
Abstract
Emerging evidence suggests that gut microbiota dysbiosis plays a role in neurodegenerative disorders. However, whether the composition and diversity of the gut microbiota are altered in tauopathies remains largely unknown. This study was aimed to examine the diversity and composition of the gut microbiota in tauopathies, as well as the correlation with pathological changes in the brain. We collected fecal samples from 32 P301L tau transgenic mice and 32 age- and gender-matched littermate mice at different ages. The 16S ribosomal RNA sequencing technique was used to analyze the microbiota composition in feces. Brain tau pathology levels were measured by immunohistochemistry. The diversity and composition of the gut microbiota significantly changed with aging. At the phylum level, the relative abundance of Bacteroidetes was increased, while Firmicutes were decreased in P301L mice compared with that in Wt mice after 3 months of age. In addition, Actinobacteria was decreased in P301L mice at 3 and 6 months of age, meanwhile Tenericutes was decreased in P301L mice at 10 months of age. Moreover, several specific macrobiota were highly associated with the levels of AT8-tau or pT231-tau protein in the brain. Our findings suggest that gut microbiota changed with aging, as well as in the tauopathy mice model. Modulation of the gut microbiota may be a potential strategy for treatment of tauopathy.
INTRODUCTION
Tauopathies are progressive neurodegenerative disorders associated with Alzheimer’s disease (AD) [1], frontotemporal lobar degeneration (FTLD) [2], Pick’s disease [3], corticobasal degeneration [4], progressive supranuclear palsy (PSP) [5], and chronic traumatic encephalopathy [6]. The accumulation of pathological tau is the hallmark of these diseases and is closely correlated with cognitive decline. Despite substantial progress in recent years, the comprehensive pathogenesis of tauopathies remains unclear. Moreover, thus far, no therapeutic interventions are currently available.
Bidirectional communication between the brain and gut has long been recognized. Recent studies have revealed the vital role of the gut microbiota in the function of the central nervous system [7, 8]. Alterations in gut microbiota composition can be related to the development of several neurological disorders, including AD [9], Parkinson’s disease [10], and amyotrophic lateral sclerosis [8]. The dyshomeostasis in the gut microbiota is proposed to be a crucial cause of the onset of AD [11]. In contrast, metabolites derived from gut microbes protect neurons from amyloid-β (Aβ)-induced neurotoxicity and paralysis in an AD invertebrate model [12]. These results indicate the complex interaction between the gut microbiota and brain dysfunction. As a complex and multisystem disease, a series of mechanisms have been identified in tauopathies. However, the alteration in gut microbial composition and diversity has not yet been clearly confirmed. This study was aimed to examine the diversity and composition of the gut microbiota in tauopathies, as well as the correlation with pathological changes in the brain. Our findings suggest that gut microbiota diversity and compositions changed with aging, as well as in the tauopathy mice model. Moreover, several specific macrobiota are highly associated with pathological changes in the brain of P301L mice, indicating that modulation of the gut microbiota may be a potential strategy for tauopathy treatment.
MATERIALS AND METHODS
Animals and sample collection
P301L tau transgenic mice, expressing the longest human tau isoform (2N4R) with the FTDP-17 mutation P301L in neurons, were provided by Professor Juergen Gotz from Queensland Brain Institute, Queensland University (Australia) and raised in Daping Hospital Animal Laboratory Center. Mice food, provided by Xietong Organism (Jiangsu, China), was sterilized with irradiation (60Coγ). The sterilized water was produced by pure water system (LT-DW1500/D, Nico, China). All mice were raised under specific pathogen-free conditions with the sterilized food and water provided. A 12-h light-dark cycle was maintained. Young mice were weaned from their mothers after 1 month and raised under the same conditions. Age- and gender-matched wild-type (Wt) littermate mice were chosen as the controls. Fresh fecal samples were collected from mice of both genders at 1, 3, 6, and 10 months of age (n = 8, per group). A total of 64 fecal samples were collected in the morning to avoid interference from any possible circadian rhythm effects and stored at -80°C until subsequent DNA extraction. This study was approved by the Animal Care and Use Committee of the Institute of Laboratory Animal Science of Daping Hospital.
DNA extraction and sequencing
Microbial DNA was extracted from 64 fecal samples using the standard E.Z.N.A. ® Stool DNA Kit (Omega Biotek, Norcross, GA, U.S.) according to standard protocols. The concentration and purification of extracted DNA were determined by a NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, USA). The V3-V4 region of the bacterial 16S ribosomal RNA gene was amplified to evaluate bacterial diversity by a thermocycler PCR system (GeneAmp 9700, ABI, USA). DNA was amplified by using the 338F and 806R primer set (338F: 5’-ACTCCTACGGGAGGCAGCAG-3’; 806R: 5’-GGACTACHVGGGTWTCTAAT-3’). PCRs were conducted according to previous protocols [13], and the resulting products were further purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) to remove failed and low quality products. Purified amplicons were pooled in equimolar concentrations and paired-end sequenced (2×300 bp) on an Illumina MiSeq platform (Illumina, San Diego, USA) by Majorbio Bio-PharmTechnology Co. Ltd. (Shanghai, China) according to the manufacturer’s instructions.
