
Abstract
Alzheimer’s disease (AD) is caused by the accumulation of neurotoxic amyloid-β (Aβ) peptides. Aβ is derived from amyloid-β protein precursor (AβPP). In the non-amyloidogenic pathway, AβPP is cleaved by α-secretase and γ-secretase at the plasma membrane, excluding Aβ production. Alternatively, AβPP in the plasma membrane is internalized via endocytosis, and delivered to early endosomes and lysosomes, where it is cleaved by β-secretase and γ-secretase. Recent studies have shown that insulin in the periphery crosses the blood-brain barrier, and plays important roles in the brain. Furthermore, impaired insulin signaling has been linked to the progression of AD, and intranasal insulin administration improves memory impairments and cognition. However, the underlying molecular mechanisms of insulin treatment remain largely unknown. To investigate the effects of insulin on AβPP processing, we tested the effects of insulin on neuroblastoma SH-SY5Y cells overexpressing AβPP, and cultured rat cortical neurons. We found that insulin increased the level of cell surface AβPP, decreasing the endocytosis rate of AβPP. Insulin reduced Aβ generation through upregulation of AβPP O-GlcNAcylation via Akt insulin signaling. Our present data suggest that insulin affects Aβ production by regulating AβPP processing through AβPP O-GlcNAcylation. These results provide mechanistic insight into the beneficial effects of insulin, and a possible link between insulin deficient diabetes and cerebral amyloidosis in the pathogenesis of AD.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease caused by the accumulation of senile plaques that consist of neurotoxic amyloid-β (Aβ) peptides. Aβ is derived from the sequential proteolytic cleavage of amyloid-β protein precursor (AβPP) [1]. Aβ is regarded as the dominant factor of AD pathogenesis and is main target of the potential treatments [2–5]. However, Aβ has recently been questioned as the main culprit of AD. Although Aβ oligomers and Aβ fibrils are present in Aβ-overexpressing mice, and Aβ deposits and senile plaques are observed, degeneration of nerve cells and neuronal loss are not observed [6, 7]. It is known that Aβ amyloidosis due to AβPP metabolic impairment leads to neuroinflammation, and overexpression of AβPP promoted the aggregation of intracellular tau in cultured cells [8]. It is also suggested that AD is triggered by impairment of AβPP metabolism, and progress through tau pathology [9]. For these reasons, Aβ is still considered as the important parts of AD pathology and development.
AβPP can undergo two metabolic pathways: non-amyloidogenic and amyloidogenic. In the non-amyloidogenic pathway, AβPP is cleaved by α-secretase and γ-secretase at the plasma membrane, excluding Aβ production [10]. First, AβPP is cleaved by α-secretase into a secreted extracellular domain (sAβPPα) and a C-terminal fragment (C83). C83 is further cleaved by γ-secretase into P3 and AβPP intracellular domain. Alternatively, AβPP at the plasma membrane is internalized via endocytosis, and delivered to early endosomes and lysosomes, where it is cleaved by β-secretase and γ-secretase. β-Secretase cleaves AβPP into a secreted extracellular domain (sAβPPβ) and a C-terminal fragment (C99), which is further cleaved by γ-secretase into Aβ peptides [11]. β-Secretase is predominantly localized in the Trans-Golgi network, endosomes, and lysosomes [12]. These endosomal compartments are at low pH condition, which provides a more favorable acidic environment for β-secretase activity [13]. Thus, AβPP endocytosis and its trafficking are considered critical for the regulation of AβPP metabolism and Aβ production [14–17].
Insulin and insulin receptors (IR) were thought to be restricted to the periphery, while the brain had been considered to be an insulin-insensitive organ. However, insulin in the periphery crosses the blood-brain barrier (BBB), and plays important roles in the brain [18]. IR and glucose transporters were also discovered in the brain. In the central nervous system (CNS), insulin signaling regulates neuronal and glial functions, including synaptogenesis, synaptic plasticity, gene expression, and cognition [19]. Furthermore, impaired insulin signaling has been linked to the progression of AD [20–25]. AD patients have defective IR expression and IR binding, reduced IR substrate-1 (IRS1) and IR substrate-2 (IRS2) expression, and increased levels of serine phosphorylation of IRS-1, indicating insulin resistance [23, 26]. Intranasal administration of insulin is an effective method that bypasses the BBB, and directly reaches the brain, thereby avoiding side effects [27]. Several clinical trials have shown that intranasal insulin primarily improves hippocampus-dependent memory function [28, 29], as well as cognitive impairment in AD patients [30]. Much evidence has indicated that insulin affects the metabolism and processing of Aβ [31–35]. However, the underlying molecular mechanisms of beneficial insulin treatment for AD remain largely unknown.
As a post-translational modification, O-GlcNAcylation is a novel type of O-glycosylation. A monosaccharide, N-acetylglucosamine, is attached to serine or threonine residues of proteins by O-GlcNAc transferase (OGT) [36]. O-GlcNAc is removed by O-GlcNAcase (OGA) [37]. O-GlcNAcylation can be found on proteins in the cytoplasmic, nuclear, mitochondrial, and plasma membrane compartments, and regulates physiological cellular functions, such as transcription, epigenetic modifications, and cell signaling [38]. Remarkably, these two enzymes regulating protein O-GlcNAcylation are expressed up to 10-times higher in the brain, compared to in peripheral tissue [39]. Therefore, O-GlcNAcylation might play important roles in neuronal physiology. There is growing evidence that O-GlcNAcylation is involved in neurological diseases, such as AD, Parkinson’s disease, and stroke. In the brains of AD patients, the levels of O-GlcNAcylation are decreased [40]. In addition, the levels of O-GlcNAcylation are regulated by glucose metabolism, which is impaired in the brains of AD patients [41]. AβPP is one of the many proteins modified by O-GlcNAcylation [42]. Increasing O-GlcNAcylated AβPP levels by the inhibition of OGA increases sAβPPα levels, and decreases Aβ levels [43]. In addition, we recently reported the role of O-GlcNAcylation in AβPP processing, and its effect on Aβ production [44]. PUGNAc, an inhibitor of OGA, decreases the AβPP endocytosis rate. Consequently, AβPP O-GlcNAcylation increases the levels of cell surface AβPP, resulting in decreased Aβ generation.
