < 0.05 and **p < 0.01 versus WT; #p < 0.05 and # #p < 0.01 versus APP23). The present study provides an upregulation of anti-inflammatory ITIH4 and a downregulation of pro-inflammatory TNFα-dependent AHSG in a novel AD plus HP mice model.
INTRODUCTION
Alzheimer’s disease (AD) is the most common dementia accounting for 69% in elderly population more than 75 years old [1]. Cerebrovascular pathologies such as cerebral amyloid angiopathy, blood-brain barrier disruption, and microvascular degeneration are confirmed in 60–90% of AD patients [2, 3]. Many studies show the involvement of oxidative stress and neuroinflammatory processes in AD pathology [4–11].
Inter-alpha-trypsin inhibitor heavy chain H4 (ITIH4) is expressed in an acute phase of several pathological situations including infections and inflammations, and is associated with cell proliferation and migration [12]. Alpha-2-HS-glycoprotein (AHSG) is a novel anti-inflammatory hepatokine under acute injuries and infections, but is regulated by pro-inflammatory TNFα [13–15]. In recent years, ITIH4 and AHSG are reported as serum biomarkers in AD patients, but have not been fully investigated in AD brain [12, 16].
In the present study, therefore, we investigated the expression changes of ITIH4 and AHSG in the brain of novel AD model mice with chronic hypoperfusion (HP) to find the relation with AD pathology.
MATERIALS AND METHODS
Experimental model
The APP23 mouse model overexpresses human APP751 carrying the Swedish double mutation (KM670/671NL) driven by a Thy1 promoter [17]. All animal experiments were performed in compliance with a protocol approved by the Animal Care and Use Committee of the Graduate School of Medicine and Dentistry of Okayama University (OKU-2014-095). This is a part of the whole project mainly focusing on inflammation in AD mice model. All used control mice were C57BL/6J. Animals were accommodated in 12/12 h light-dark cycle with a controlled temperature around 23°C and free access to food and water.
Three groups were designed in this study: wild type mice (WT + sham surgery, n = 10), APP23 group (APP23 + sham surgery, n = 10), and APP23 with chronic hypoperfusion (HP) group (APP23 + HP, n = 12). Groups comprised approximately equal numbers of male and female mice.
Ameroid constrictors with an inner diameter (D) of 0.75 mm (Research Instruments NW, Lebanon, OR, USA) were applied to induce chronic cerebral hypoperfusion. For the CCH group, a cervical incision was made and ameroid constrictors were applied to bilateral common carotid arteries at 4 months of age. At the time points of 1, 3, 7, 14, and 28 days after surgery, a laser-Doppler flowmeter (FLO-C1, Omegawave, Tokyo, Japan) was used to measure cerebral blood flow as our previous report [11].
Tissue preparation
At 12 months of age, mice were deeply anesthetized and then perfused with 20 ml of ice-cold phosphate-buffered saline (PBS, pH 7.4), followed by 20 ml of ice-cold 4% paraformaldehyde in 0.1 mol/L phosphate buffer. The brains were removed and post-fixed in the same fixation overnight at 4°C. After washing with PBS, the fixed tissues were transferred into 10, 20, and 30% (wt/vol) sucrose in PBS, each sucrose incubation step was for 24 h at 4°C. Then, the brains were frozen in dry ice and kept at – 80°C. 20– μm thick coronal sections were prepared on a cryostat at – 20°C and mounted on silane-coated glass slides.
Single immunohistochemistry analysis
For single immunohistochemistry, after incubation in 0.3% hydrogen peroxide/methanol for 30 min followed by 5% bovine serum albumin (BSA) in PBS with 0.1% triton for 1 h, the sections were stained overnight at 4°C with the following primary antibodies: rabbit anti-inter-alpha-trypsin inhibitor heavy chain H4 (ITIH4) antibody (1:50, Proteintech Group, Chicago, IL); rabbit anti-alpha-2-HS-glycoprotein (AHSG) antibody (1:50, Cloud-Clone Corp, Houston, TX, USA). After being washed with PBS, brain sections were treated with suitable biotinylated secondary antibodies (1:500; Vector Laboratories, Burlingame, CA) for 2 h at room temperature. Then the sections were incubated with the avidin-biotin-peroxidase complex (Vectastain ABC Kit; Vector) for 30 min and visualized with 3, 3′-diaminobenzidine. Negative control sections were stained in the same manner as described above except for the primary antibodies.