Processing of sequencing data
Raw fastq files were demultiplexed and quality filtered by Trimmomatic and then merged by FLASH. The criteria were followed as previously described [14]. The obtained sequences were clustered into operational taxonomic units (OTUs) at the 97% sequence similarity level using UPARSE (version 7.1). Then, UCHIME (version 4.2.40) was used to identify and remove chimeric sequences. The taxonomy of each 16S rRNA gene sequence was analyzed by the Ribosomal Database Project Database Classifier algorithm (http://rdp.cme.msu.edu/) using a confidence threshold of 70%. All of the sequencing data in the present study can be accessed from NCBI with the accession number SRP188916.
Bioinformatic analysis
Alpha diversity analysis, including the Shannon index, Simpson, Chao1, ACE index and Sobs index, was carried out in QIIME on rarefied OTU tables. Beta diversity between samples was generated on the basis of bray_curtis or weighted_unifrac algorithms and reported according to principal coordinate analysis (PCoA) using the R package (Version 2.15.3). LEfSe (http://huttenhower.sph.harvard.edu/galaxy/root?tool_id=lefse_upload) was used to identify the taxa that were significantly different between groups. The area under the receiver operating characteristic curve (ROC) of the random forest (RF), namely AUC-RF, was carried out to identify and validate key predictive biomarkers using the R package. Briefly, AUC-RF used the ROC curve and the AUC as the predictive accuracy of RF and then selected the set of variables with the highest AUC value. The process of AUC-RF for key predictive biomarkers selection consists of the following steps: step 1) iterative elimination process; step 2) visual representation of the elimination process; step 3) selection of the optimal set of predictor variables; and step 4) predictive accuracy and probability of selection.
Immunohistochemistry
Free-floating immunohistochemistry methods were used for phosphorylated tau immunohistochemical staining. Phospho-tau at Thr231 (pT231) antibody was used to stain phosphorylated tau in the pre-neurofibrillary tangle (NFT) stage, while AT8 (phospho-tau at Ser202 and Thr205) antibody was used to stain hyperphosphorylated-tau deposited in intra-NFTs and extra-NFTs at the later stage of tangles [15]. Brain tissue sections were treated with 0.5% Triton X-100 (Sangon Biotech, Shanghai, China) mixed with 0.3% H2O2 in PBS for 30 min and then blocked with 3% BSA (Solarbio, Beijing, China) for 30 min at room temperature. The primary antibody was diluted with 3% BSA in a proper proportion (pT231 1:200, Signalway Antibody, USA; AT8 1:100, Invitrogen, USA.) and incubated overnight at 4°C. Secondary antibodies conjugated with HRP were diluted 1:200 with PBST to incubate the sections for 1 h at 37°C the next day, followed by visualization with DAB solution. Three brain slices (bregma –2.1 mm) per mouse were selected for pT231-tau staining, and another three brain slices (bregma -2.1 mm) per mouse were selected for AT8-tau staining in different groups of P301L mice. A 40-fold objective was used to capture images for pT231-tau staining from hippocampal CA1 and AT8-tau staining from amygdala. ImageJ software (National Institutes of Health, Bethesda, USA) was used to quantify the positive staining fraction of each image by another experimenter manually. Tau levels in the brain were quantified by the average positive staining fraction of the three brain slices and used for subsequent correlation analysis.
Statistical analysis
All data are represented as the mean±SD. One-way ANOVA and Tukey’s test were used to compare observed species among different groups. Two-tailed Student’s t test or a Mann-Whitney U test was used to compare the differences in bacterial communities between P301L and Wt as applicable. Correlations between brain pathology and the abundance of microbiota were calculated using Spearman’s rank correlation. To correct for multiple testing, p values were adjusted using Benjamini-Hochberg false discovery rate, and significant association was considered below a threshold of 0.05. All analyses were conducted with GraphPad Prism 6.0 software (GraphPad Software, Inc., San Diego, USA). p < 0.05 was considered statistically significant.
RESULTS
Characteristics of high-throughput sequencing data
A total of 2,995,005 qualified reads were filtered for downstream analysis. An average of 46797 reads per sample was obtained for this study (the minimum of one sample was 30920, and the maximum was 72203). The gradually flattening rarefaction curves and coverage index (99.78%) indicated an adequate depth of sequencing (Supplementary Figure 1a).