It is reported that insulin increases the O-GlcNAcylation of proteins by increasing the expression of OGT [45]. However, it is not known whether insulin affects the level of O-GlcNAcylated AβPP. The underlying insulin signaling for AβPP O-GlcNAcylation is also not known. In this study, we used neuroblastoma SH-SY5Y and embryonic cortical neurons of Sprague Dawley rats. We showed that insulin increases the levels of AβPP O-GlcNAcylation, and decreases the rate of AβPP endocytosis. As a result, the level of cell surface AβPP is increased, and Aβ generation is reduced. These results are also confirmed in primary cultured cortical neurons. Consequently, our present data could explain the molecular mechanism of insulin effects on AβPP processing and metabolism, and provide a possible link between insulin deficient diabetes and cerebral amyloidosis in the pathogenesis of AD.
MATERIALS AND METHODS
Cell culture and experimental treatments
SH-SY5Y cells transfected with wild type AβPP and BACE1 (SH-SY5Y-AβPP/BACE1) were cultured at 37°C in an atmosphere of 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) containing 100 units/ml penicillin and 260 μg/ml Zeocin. Insulin (Insulin, Human Recombinant Zinc, Gibco) was added to the culture medium for 2 h. Akti (Akt inhibitor, Calbiochem) and OSMI-1 (OGT inhibitor, Sigma-Aldrich) were treated in the culture medium for 2 h. Dynasore (Tocris Bioscience) and chlorpromazine (Sigma-Aldrich) were treated in the culture medium with 1 μM insulin for 2 h.
Immunoprecipitation
After washing in ice-cold phosphate buffered saline (PBS), cells were lysed in a buffer composed of 50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1% NP-40, 0.5% sodium deoxycholate, and a protease inhibitor mixture (pH 8.0). Cell lysates were centrifuged at 10,000×g at 4°C for 10 min, and protein concentrations were measured using the Bradford protein assay (Bio-Rad). Total protein (300 μg) was incubated with 4 μl of antibody against O-GlcNAc (Biolegend) and Protein G agarose, Fast Flow (Millipore) for 2 h at room temperature (RT), and then the immunoprecipitated samples were washed three times with PBS, and detected by western blot using AβPP antibody.
Protein extraction and Western blotting
Cells were lysed in buffer composed of 50 mM HEPES, 100 mM NaCl, 1% Triton X-100, 1 mM sodium orthovanadate, and a protease inhibitor mixture (pH 7.2), using a 23-gauge needle. Cell lysates were obtained by centrifugation at 10,000×g at 4°C for 10 min, and protein concentrations were determined by Bradford protein assay (Bio-Rad). Protein from each sample was resolved by 6 or 8% SDS-PAGE, and transferred to a nitrocellulose membrane. The membrane was placed in blocking solution containing 5% non-fat milk powder in Tris-buffered saline/Tween 20 for 1 h at RT, prior to incubation with a 1:2,000 dilution of anti-AβPP (6E10; SIG-39320, Covance) and 1:2,000 dilution of mouse anti β-actin (T2200, Sigma-Aldrich) overnight at 4°C. Blots were washed with Tris-buffered saline/Tween 20, and incubated with 1:2,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit IgG antibody or goat anti-mouse IgG antibody (both from Invitrogen) for 1 h at RT, and washed. Peroxidase activity was measured with enhanced chemiluminescence. The detected signals were analyzed with Multi Gauge software using a LAS-3000 system (Fuji Film).
Cell surface biotinylation
Cells were washed with PBS, and incubated in PBS supplemented with 0.25 mg/ml Sulfo-NHS-SS-biotin (Thermo) for 10 min at 4°C. To remove excessive biotin reagent, cells were washed three times with PBS. Cells were lysed with lysis buffer (50 mM HEPES, 100 mM NaCl, 1% Triton X-100, 1 mM sodium orthovanadate, with protease inhibitor mixture, pH 7.2) for 1 h at 4°C. Biotinylated proteins were pulled down using streptavidin-agarose slurry (Millipore) at 4°C for 3 h. After washing, the bound material was analyzed by western blot.
Primary antibody uptake
Cells were grown on glass cover slips coated with poly-D-lysine in 35 mm culture dishes. The following day, the cells were washed three times with PBS for 5 min. Then, cells were incubated in PBS with 6E10 antibody (1:100 dilution) for 45 min at 4°C for the labeling of surface AβPP. Next, cells were washed in ice-cold PBS, and incubated at 37°C for 0, 5, 10, and 30 min. Cells were fixed in 4% paraformaldehyde at RT for 15 min, washed, and then permeabilized with 0.1% Triton X-100 for 5 min. Cells were blocked with 2% goat serum in PBS for 1 h at RT. Then, cells were washed, and incubated with goat anti-mouse antibodies conjugated with Alexa Fluor 488 in blocking buffer for overnight. Finally, cells were washed in PBS, mounted with mounting medium (S3025, DakoCytomation), and left overnight at 4°C to dry. Immunofluoresence staining was captured by confocal microscopy (LSM510, Zeiss). Images were analyzed with the Image J program to measure mean fluorescence intensity values from the region of interest according to the cell shape.
To separate the cell surface AβPP from the internalized AβPP, we used two different secondary antibodies conjugated with Alexa 488 and Alexa 647. First, cells were washed three times with PBS for 5 min, followed by incubating with 6E10 antibody (1:100 dilution) for 45 min at 4°C for the labeling of surface AβPP. Next, cells were washed in ice-cold PBS, and incubated at 37°C for 5, 10, and 30 min to allow endocytosis. Then, cells were washed, and incubated with goat anti-mouse antibodies conjugated with Alexa Fluor 647 at 4°C for the labeling of remaining AβPP in the cell surface. Cells were fixed in 4% paraformaldehyde at RT for 15 min, washed, and then permeabilized with 0.1% Triton X-100 for 5 min. Cells were blocked with 2% goat serum in PBS for 1 h at RT. Then, cells were washed, and incubated with goat anti-mouse antibodies conjugated with Alexa Fluor 488 in blocking buffer for overnight to detect internalized AβPP.