Double immunofluorescence analysis
To analyze the localizations of ITIH4 and AHSG in neuronal cells, double immunofluorescent stainings for ITIH4 plus neuronal nuclear antigen (NeuN) and AHSG plus NeuN were performed. NeuN is used as a biomarker for neurons. Brain sections were incubated with 5% BSA in PBS for 1 h at room temperature. The following primary antibodies were used: rabbit anti-ITIH4 antibody (1:50, Proteintech Group, Chicago, IL); rabbit anti-AHSG antibody (1:50, Cloud-Clone Corp, Houston, TX); mouse anti-NeuN antibody (1:200; Millipore, Burlington, MA). After rinsing in PBS, the sections were incubated with secondary antibodies conjugated to Alexa 488 and 555 (1:500, Molecular Probes, Eugene, OR) for 1 h at room temperature. The sections were then mounted using Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, CA), and scanned with a confocal microscope equipped with argon and HeNe1 laser (LSM-780; Zeiss, Jena, Germany).
Quantitative analysis
For each measurement, 3 sections per brain and 4 random selected regions were then analyzed with a light microscope (Olympus BX-51, Tokyo, Japan). The pixel intensity of ITIH4 and AHSG were measured at cerebral cortex (CTX), hippocampus (HI), and thalamus (TH) by an image processing software (Scion Image, Scion Corporation, Frederick, MD).
Statistical analysis
All data are presented as mean±SD. Statistical comparisons were performed using one-way analysis of variance based on a Tukey-Kramer post comparison. p < 0.05 was considered statistically significant.
RESULTS