Changes in bacterial communities in mice with aging
The Chao1 index showed higher diversity and richness in 3-month-old and 6-month-old Wt mice than in 1-month-old Wt mice, and no significant difference was observed in P301L between different ages (Fig. 1a). The relationships between the community structures were examined by PCoA and bray_curtis analysis. The community structures of the gut microbiota differed as a function of age in both Wt (Fig. 1b, ANOSIM r = 0.4568, p < 0.001) and P301L mice (Fig. 1c, ANOSIM r = 0.2281, p < 0.001).

Diversity of the gut microbiota between P301L and Wt mice. a) The Chao1 index displays the microbial diversity of each group. Box represent 5-95 percentile. Principal coordinate analysis (PCoA) for the gut microbiota composition in the Wt (b) and P301L groups (c) at 1, 3, 6, and 10 months of age. d) PCoA for the gut microbiota composition between P301L and Wt mice at 3 months of age. The first two principal components are plotted (n = 8 per group). One-way ANOVA and Turkey’s test, **p < 0.01.
The difference in bacterial communities between P301L and Wt mice during the course of tauopathy development
Alpha diversity, including the Shannon index, Simpson index, ACE index, and Sobs index, showed no significant difference between P301L and Wt mice at any age (Supplementary Figure 2). Based on the distribution of the sample points, weighted_unifrac analysis showed no significant difference in PCoA between P301L and Wt mice at 1 month (ANOSIM r = -0.0184, p = 0.516, Supplementary Figure 1b). By contrast, the P301L and Wt groups were obviously distinct after 3 months (Fig. 1d, ANOSIM r = 0.6356, p < 0.001, Supplementary Figure 1c and d), and the total explanation rate of PC1 and PC2 reached 56.61% at 3 months.
At the phylum level, no significant difference was observed in the gut microbiota at 1 month between P301L and Wt mice (Fig. 2a). After 3 months of age, the relative abundance of Bacteroidetes was increased, while Firmicutes was decreased in P301L mice compared with that in Wt mice (Fig. 2b-d). In addition, Actinobacteria was decreased in P301L mice at 3 and 6 months of age (Fig. 2b, c); meanwhile Tenericutes was decreased in P301L mice at 10 months of age (Fig. 2d). From the phylum to genus level, the statistically significant differences in microbiota between P301L and Wt mice at 3, 6, and 10 months of age are presented in Figures 3, 4, and 5, respectively. At 3 months of age, the relative abundance of norank_f_Bacteroidales_24_7_group, Phascolarctobacterium, Parabacteroides, and unclassified_o_Bacteroidales were increased in P301L mice, while a total of 22 bacteria, including Lactobacillus, Enterorhabdus, and Staphylococcus were significantly decreased in P301L mice compared with those in matched Wt littermates at the genus level. At 6 months of age, the relative abundance of Ruminococcaceae_NK4A214_group, Bacteroides, Anaeroplasma, and norank_f_Clostridiales_vadinBB60_group were increased in P301L mice, while Staphylococcus, Bifidobacterium, and Gemella were significantly decreased in P301L mice at the genus level. At 10 months of age, the relative abundance of norank_f_Bacteroidales_24_7_group Anaerovorax, Peptococcus, unclassified_o_Bacteroidales, Caproiciproducens, Ruminiclostridium, norank_f_Ruminococcaceae, norank_f_Clostridiales_vadinBB60_group, and Oscillibacter were increased in P301L mice, while Roseburia, norank_f_Mycoplasmataceae, Lachnoclostridium, Klebsiella, Streptococcus, and unclassified_f_Coriobacteriaceae were significantly decreased in P301L mice at the genus level.

Different bacterial communities between P301L and Wt mice at different months of age. a) Different bacterial communities on the phylum level at 1 month of age, (b) 3 months of age, (c) 6 months of age, and (d) 10 months of age. (n = 8 per group). Mann-Whitney U test, *p < 0.05, **p < 0.01, ***p < 0.001.