Transferrin internalization
To measure transferrin internalization using fluorescence microscopy, cells were incubated in PBS with 0.1% bovine serum albumin (BSA) containing 25 μg/ml Alexa Fluor 488-conjugated transferrin (T-13342, Invitrogen) for various time periods at 37°C. Then, transferrin was stripped from plasma membrane by pH 5.5 buffer (100 mM sodium acetate, 50 mM NaCl) for 5 min. Cells were washed with PBS, and fixed in 4% paraformaldehyde for 15 min. Immunofluorescence staining was captured by confocal microscopy (LSM510, Zeiss). Images were analyzed with the Image J program to measure mean fluorescence intensity values from the region of interest according to the cell shape.
To measure the fluorescence intensity of internalized transferrin, cells were incubated in PBS with 0.1% BSA containing 25 μg/ml Alexa Fluor 488-conjugated transferrin for various time intervals at 37°C. Next, cells were washed with PBS, and the extracellular Alexa488-transferrin was removed by acidic treatment. The cells were lysed in lysis buffer (50 mM Hepes, 100 mM NaCl, 1% Triton X-100, 1 mM sodium orthovanadate, with protease inhibitor mixture, pH 7.2) and cell lysates were resolved in SDS-PAGE. The fluorescence intensity of the bands was detected by laser fluorescence scanner (Fluor Chem Q, Alpha Innotech). For comparison, fluorescence intensity of bands was normalized to the control cells at 30 min (control at 30 min = 100%).
Preparation of the primary cortical neuron
Primary neuronal cultures were prepared from the 18-day-old embryo of Sprague Dawley rats. The animal protocols used in this study were accordance with and granted by the Sungkyunkwan University Animal Care and Ethics Committee. Briefly, dissociated cortical cells were plated onto poly-D-lysine-coated coverslips in DMEM, supplemented with 10% FBS containing 100 units/ml penicillin. After 3 h, neurons were maintained in Neurobasal medium with B27 supplement (Invitrogen) at 37°C with 5% CO2 for up to one week. Experiments were performed in fresh Neurobasal medium.
sAβPPα, sAβPPβ, Aβ42 peptide assay
Cells at 80% confluence were incubated with insulin or DMSO for control in DMEM culture medium for 2 h. The medium was analyzed by specific ELISA (IBL) for the detection of sAβPPα and sAβPPβ-w, according to the supplier’s instructions. The levels of Aβ42 were measured from conditioned medium using specific sandwich ELISA (Millipore).
Statistical analysis
Data are expressed as mean±SEM. We conducted statistical analysis using one way ANOVA between the controls and the drug-treated groups. p < 0.05 was considered as statistically significant.
RESULTS
Insulin increased O-GlcNAcylated proteins including AβPP
It is reported that insulin increases the O-GlcNAcylation of proteins by increasing the expression of OGT [45]. Since AβPP is one of the many proteins modulated by O-GlcNAcylation, and GlcNAcylation of AβPP leads to non-amyloidogenic pathway [44], we hypothesized that insulin affects AβPP processing or trafficking. To test this possibility, we used SH-SY5Y cells stably transfected with wild type AβPP and BACE1 (SH-SY5Y-AβPP/BACE1). Cells were incubated with insulin for 2 h, and the levels of O-GlcNAcylated proteins were measured by western blotting as shown for a typical result in Figure 1A. The densitometric analysis of western blots shows that the levels of O-GlcNAcylated proteins were significantly increased by insulin in a dose-dependent manner (Fig. 1B; n = 8). We also confirmed that insulin increased the level of OGT by 77% in our experimental condition (Supplementary Figure 1). Then, we examined whether the effect of insulin on O-GlcNAcylation of proteins was mediated by OGT. SH-SY5Y-AβPP/BACE1 cells were treated with 1 μM insulin for 2 h in the presence of 50 μM OSMI-1, an inhibitor of OGT [46]. OSMI-1 alone decreased levels of O-GlcNAcylated proteins by 38 ± 13.4%, which might indicate that OSMI-1 inhibited the basal OGT activity (Supplementary Figure 2). OSMI-1 completely inhibited the effect of insulin on levels of O-GlcNAcylated proteins. Thus, these results suggested that insulin increased the levels of O-GlcNAcylated proteins via OGT.