ITIH4 and AHSG expressions in cortex
Both ITIH4 and AHSG were obviously stained in neurons of CTX of WT mice (Fig. 1, top left). Although no significant difference of the pixel intensity of both ITIH4 and AHSG was detected between WT and APP23 group in the CTX, the pixel intensity of ITIH4 significantly increased in the APP23 + HP group as compared with the WT and APP23 group (Fig. 1, **p < 0.01 versus WT; #p < 0.05 versus APP23). In contrast, the pixel intensity of AHSG significantly decreased in the APP23 + HP group as compared with the WT group (Fig. 1, *p < 0.05 versus WT).
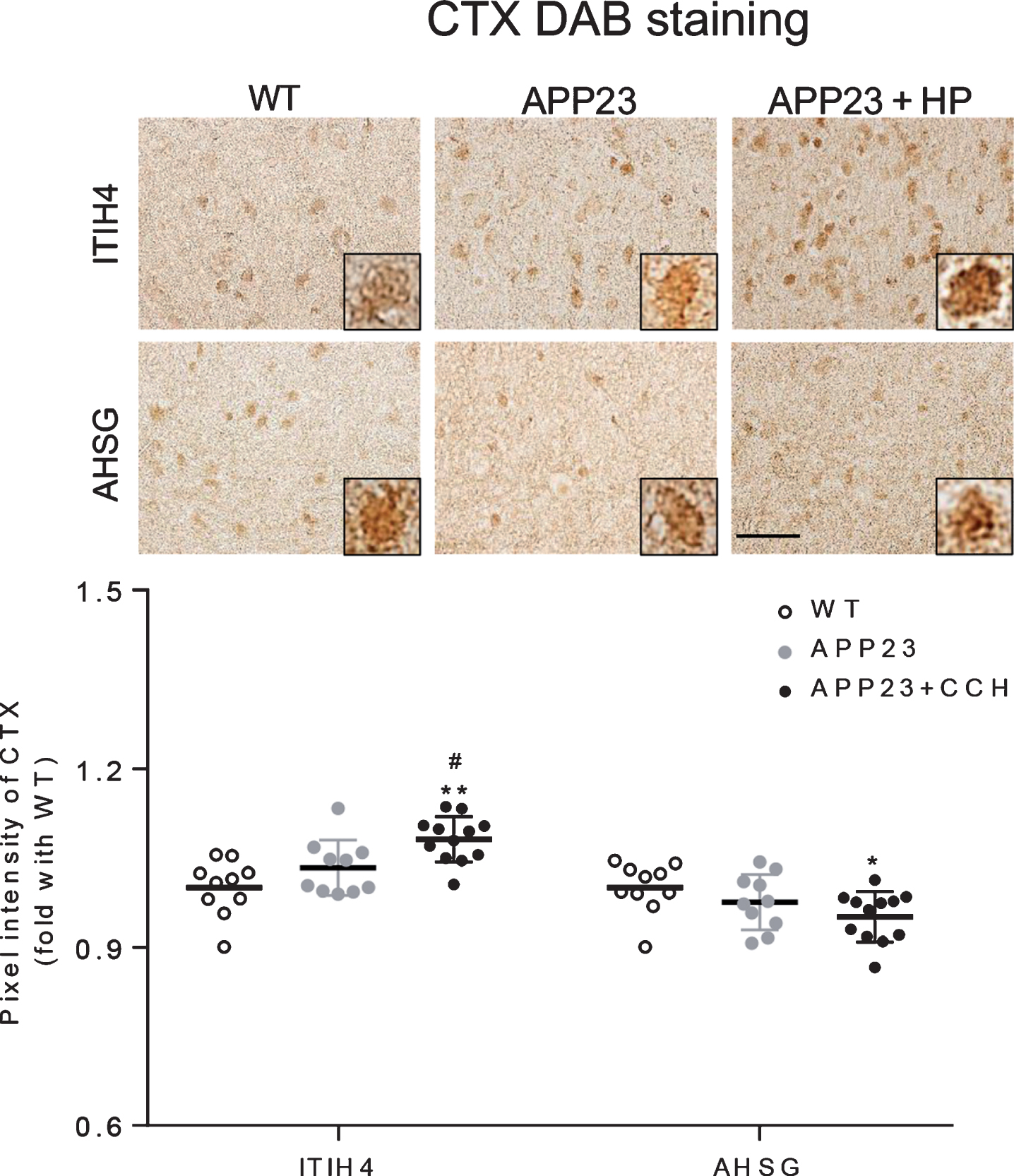
Immunohistochemical stainings of ITIH4 and AHSG in the CTX of WT, APP23 and APP23 + HP groups (top), showing an increased pixel intensity of ITIH4 and a decreased pixel intensity of AHSG in APP23 + HP group (*p < 0.05 and **p < 0.01 versus WT; #p < 0.05 versus APP23). Scale bar = 50μm.
ITIH4 and AHSG expressions in hippocampus
The ITIH4 was slightly stained in neurons of CA1, CA2, and CA3 (Fig. 2, top). Compared with the WT and APP23 group, the pixel intensity of ITIH4 significantly increased in the CA1, CA2, and CA3 of APP23 + HP group (Fig. 2, *p < 0.05 and **p < 0.01 versus WT; #p < 0.05 and # #p < 0.01 versus APP23). The AHSG was stained detected in the neurons of CA1, CA2, and CA3 of WT and APP23 mice (Fig. 3, top). While, the pixel intensity of AHSG significantly decreased in the CA1, CA2, and CA3 of APP23 + HP group compared with WT and APP23 group (Fig. 3, **p < 0.01 versus WT; # #p < 0.01 versus APP23).