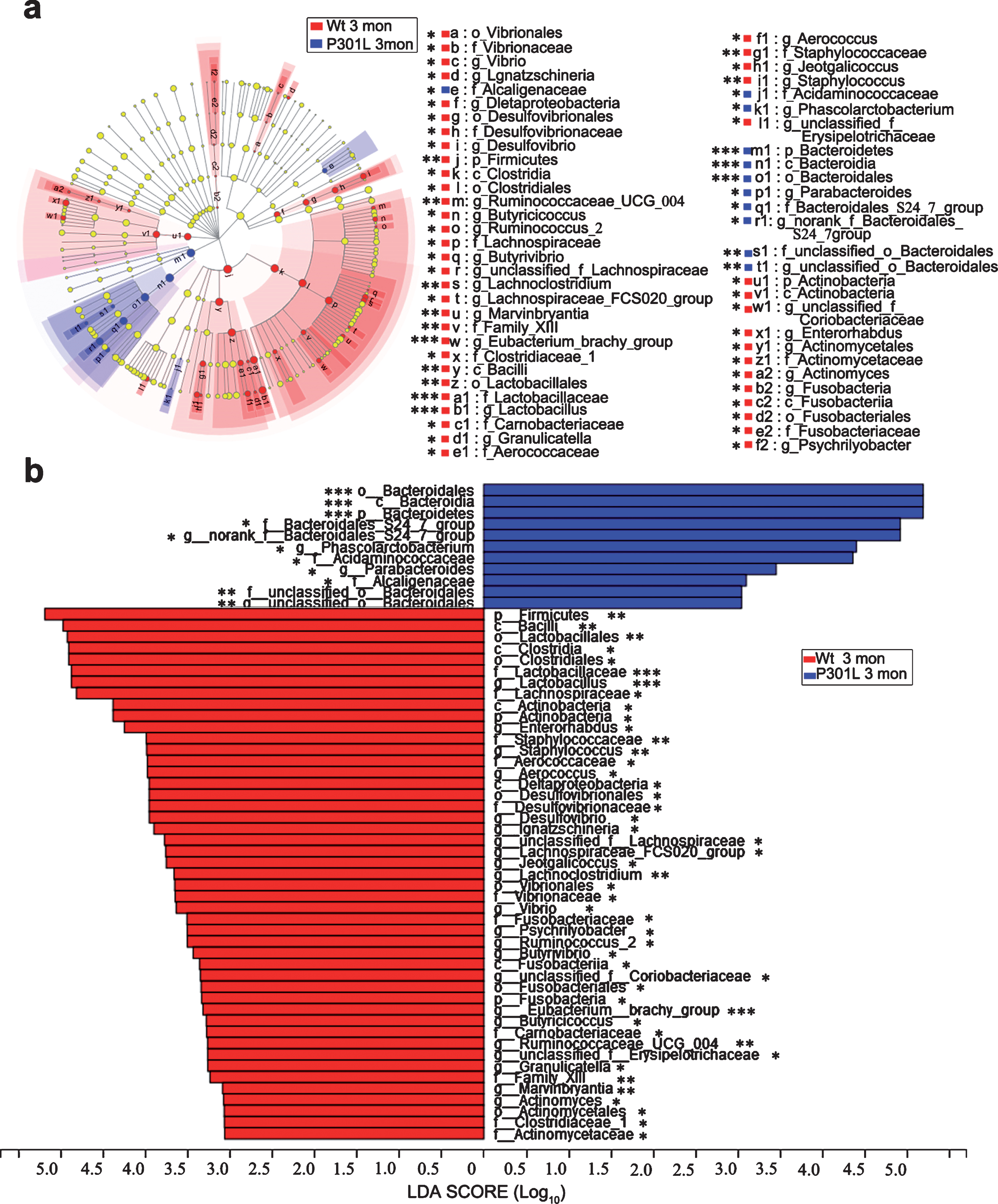
Taxonomic differences of microbial communities between P301L and Wt mice at 3 months of age. a) LEfSe identified the most differentially abundant taxons between P301L and Wt mice from the phylum to genus level. (Blue) taxa enriched in P301L mice; (Red) taxa enriched in Wt mice; (Yellow) no significant difference in taxa between P301L and Wt mice. The size of each dot is proportional to its effect size. (n = 8 per group). b) P301L mouse-enriched taxa are indicated with a positive LDA score (blue), and taxa enriched in Wt mice are indicated with a negative score (red). The LDA score threshold is≥2. (n = 8 per group), non-parametric factorial Kruskal-Wallis sum-rank test, *p < 0.05, **p < 0.01, ***p < 0.001.