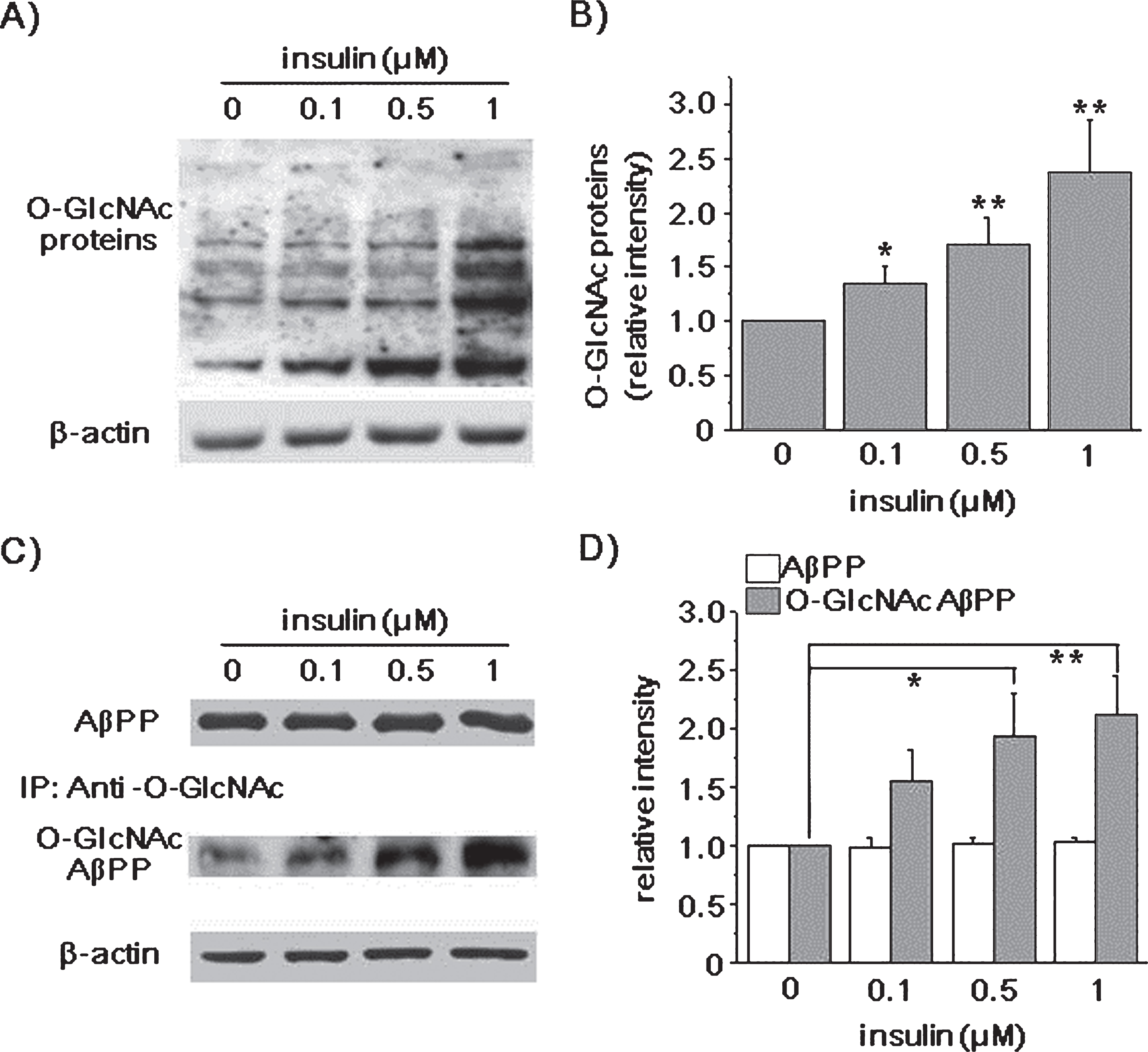
Insulin increased the levels of O-GlcNAcylated AβPP. SH-SY5Y-AβPP/BACE1 cells were incubated with 0.1, 0.5, and 1 μM insulin for 2 h in the presence of 10% FBS, and cell lysates were prepared. A) A representative western blot shows the levels of O-GlcNAcylated proteins detected using antibody against O-GlcNAc. β-actin was used as a loading control. B) Bars correspond to the densitometric analysis of O-GlcNAc protein levels (0.1 μM insulin relative density [RD], 1.34; 95% confidence interval [CI], 0.92–1.49; p = 0.023;0.5 μM insulin RD, 1.61; 95% CI, 1.41–1.88; p < 0.01; 1 μM insulin RD, 2.19; 95% CI, 1.77–2.60; p < 0.01; n = 8). C) A representative western blot shows the levels of AβPP. Also shown is O-GlcNAcylated AβPP, which was obtained by immunoprecipitation with O-GlcNAc antibody, and probing with AβPP antibody. Insulin increased levels of O-GlcNAcylated AβPP, while AβPP levels were not changed. D) The levels of AβPP and O-GlcNAcylated AβPP were quantified by densitometric analysis of the bands in (C) (0.5 μM insulin RD, 1.93; 95% CI, 1.45–2.04; p = 0.03; 1 μM insulin RD, 2.12; 95% CI, 1.71–2.76; p < 0.01; n = 9). One-way ANOVA: *p < 0.05; **p < 0.01.
We next used immunoprecipitation method to detect O-GlcNAcylated AβPP, and tested the effect of insulin. Since a specific antibody against O-GlcNAcylated AβPP was not available, we pulled down O-GlcNAcylated proteins using antibody against O-GlcNAc, and AβPP antibody was used to measure O-GlcNAcylated AβPP. We found that the level of full length AβPP was not changed by insulin, as shown in (Fig. 1C and D). However, the levels of O-GlcNAcylated AβPP were increased in the presence of 0.5 and 1 μM insulin by 93% and 112% (n = 9), respectively. The specificity of the O-GlcNAc antibody used for immunoprecipitation was tested using O-GlcNAc competition assay. The detection of O-GlcNAcylated AβPP was blocked by the presence of GlcNAc, validating the specificity of the O-GlcNAc antibody we used (Supplementary Figure 3). As another way to measure the levels of AβPP O-GlcNAcylation, we used the analogue of natural substrate, in which N-acyl side chain has been modified to bear a bio-orthogonal azide moiety, N-azidoacetyl glucosamine (GlcNAz) [47]. The metabolic incorporation of the azido sugar into the O-GlcNAcylation site of AβPP was covalently derivatized to phosphine-PEG3-biotin (Supplementary Figure 4). We found that insulin increased the levels of GlcNAz-AβPP, supporting that insulin increased AβPP O-GlcNAcylation.
Insulin promoted non-amyloidogenic pathway of AβPP
Some post-translational modifications, including phosphorylation, affect AβPP processing and trafficking both in vitro and in vivo [48-50]. The O-GlcNAcylation of AβPP is shown to decrease amyloidogenic pathway of AβPP [44]. We tested whether the upregulation of AβPP O-GlcNAcylation by insulin also affects the processing of AβPP by measuring α-, β-, and γ-secretase products from the conditioned media. For this purpose, SH-SY5Y-AβPP/BACE1 cells were incubated with insulin for 2 h, and the levels of sAβPPα, sAβPPβ, and Aβ42 were measured using ELISA kits. Insulin significantly increased sAβPPα and decreased sAβPPβ levels in a dose-dependent manner (Fig. 2A; n = 7). Insulin at 1 μM reduced the secreted Aβ42 levels by 20 ± 2.7% (n = 7; Fig. 2B). Thus, these results suggest that insulin decreased the products of β-secretase, and promoted non-amyloidogenic pathway.