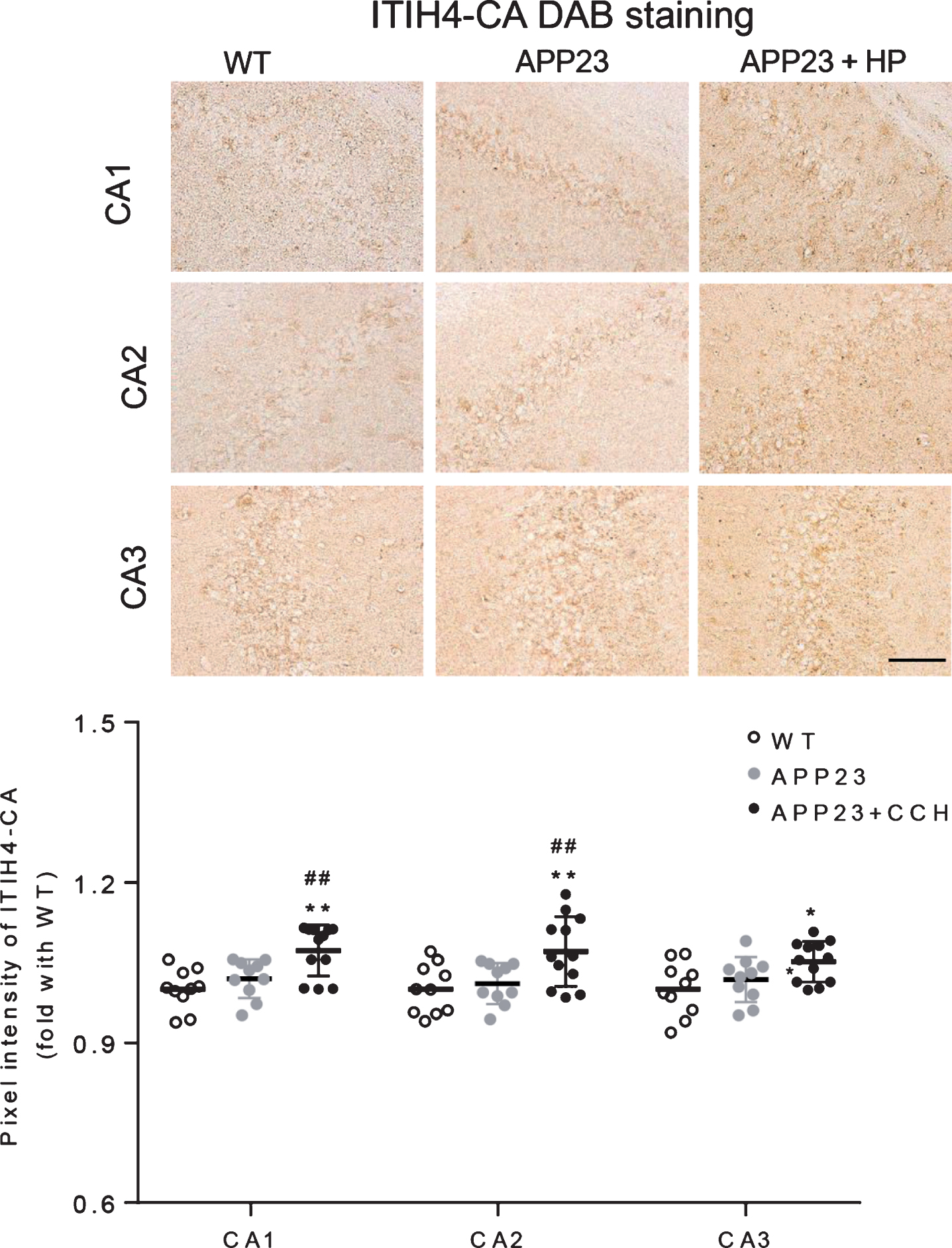
Immunohistochemical stainings of ITIH4 in the hippocampal CA1, CA2, and CA3 of WT, APP23, and APP23 + HP groups (top), showing increased pixel intensities in CA1, CA2, and CA3 of APP23 + HP group (*p < 0.05 and **p < 0.01 versus WT; #p < 0.05 and # #p < 0.01 versus APP23). Scale bar = 50μm.

Immunohistochemical stainings of AHSG in the hippocampal CA1, CA2, and CA3 of WT, APP23, and APP23 + HP groups (top), showing decreased pixel intensities in CA1, CA2, and CA3 of APP23 + HP group (**p < 0.01 versus WT; # #p < 0.01 versus APP23). Scale bar = 50μm.
Both ITIH4 and AHSG were stained in the neurons of dentate gyrus (Fig. 4, top). Although no significant difference was observed in the pixel intensity of both ITIH4 and AHSG between WT and APP23 group, the pixel intensity of ITIH4 significantly increased in APP23 + HP group compared with WT and APP23 group (Fig. 4, **p < 0.01 versus WT; # #p < 0.01 versus APP23). In contrast, the intensity of AHSG significantly decreased in APP23 + HP group compared with the WT and APP23 group (Fig. 4, **p < 0.01 versus WT; # #p < 0.01 versus APP23).

Immunohistochemical stainings of ITIH4 and AHSG in the hippocampal dentate gyrus of WT, APP23, and APP23 + HP groups (top), showing an increased pixel intensity of ITIH4 and a decreased pixel intensity of AHSG in APP23 + HP group (**p < 0.01 versus WT; # #p < 0.01 versus APP23). Scale bar = 50μm.