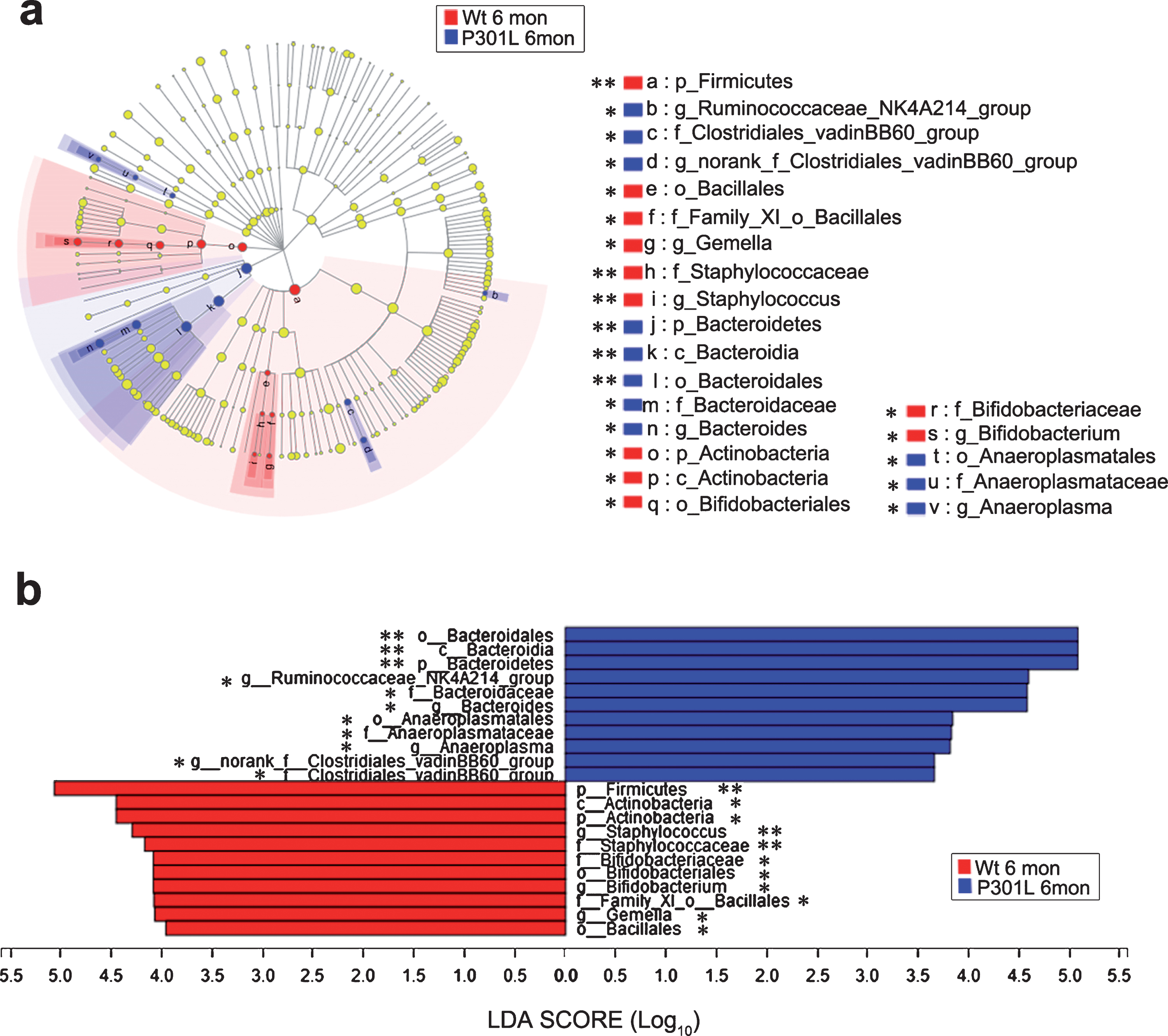
Taxonomic differences of microbial communities between P301L and Wt mice at 6 months of age. a) LEfSe identified the most differentially abundant taxons between P301L and Wt mice from the phylum to genus level. (Blue) taxa enriched in P301L mice; (Red) taxa enriched in Wt mice; (Yellow) no significant difference in taxa between P301L and Wt mice. The size of each dot is proportional to its effect size. (n = 8 per group). b) P301L mouse-enriched taxa are indicated with a positive LDA score (blue), and taxa enriched in Wt mice are indicated with a negative score (red). The LDA score threshold is≥2. (n = 8 per group), non-parametric factorial Kruskal-Wallis sum-rank test, *p < 0.05, **p < 0.01, ***p < 0.001.

Taxonomic differences of microbial communities between P301L and Wt mice at 10 months of age. a) LEfSe identified the most differentially abundant taxons between P301L and Wt mice from the phylum to genus level. (Blue) taxa enriched in P301L mice; (Red) taxa enriched in Wt mice; (Yellow) no significant difference in taxa between P301L and Wt mice. The size of each dot is proportional to its effect size. (n = 8 per group). b) P301L mouse-enriched taxa are indicated with a positive LDA score (blue), and taxa enriched in Wt mice are indicated with a negative score (red). The LDA score threshold is≥2. (n = 8 per group), non-parametric factorial Kruskal-Wallis sum-rank test, *p < 0.05, **p < 0.01, ***p < 0.001.