Insulin decreased levels of β-secretase products. SH-SY5Y-AβPP/BACE1 cells were incubated with 0.1, 0.5, and 1 μM insulin for 2 h in the presence of 10% FBS. The levels of secreted sAβPPα, sAβPPβ, and Aβ42 were measured from the conditioned media using ELISA methods. A) Insulin increased the levels of sAβPPα (0.1 μM insulin, 108.41%; 95% CI, 101.28–111.28; p = 0.02; 0.5 μM insulin, 120.61%; 95% CI, 112.61–120.92; p < 0.001; 1 μM insulin, 127.21%; 95% CI, 122.57–136.49; p < 0.001; n = 7),while decreased the levels of sAβPPβ (0.5 μM insulin, 89.42%; 95% CI, 88.23–94.70; p < 0.01; 1 μM insulin, 85.39%; 95% CI, 78.62–90.05; p < 0.01; n = 7). B) Insulin also reduced the secreted Aβ42 levels (0.1 μM insulin, 81.49%; 95% CI, 81.45–92.83; p < 0.001; 0.5 μM insulin, 68.68%; 95% CI, 76.03–86.61; p < 0.001; 1 μM insulin, 80.93%; 95% CI, 65.80–83.39; p < 0.001; n = 7). One-way ANOVA: *p < 0.05; **p < 0.01; ***p < 0.001.
Since we observed that AβPP O-GlcNAcylation by insulin treatment affects the AβPP metabolism, we tested whether insulin alters the expression levels of α- and β-secretase, such as ADAM9, ADAM17 (α-secretases), and BACE1 (β-secretase). Insulin showed no effects on the expression levels of ADAM9, ADAM17, and BACE1 in our experimental condition (lementary Figure 5), indicating that the effects of insulin on AβPP processing were not mediated by the expression changes of these secretases.
Then, we examined whether the effects of insulin on AβPP O-GlcNAcylation and Aβ42 were mediated by OGT. SH-SY5Y-AβPP/BACE1 cells were pre-treated with OGT inhibitor, and were incubated with insulin for 2 h. As expected, insulin significantly increased the level of AβPP O-GlcNAcylation without changes in AβPP level, as shown for a typical experiment (Fig. 3A), and the densitometric analysis of western blots (Fig. 3B, C; n = 8). OGT inhibitor (OSMI-1) alone reduced the level of AβPP O-GlcNAcylation, which was similar to the effect of OSMI-1 on O-GlcNAcylated proteins (Supplementary Figure 2). Also, OSMI-1 alone increased Aβ42 level by 34 ± 8.5% (n = 8), compared to control (Fig. 3D). This might be due to the inhibitory effect of OSMI-1 on the basal OGT activity for AβPP O-GlcNAcylation. More importantly, the presence of OSMI-1 completely blocked the effects of insulin on AβPP O-GlcNAcylation, and Aβ42 level (Fig. 3 C, D; n = 8). Thus, these results suggest that the effects of insulin on AβPP O-GlcNAcylation and Aβ42 production were mediated by OGT.

The effects of insulin on AβPP O-GlcNAcylation and Aβ42 were mediated via OGT. An OGT inhibitor, OSMI-1, was added to the culture media of SH-SY5Y-AβPP/BACE1 cells. Cells were incubated for an additional 2 h with or without 1 μM insulin. A) The representative western blot shows the levels of total AβPP and O-GlcNAcylated AβPP in different conditions. β-actin was used as a loading control. B) The expression levels of total AβPP were not changed (n = 8). The effects of insulin on O-GlcNAcylated AβPP (C) (insulin RD, 1.39; 95% CI, 1.12–1.67; p = 0.0049; OSMI-1 RD, 0.86; 95% CI, 0.76–0.95; p = 0.03; insulin and OSMI-1 RD, 0.83; 95% CI, 0.76–0.90; p < 0.01; n = 8) and Aβ42 (D) were inhibited in the presence of OSMI-1 (insulin, 85.36%; 95% CI, 85.22–90.85; p < 0.001; OSMI-1, 133.91%; 95% CI, 123.14–143.12; p < 0. 01; insulin and OSMI-1, 132.91%; 95% CI, 108.89–147.06; p = 0.02; n = 8). One-way ANOVA: *p < 0.05; **p < 0.01; ***p < 0.001.
Insulin promoted non-amyloidogenic pathway via Akt signaling
It is well known that insulin has various signal pathways, such as PI3K/Akt and mitogen-activated protein kinase (MAPK) signaling. Akt, also known as protein kinase B (PKB), is a serine- and threonine-specific protein kinase that plays a key role in many cellular processes, such as glucose metabolism, apoptosis, cell proliferation, transcription, and cell migration [51]. It has been reported that insulin controls the expression and localization of OGT via Akt signaling [45]. If Akt is involved in the effects of insulin on AβPP O-GlcNAcylation and AβPP processing, we expected that Akti, a known Akt inhibitor [52], might inhibit the effects of insulin on AβPP O-GlcNAcylation, as well as Aβ generation. First, we tested the effect of Akti on OGT expression. The effect of insulin on OGT expression was blocked by Akti (Supplementary Figure 1), confirming that insulin controls the expression of OGT via Akt signaling [45]. Consistent with our previous results, insulin treatment significantly increased the level of AβPP O-GlcNAcylation without changes in AβPP levels, as shown for a typical result (Fig. 4A), and the densitometric analysis of western blots (Fig. 4B, C; n = 6). Also, insulin decreased Aβ42 level (Fig. 4D; n = 6). Akti alone reduced the level of AβPP O-GlcNAcylation by 26 ± 10.7%, while Aβ42 level was not changed. Akti might inhibit the basal OGT activity toward AβPP O-GlcNAcylation, without affecting Aβ42 level. However, the presence of Akti completely inhibited the effects of insulin on AβPP O-GlcNAcylation and Aβ42 level (Fig. 4C, D; n = 6). Thus, these results strongly suggest that the effects of insulin on AβPP O-GlcNAcylation and Aβ42 level were via Akt signaling, which controls the expression of OGT.