ITIH4 and AHSG expression in thalamus
In the TH, the ITIH4 was scarcely detected in the neurons of WT mice (Fig. 5, top left), which was enhanced in both APP23 and APP23 + HP mice. Although no significant difference of the pixel intensity was detected between WT and APP23 group, the pixel intensity of ITIH4 significantly increased in the APP23 + HP group as compared with WT and APP23 mice (Fig. 5, **p < 0.01 versus WT; # #p < 0.01 versus APP23). The AHSG was stained in the neurons of TH of WT mice. The pixel intensity of AHSG decreased in APP23 group compared with WT group, which was further emphasized in APP23 + HP group (Fig. 5, *p < 0.05 and **p < 0.01 versus WT; # #p < 0.01 versus APP23).

Immunohistochemical stainings of ITIH4 and AHSG in the TH of WT, APP23, and APP23 + HP groups (top), showing an increased pixel intensity of ITIH4 and a decreased pixel intensity of AHSG in APP23 + HP group (*p < 0.05 and **p < 0.01 versus WT; # #p < 0.01 versus APP23). Scale bar = 50μm.
Localizations of ITIH4 and AHSG
Double immunofluorescence analysis showed that both ITIH4 and AHSG were expressed in neurons of both CTX and HI (Figs. 6 and 7), and mainly localized in the cytoplasm of neurons (Fig. 8).

Double immunofluorescent stainings of ITIH4 and NeuN in the CTX and HI of WT, APP23, and APP23 + HP groups, showing the localization of ITIH4 in neurons. Scale bar = 50μm.
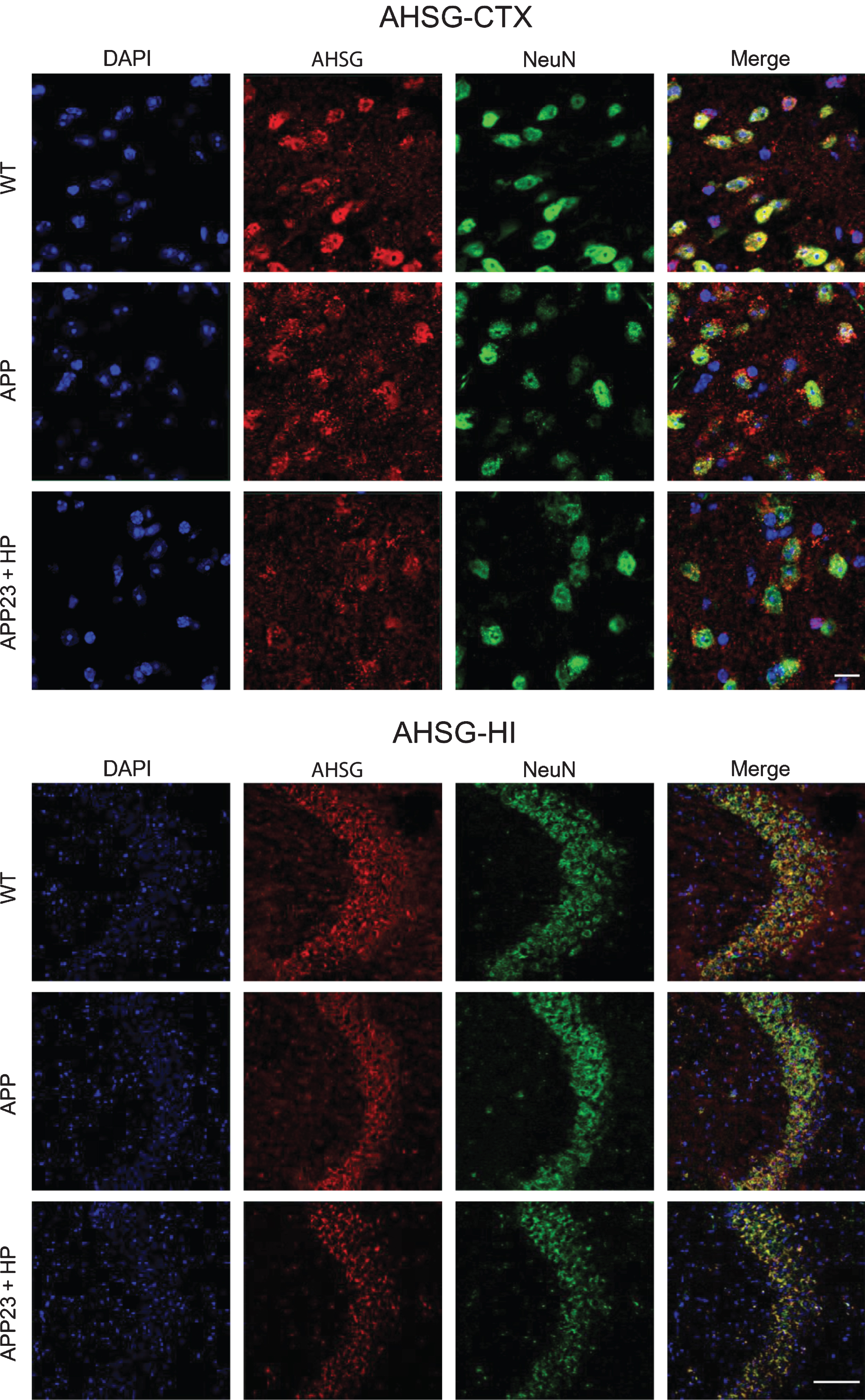
Double immunofluoresecnt stainings of AHSG and NeuN in the CTX and HI of WT, APP23, and APP23 + HP groups, showing the localization of AHSG in neurons. Scale bar = 50μm.
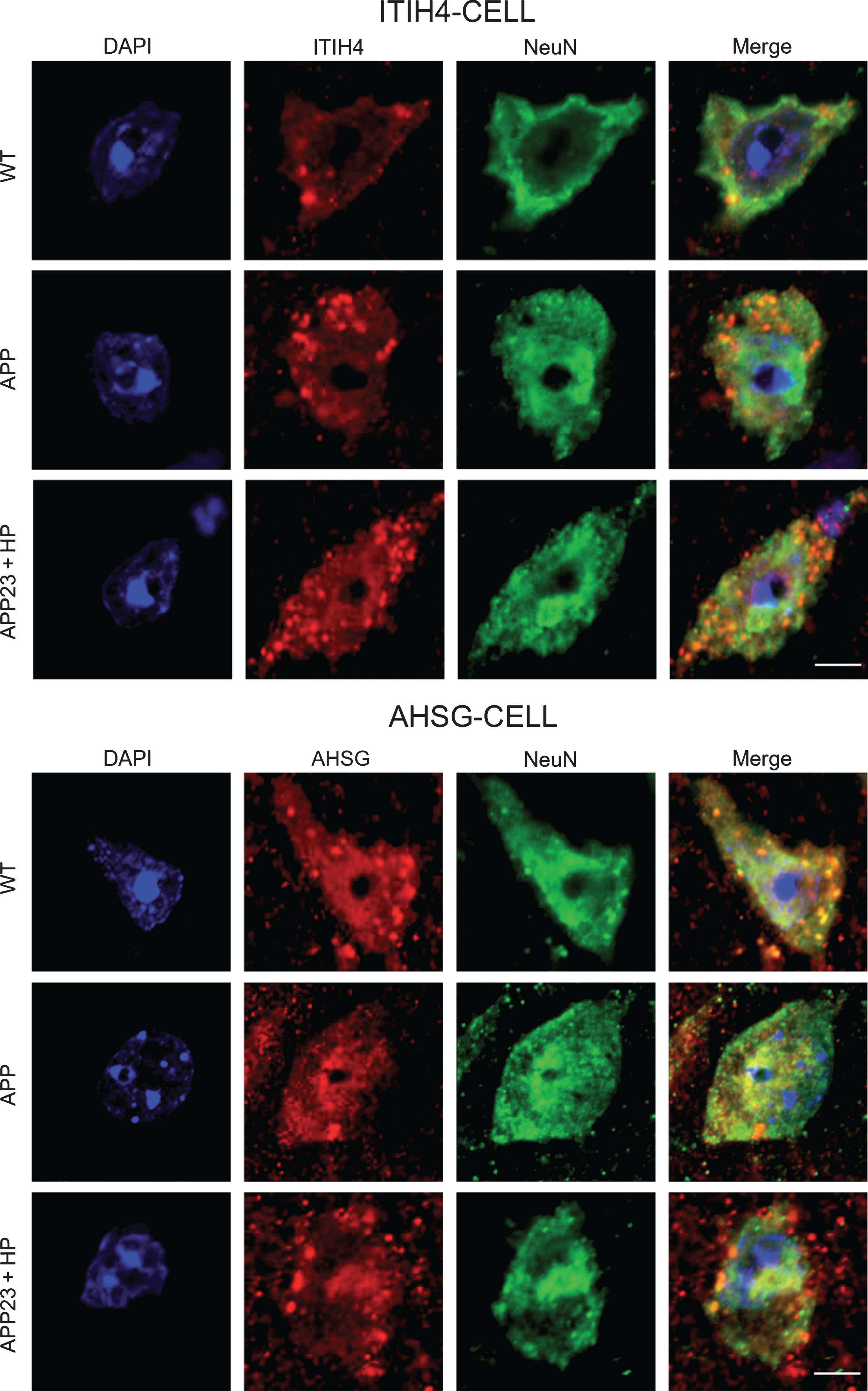
Double immunofluoresecnt stainings of ITIH4 or AHSG plus NeuN in the CTX of WT, APP23, and APP23 + HP groups, showing the main localizations of both markers in the cytoplasm of neurons. Scale bar = 20μm.