Association between the gut microbiota and tau pathologies in the brain
To investigate whether gut microbiota are associated with tau pathologies in the brain of P301L mice, we used pT231 and AT8 to investigate the levels of different types of phosphorylated tau proteins. According to immunohistochemical assays, pT231-positive staining was observed as early as 1 month in p301L mice, while AT8-positive staining was observed until the animal reached 3 months (Supplementary Figure 3). The correlations among the abundance of microbiota and tau pathologies in the brain of P301L mice at different months of age were presented in Table 1. At the genus level, no statistically significant correlation was observed between gut microbiota and tau pathologies (pT231-tau) in mice at 1 month of age, while statistically significant correlations were observed between specific gut microbiota and tau pathology in mice at 3, 6, and 10 months of age. Marvinbryantia was negatively correlated with the abundance of pT231-tau in P301L mice at 3 months of age. For P301L mice at 6 months of age, Lactobacillus and Desulfovibrio were negatively correlated with the abundance of pT231-tau, meanwhile Streptococcus, Anaerotruncus, Lactococcus, unclassified_f_Erysipelotrichaceae, and Eubacterium_brachy_group were negatively correlated with the abundance of AT8-tau. For P301L mice at 10 months of age, Candidatus_Saccharimonas, Alistipes, Rikenella, Odoribacter, Blautia, Ruminococcaceae_UCG_014, Eubacterium_xylanophilum_group, Paraprevotella, Butyricicoccus, Ruminiclostridium_6, and Parvibacter were negatively correlated with the abundance of pT231-tau, meanwhile Bacteroides, Parabacteroides, Escherichia-Shigella, and Clostridium_innocuum_group were positively correlated with the abundance of pT231-tau. These results suggest that changes in bacterial communities may be associated with tau pathologies in the brain of P301L mice.
Correlations between the abundance of gut microbiota and tau pathologies in the brain of P301L mice at different months of age*
*r values: negative value, microbiota was negatively correlated with tau pathology; positive value, microbiota was positively correlated with tau pathology. # q value, adjusted by Benjamini – Hochberg false discovery rate.
Predictive value of microbial markers for tauopathies
Based on the AUC-RF algorithm [16], the optimal model with the maximum area under the ROC curve utilized 3 crucial bacterial populations (Eubacterium_brachy_group, Staphylococcus, and Prevotellaceae_UCG_001). For the whole cohort, the AUC for a single bacterium ranged from 0.641 to 0.781 (Supplementary Figure 4a), while for multi-biomarker analysis, the ROC curve for the model had an AUC of 0.835 (Supplementary Figure 4b). For the young cohort (1 month), the maximum AUC was 0.703 (Supplementary Figure 4c), while for the adult cohort (3–10 months), the AUC reached 0.872 (Supplementary Figure 4d).
DISCUSSION
In the present study, gut microbiota composition and diversity changed with aging. Moreover, significant differences were observed in gut microbiota composition between P301L tau transgenic mice and Wt littermates from 3 months of age onward. The relative abundance of Bacteroidetes was increased, while Firmicutes were decreased in P301L mice compared with that in Wt ones at 3, 6, and 10 months of age. In addition, Actinobacteria was decreased in P301L mice at 3 and 6 months of age, meanwhile Tenericutes was decreased in P301L mice at 10 months of age. Notably, several specific bacterial genera were highly correlated with the tau pathology in the brain of P301L mice at 3, 6, and 10 months of age.
Emerging evidence indicates a link between the gut microbiome and aging and age-related diseases, although correlations between specific patterns of the gut microbiota and specific disease entities are undisputed [9, 17]. Recent investigations indicate that neurodegeneration and cognitive disorders might directly interfere with the neuronal network of the gut-enteric nervous system [18, 19] or may even arise from the enteric nervous system via the microbiota-gut-brain axis [20], indicating the complexity of the bidirectional interaction network between the central nervous system and gut microbiota. Based on this study, Bacteroidetes, Firmicutes, and Proteobacteria are the dominant components of the gut microbiota in mice, consistent with previous reports [17, 18]. Clear differences were observed in gut microbiota composition between mice of different ages, suggesting some parallel shifts during aging. Indeed, the gut bacterial composition and ecological network change with aging and is proposed to play a crucial role in basic neurogenerative processes and maintain normal functions at different life stages [21]. For term infants, most of the early gut microbiota colonizers are derived from the mother; the early gut microbiota is mainly affected by the mode of delivery and feeding patterns [22, 23] and then gradually matures into an adult-like structure until the age of 1 to 3 years old [23, 24]. Likewise, gut microbiota compositions in an AD mouse model were also proved similar to Wt mice at the young age (3 months old) and started to diverge with aging [25]. Similar to these previous studies, no significant difference was observed in the gut microbiota between the matched littermate mice at 1 month in this study, which might be due to the same delivery mode and feeding patterns.