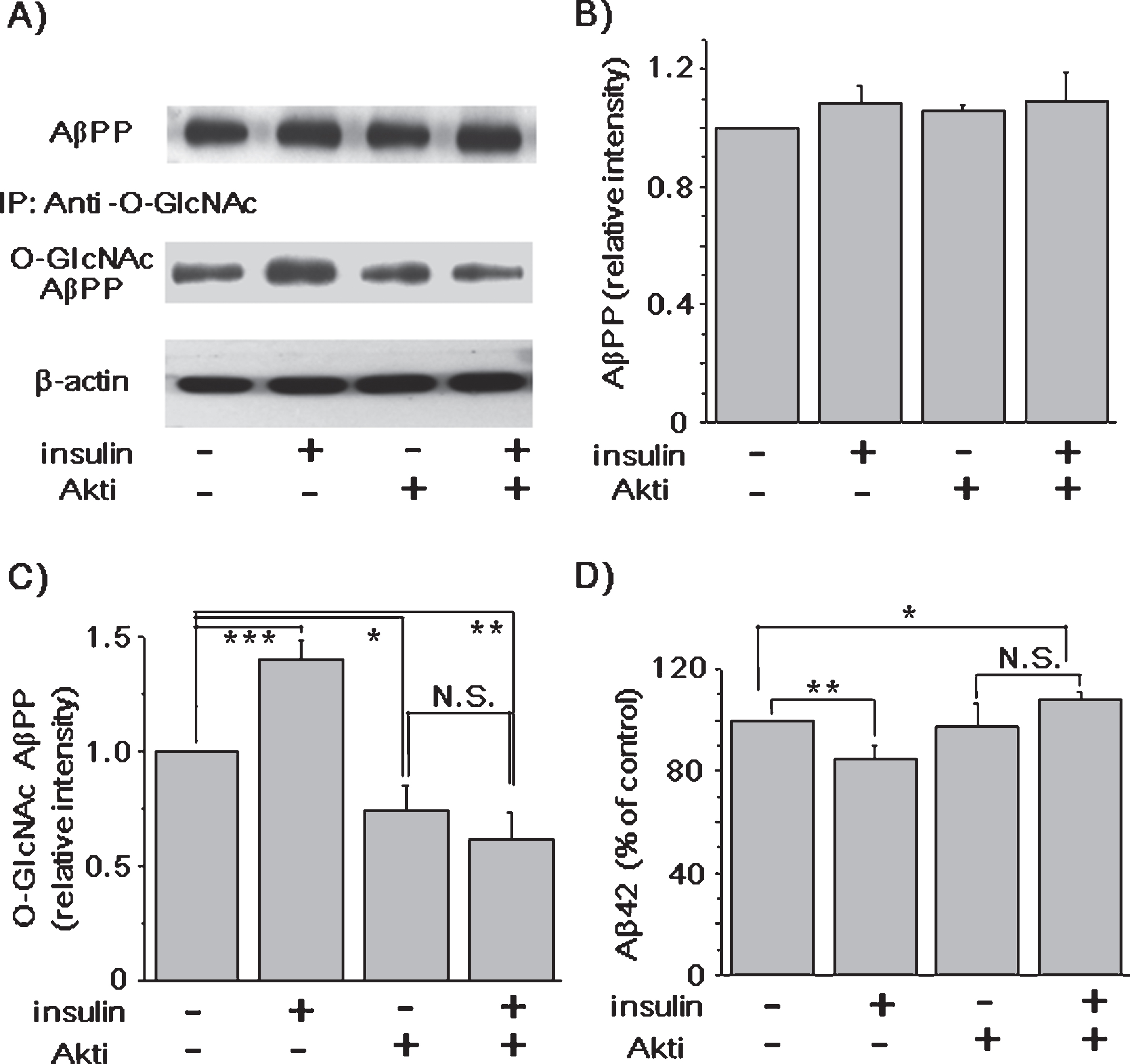
The effects of insulin on AβPP O-GlcNAcylation and Aβ42 were mediated via Akt signaling. An Akt inhibitor (Akti) was added to the culture media of SH-SY5- AβPP/BACE1 cells. Cells were incubated for an additional 2 h with or without 1 μM insulin. A) A representative western blot shows the levels of total AβPP and O-GlcNAcylated AβPP. β-actin was used as a loading control. B) The levels of total AβPP were not changed in different conditions. The effects of insulin on the levels of O-GlcNAcylated AβPP (C) (insulin RD, 1.40; 95% CI, 1.26–1.53; p < 0.001; Akti RD, 0.74; 95% CI, 0.58–0.91; p = 0.03; insulin and Akti RD, 0.62; 95% CI, 0.44–0.80; p < 0.01; n = 6) and Aβ42 (D) were inhibited in the presence of Akti (insulin, 85.07%; 95% CI, 77.08–93.07; p < 0. 01; insulin and Akti, 107.78%; 95% CI, 103.01–112.55; p = 0. 03; n = 6). One-way ANOVA: *p < 0.05; **p < 0.01; ***p < 0.001.