DISCUSSION
In the present study, we showed the upregulated expression of ITIH4 and the downregulation of AHSG in the neurons of CTX, HI, and TH of APP23 + HP mice model (Figs. 1–8).
ITIH4 belongs to the family of inter-alpha inhibitor proteins (IAIPs), which is expressed in a lot of tissues including liver, intestine, kidney, stomach, placenta, and brain to signify their diverse biological functions by inhibiting serine proteases [13, 18–21]. ITIH4 is related to cell proliferation and migration during the development of the acute-phase inflammatory response and plays a role in the response for the inflammation of trauma and infection independent from pro-inflammatory TNFα [12, 22–24]. ITIH4 is a member of liver-derived ITI family having an anti-apoptotic and matrix-stabilizing functions. However, the role of ITIH4 has not been examined in cerebral ischemia or AD. Therefore, we used a mouse model of AD with HP to examine the pathological changes of ITIH4 in mice brain. Our previous studies showed that HP dramatically accelerated the progression of AD pathology accompanied with inflammation [25]. In addition, ITIH4 is regulated by IL-6, which regulates inflammation [26–28]. In the present study, HP significantly increased the expression of ITIH4 in the CTX, HI, and TH of APP23 mice (Figs. 1, 2, 4–8), suggesting that HP promoted the neuroinflammation in the brains of AD model mice and ITIH4 may serve as a potential indicator of the progression of AD pathology.
In the response to injury and infection, the liver releases many acute phase proteins including AHSG. AHSG is a cysteine protease inhibitor secreted from the liver to inhibit vascular calcification by preventing spontaneous mineral precipitation in the vasculature [29–31]. AHSG is regulated by several pro-inflammatory mediators [32], and produced as anti-inflammatory protein primarily in the liver under an acute inflammation phase [32]. However, AHSG is a negative acute phase reactant protein under the regulation of pro-inflammatory TNFα, attenuating inflammatory responses to injury and infection [13, 14]. In the present study, AHSG expression decreased in brains of APP23 + HP group (Figs. 1, 3, 4, 5), probably due to suppressions by pro-inflammatory TNFα, IL-1, and IL-6 [13, 14]. A previous paper showed the lower serum level of AHSG was related to the cognitive impairment in the mild-to-moderate AD patients accompanied by the higher concentration of TNF-α [16]. The decreased AHSG level may indicate the severity of AD progression in the present study (Figs. 1, 3, 4–8).
In summary, the present study demonstrated the expression changes of ITIH4 and AHSG in the brains of AD model mice accompanied by HP, suggesting the potential roles of ITIH4 and AHSG as candidate biomarkers to predict the severity of AD progression.
Footnotes
ACKNOWLEDGMENTS
This work was partly supported by a Grant-in-Aid for Scientific Research (B) 17H0419619, (C) 15K0931607, 17H0419619 and 17K1082709, and by Grants-in-Aid from the Research Committees (Kaji R, Toba K, and Tsuji S) from Japan Agency for Medical Research and development (AMED) 7211700176, 7211700180 and 7211700095. X. S. would like to thank Otsuka Toshimi Scholarship Foundation for a scholarship support (18-235).