Microtubule associated tau protein is abnormally phosphorylated in AD and aggregates as paired helical filament (PHF)-tau in NFTs. In mice expressing normal human tau isoforms (Htau), young Htau mice with early stages of tau pathology showed no cognitive deficits, while in old Htau mice, with tau accumulation progresses and the presence of NFTs, cognitive function also declines [26], suggesting highly association between PHF-tau and cognitive impairment. In AD patients, the burden of PHF-tau measured by [F-18]-T807-PET significantly elevated and correlated with the severity stages defined by Braak staging. Interestingly, in a small normal cohort, aged subjects also presented a slightly higher PHF-tau burden than that in younger subjects, suggesting that PHF-tau may be associated with aging and cognitive damage [27]. In this study, pT231-tau protein was observed at 1 month in P301L mice, which is earlier than that of the alteration in the gut microbiota. However, AT8-tau protein was observed at 3 months of age, which was consistent with a previous report [28].
Homeostasis between Bacteroidetes and Firmicutes reportedly changes with aging [29]. According to a previous report, the relative abundance of Bacteroidetes decreased in patients with AD [30]. In an AD mouse model (APP/PS1), the ratio of Firmicutes to Bacteroidetes dynamically changing with aging, although no significant difference was observed when compared with their Wt littermates [17]. However, in another AD mouse model (5xFAD), the amount of Firmicutes was increased while Bacteroidetes was decreased in comparison with the Wt littermates at the age of nine weeks [18]. Undoubtedly, these composition changes are due to various factors. In our study, Bacteroidetes were increased while Firmicutes were decreased in P301L mice compared with those in their matched littermates at 3, 6, and 10 months of age. This difference may be partly due to the tau transgenic background. In mThy1.2 promoter-driven P301L mice, NFTs mainly accumulate in the amygdala, which is involved in mediating the effects of emotion and stress on learning and memory [31], as well as increased exploratory behavior [32]. In our previous study, significantly increased total distance and average speed were confirmed in P301L mice [2]. In fact, exercise has a complex impact on gut microbiota diversity and abundance. In mice, exercise could significantly increase the percentage of Bacteroidetes and decrease Firmicutes through several metabolic pathways [33]. In this sense, these species-disparate findings may be partly due to the behavioral dysfunction in P301L mice caused by tau pathology in the brain, indicating that tau pathology in the brain may affect the homeostasis of the gut microbiota. One possible mechanism may be the intestinal dysfunction caused by the presence of tau in the gut, resulting in gut microbiota disorder ultimately. As we know, enteric nervous system (ENS) is modulated by central nervous system. In APP transgenic AD mice, ENS alterations in molecule and function have been proved. The ENS undergoes significant changes during the early onset of APP expression. Notably, the high amount of Aβ within the ENS leads to neuronal tissue losses and significant disturbances in the gut wall [34]. It is reported that certain tau species are present in peripheral tissues, including skin, liver, and sigmoid colon, but the significance of its presence is still unclear [35, 36]. Thus, pathological changes in the brain may affect gut microbiota by regulating ENS. In addition, amygdala appears to play a prominent role in the development of stress-related gastrointestinal disorders [37]. In this study, accumulation of the PHF-tau in the area of amygdala may cause intestinal dysfunction and eventually lead to alteration of gut microbiota. Another possible mechanism may be infections associated with tauopathies. In AD patients, tissue from the brain was proved to exist fungal infection [38]. The dysbiosis of fungal was also proved to be correlated with intestinal dysfunction [39], suggesting that the existence of brain infection may be an ignored mechanism underlying the dyshomeostasis of the gut microbiota.
Notably, increasing evidence proposed that gut microbiota may be involved in the pathogenesis of brain diseases, especially in neurodegenerative diseases and aging [40–42]. Germ-free AD mice showed a drastic reduction of Aβ pathology in the brain when compared to those with gut microbiota, and implantation of microbiota from conventionally-raised AD mice increased brain Aβ levels [43].The results suggested that gut microbiota may contribute to the development of AD. Moreover, the germ-free condition may be another influencing factor for the reduction of Aβ pathology in the brain. In our study, all mice were raised under specific pathogen-free conditions to minimize the influences of other factors on the results as much as possible. This may lead to a decrease of tau pathological level in the brain, and the influence of the cleanliness of the living environment to tauopathies should be further explored.