AβPP O-GlcNAcylation by insulin increased the level of cell surface AβPP and reduced its endocytosis rate
Only a small portion of AβPP is detected at the plasma membrane, and the majority of AβPP is internalized within 10 min [53, 54]. α-Secretase is particularly enriched at the cell surface, and competes with BACE1 for AβPP processing [55], while β-secretase is rapidly internalized from the cell surface [56, 57], and predominantly localized in the Trans-Golgi network and endosomes [12]. Since the enzyme activity of BACE1 is greater in acidic compartments than in neutral pH environments, Aβ generation would occur favorably in late endosomes or lysosomes [58]. Therefore, the majority of cell surface AβPP is processed via the non-amyloidogenic pathway, while intracellular AβPP processing predominantly involves the amyloidogenic pathway [59, 60]. These studies indicate that both AβPP localization and AβPP trafficking play a key role in AβPP metabolism. Consistent with these studies, we recently reported that the O-GlcNAcylation of AβPP increased the steady-state levels of cell surface AβPP, and promoted non-amyloidogenic pathway [44]. We examined whether AβPP O-GlcNAcylation by insulin also affected the level of AβPP at the plasma membrane. SH-SY5Y-AβPP/BACE1 cells were treated with insulin for 2 h, and cell surface proteins were biotinylated at 4°C. After lysis, cell surface proteins were isolated by using streptavidin beads, and analyzed by western blot using AβPP antibody. Insulin significantly increased the steady-state level of cell surface AβPP by 73% and 77% in the presence of 0.5 and 1 μM insulin, whereas the level of total AβPP was not changed, as shown for a typical result in (Fig. 5A). Results from the densitometric analysis of western blots are shown in (Fig. 5B) (n = 8). These results clearly show that insulin increased AβPP level in the plasma membrane.
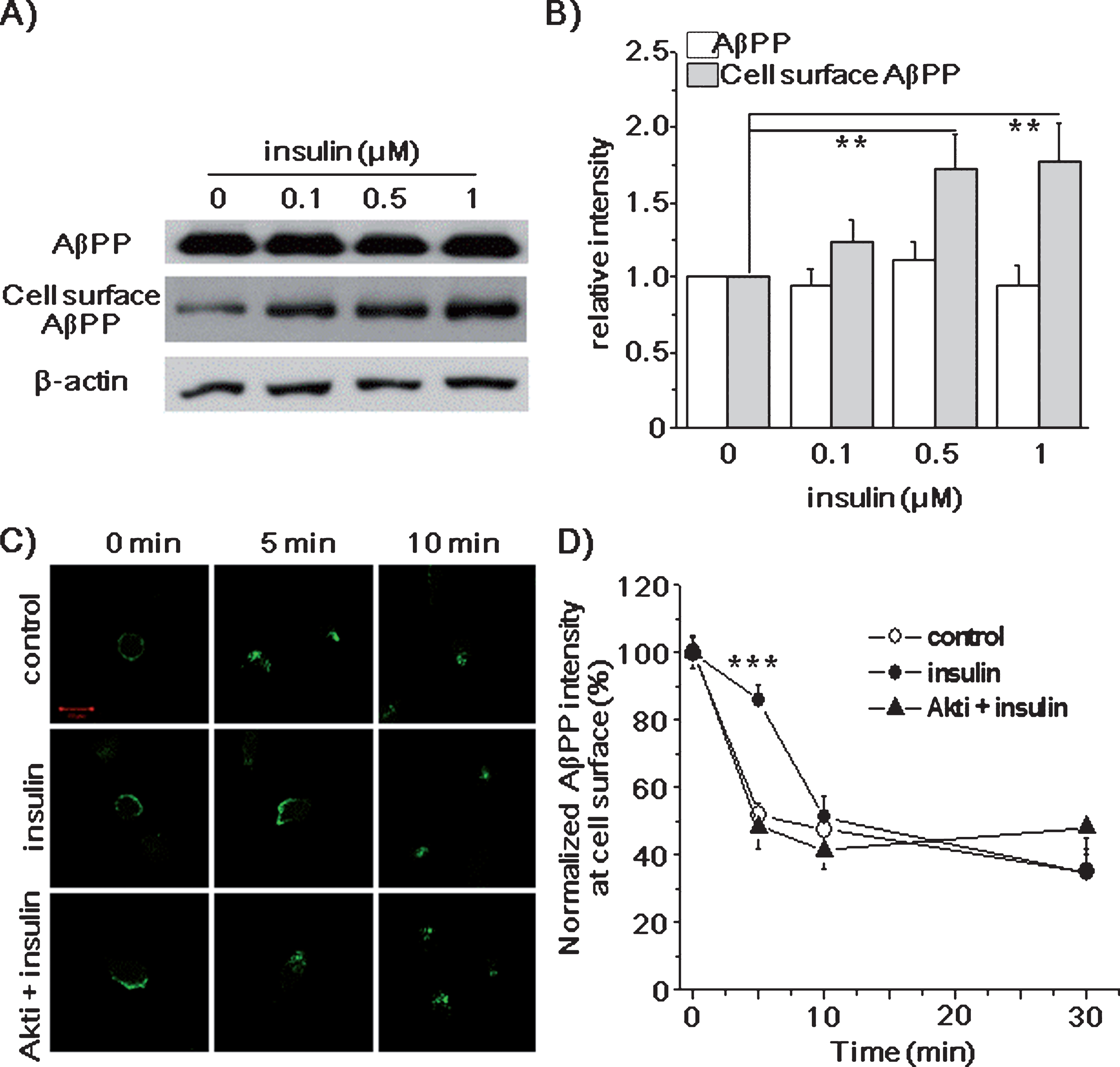
Insulin increased the level of cell surface AβPP and decreased its endocytosis rate. SH-SY5Y-AβPP/BACE1 cells were treated with 0.1, 0.5, and 1 μM insulin for 2 h. A) Levels of AβPP were detected using western blot from the prepared cell lysates. The levels of cell surface AβPP was measured by using cell surface biotinylation method. β-actin was used as a loading control. B) Densitometric analysis of the bands in (A) shows that the levels of total AβPP were not changed, while the levels of cell surface AβPP were increased by insulin (0.5 μM insulin RD, 1.73; 95% CI, 1.37–1.78; p < 0.01; 1 μM insulin RD, 1.77; 95% CI, 1.64–2.37; p < 0.01; n = 8). C, D) SH-SY5Y-AβPP/BACE1 cells were incubated with 1 μM insulin with or without 10 μM Akt inhibitor (Akti) for 2 h. Then cells were incubated at 4°C with the 6E10 antibody to label cell surface AβPP. Cells were transferred to 37°C for 0, 5, 10, and 30 min to permit endocytosis. Then, cells were fixed, permeabilized, and incubated with fluorescent conjugated secondary antibody. C) AβPP located at the cell surface before starting endocytosis (0 min). After 5 min at 37°C, AβPP was rapidly internalized in control cells, while most AβPP remained at the cell surface in insulin-treated cells. The effects of insulin on AβPP endocytosis were blocked by Akti. Scale bar is 10 μm. D) Fluorescence intensities of cell surface AβPP in (C) were analyzed by using Image software (intensity of insulin at 5 min, 85.61%; 95% CI, 83.85–86.92; p < 0.001; n = 20). One-way ANOVA: **p < 0.01; ***p < 0.001.