Recently, increasing interest has been focused on the phyla Actinobacteria because of its proven involvement in the modulation of gut homeostasis, inflammatory immune changes, autoimmune responses, metabolism, and the gut-brain axis [44]. Potent metabolic activity against pathogens, especially its role in systemic diseases as well as its possible therapeutic applications, has also been proposed. In this study, Actinobacteria was decreased in P301L mice at 3 and 6 months of age. In an AD mouse model, the relative abundance of Actinobacteria was also proved to be decreased with age [17]. Bifidobacterium is a crucial class of this phylum that can produce γ-aminobutyric acid (GABA) in the human intestine [45]. As mentioned earlier, GABA, the major inhibitory neurotransmitter in the human CNS, has been implicated in a number of tauopathies, including AD [46], FTLD [47], and PSP [48]. The levels of GABA were decreased in the brains of patients with AD, FTLD, and PSP compared to those in normal controls [48–51]. This dysfunction of the brain GABAergic system has also been associated with cognitive impairment [52]. In fact, GABA concentrations in the CNS and gastrointestinal tract are linked. Decreased amounts of enteric Bifidobacterium can decrease the levels of GABA in the gut, ultimately leading to decreased GABA in the CNS [46]. However, the number of Bifidobacteria substantially decreases after weaning and continues to decrease with age [53, 54]. Similar changes were observed in Lactobacilli, a pivotal component of the Firmicutes phylum; together with Bifidobacteria, these changes represent the probiotic therapeutic approach. In middle-aged rats, Lactobacillus and Bifidobacterium dietary supplementation could enhances robust improvements in long-term object recognition memory by altering brain metabolites [55]. In this study, all mice were raised under specific pathogen-free conditions with free feeding and drinking water. The food and water provided were sterilized. No significant differences were observed in body weight, food intake, and water intake between the two groups of mice, indicating that the differences in gut microbiota may not be caused by abnormal feeding behavior or body weight.
In the current study, Lactobacillus and Bifidobacterium were significantly decreased in P301L mice at 3 and 6 months of age, respectively. Moreover, Lactobacillus was negatively correlated with the pathological changes in the brain of P301L mice at 6 months of age. These reduced Bifidobacterium and Lactobacilli in the gut might contribute to tau pathogenesis and cognitive impairment, suggesting that dyshomeostasis in the gut microbiota may contribute to the progression of tauopathy. The neuronal degeneration caused by gene mutation may be a crucial reason for the reduction of Bifidobacterium and Lactobacilli mediated by ENS as discussed above. The dysfunction of amygdala caused by the abnormal aggregation of tau protein may be another ignored mechanism. In addition, tau pathology caused by gene mutation may also cause dietary dysfunction and lead to the reduction of Bifidobacterium and Lactobacilli ultimately.
Staphylococcus, a genus of the Firmicutes phylum, has traditionally been considered harmful to the health of the host. Interestingly, Staphylococcus is an early and enriched gut colonizer of infants, especially in those residing in high hygienic areas recently. Additionally, this high colonization of Staphylococcus was proposed to have a potential role in the development of the microbiota and immune system [56]. In our study, Staphylococcus was decreased in P301L mice at 3 and 6 months of age when compared with that in Wt mice. Thus, it is becoming apparent that the abundance and physiological function of Staphylococcus in the gut microbiota is probably more complex and diverse than previously thought.
In this study, most of the bacteria taxa, such as Marvinbryantia, Lactobacillus, Streptococcus, unclassified_f_Erysipelotrichaceae, and Eubacterium_brachy_group were negatively correlated with tau pathology in the brain, suggesting the decrease of these microbiota may be closely related to the severity of tauopathies. However, the mechanism needs to be further studied.
Tauopathies encompass different disorders that may manifest with various clinical syndromes. Differential diagnosis with other proteinopathies remains challenging. In this study, a significant difference was observed in the gut microbiota between P301L and Wt mice, suggesting that the alteration in the gut microbiota may have potential value in the differentiation and early prediction of tauopathies. When used as a single microbial marker to predict tauopathies, Eubacterium_brachy_group showed higher AUC in comparison with Staphylococcus and Prevotellaceae_UCG_001. However, combination of Eubacterium_brachy_group, Staphylococcus, and Prevotellaceae_UCG_001 substantially represent the highest AUC for prediction of tauopathies among multiple markers. Early diagnosis and differentiation are of great clinical value for the prevention and treatment of tauopathies. For the young cohort at the early stage of tauopathies, combinations of Eubacterium_brachy_groupaphylococcus with Prevotellaceae_UCG_001 represent the highest AUC for prediction of tauopathies. Although more human-based studies should be explored, the above results suggested that the alteration in the gut microbiota may have potential value in the differentiation and early prediction of tauopathies.
This study was based on a mouse model; further investigations based on human tauopathy should be conducted to confirm our findings. Nevertheless, by recruiting well-matching subjects and controlling the conditions, our study identified alterations in the gut microbiota composition in a tauopathy model that were highly associated with tau pathology in the brain, although the causality remains unclear. As genome-wide association studies have revealed that genetic locus variation and epigenetics are highly associated with both familial and sporadic tauopathy [57], our study still has important value for human sporadic tauopathy research. Additionally, modulation of the gut microbiota may be a potential strategy for tauopathy treatment.