Since the effect of insulin on AβPP O-GlcNAcylation increased the level of cell surface AβPP, insulin might affect the endocytosis rate of AβPP, as we reported previously [44]. To test this possibility, we first measured the endocytosis rate of AβPP by using antibody uptake method. SH-SY5Y-AβPP/BACE1 cells were pre-treated with 1 μM insulin for 2 h, and then treated with the AβPP antibody at 4°C. After washing, cells were incubated at 37°C for 5, 10, and 30 min to allow endocytosis. Then, cells were fixed, permeabilized, and visualized by using a fluorescent conjugated secondary antibody. Figure 5 C shows typical immunoreactivities for AβPP. Fluorescence intensities at the plasma membrane were analyzed as shown in Figure 5D from 5 different experiments (n = 20). AβPP was observed at the cell surface membrane before endocytosis was initiated in both control and insulin-treated cells (0 min). At 5 min, only 52±3.5% of AβPP was localized at the plasma membrane in control cells, indicating the rapid internalization of AβPP [59]. However, in insulin-treated cells, 86±4.8% of AβPP remained at the plasma membrane at 5 min. These results indicate that the AβPP endocytosis rate was significantly reduced by insulin. After 10 and 30 min, no difference in AβPP endocytosis rate between control and insulin-treated cells was observed. This may be due to the relatively fast cleavage of AβPP, once it is internalized [61]. As another way to measure the AβPP endocytosis rate, we distinguished the cell surface AβPP from the internalized AβPP by using two different secondary antibodies conjugated with Alexa 488 and Alexa 647 (Supplementary Figure 6). Unlike in control cells, AβPP remained at the plasma membrane at 5 min in the insulin-treated cells, indicating that the AβPP endocytosis rate was significantly reduced by insulin.
Next, we tested the effect of insulin on the endocytosis rate of AβPP from Akt inhibitor-treated or OGT inhibitor-treated cells. The effect of insulin on AβPP endocytosis rate was inhibited by the presence of Akti (Fig. 5 C, D; n = 20). Similar results were obtained when we used OGT inhibitor (Supplementary Figure 7). Taken together, these data indicate that the effect of insulin on AβPP endocytosis is mediated by OGT and Akt signaling.
Many proteins are O-GlcNAcylated, such as cytoplasmic, nuclear, and mitochondrial proteins. Also, it has been reported that proteins involved in endocytosis, such as adaptor protein 80 (AP-180) and adaptor protein 3 (AP-3), are O-GlcNAcylated [62, 63]. Since transferrin is internalized through clathrin-dependent endocytosis similar to AβPP [64], we tested the effect of insulin on transferrin. SH-SY5Y-AβPP/BACE1 cells were treated with insulin for 2 h, and then incubated with Alexa 488-transferrin at 37°C to measure the endocytosis rate by using fluorescence microscopy. We also used the biochemical method to quantify the effect of insulin on transferrin internalization. Using these two different experimental approaches, the endocytosis rate of transferrin was not altered by insulin treatment (Supplementary Figure 8). These results suggest that the effect of insulin is specific for AβPP endocytosis, and that insulin does not inhibit clathrin-dependent endocytosis itself.
Next, to confirm that the effect of insulin on Aβ42 generation was mainly mediated by the inhibition of AβPP endocytosis, we used inhibitors for endocytosis, dynasore and chlorpromazine (CPZ). Dynasore is an inhibitor of dynamin, a component for clathrin-dependent endocytosis [65], while CPZ induces a redistribution of clathrin coated vesicle component, leading to the disruption of endocytosis [66]. Insulin significantly decreased Aβ42 levels, consistent with our previous result. However, the presence of dynasore or CPZ completely prevented the insulin effect on Aβ42 level (Supplementary Figure 9). These results support the conclusion that the reduced endocytosis rate of AβPP by insulin may be the underlying mechanism for the effects of insulin on Aβ42 generation.
Insulin promotes the non-amyloidogenic processing of AβPP from cultured cortical neurons
To test the effects of insulin on the primary cortical neurons, we used Sprague Dawley rats. Since we used non-transgenic normal rats, both AβPP and Aβ42 were endogenous. Cortical neurons were treated with insulin for 2 h, and cell lysates were immunoprecipitated with O-GlcNAc antibody (Fig. 6A). The levels of O-GlcNAcylated AβPP were significantly increased by insulin (Fig. 6B; n = 4). Next, we examined whether insulin affected the level of cell surface AβPP of cortical neurons by using biotinylation method. The levels of cell surface AβPP were increased in a dose-dependent manner (Fig. 6C; n = 4). The levels of endogenous sAβPPα and sAβPPβ were too low to detect in the neuronal culture media. The levels of Aβ42 were reduced by 28±2.6% in the presence of 1 μM insulin (Fig. 6D; n = 8). Thus, these results are consistent with the conclusion that the O-GlcNAcylation of AβPP by insulin promoted non-amyloidogenic AβPP processing via the inhibition of endocytosis of AβPP.
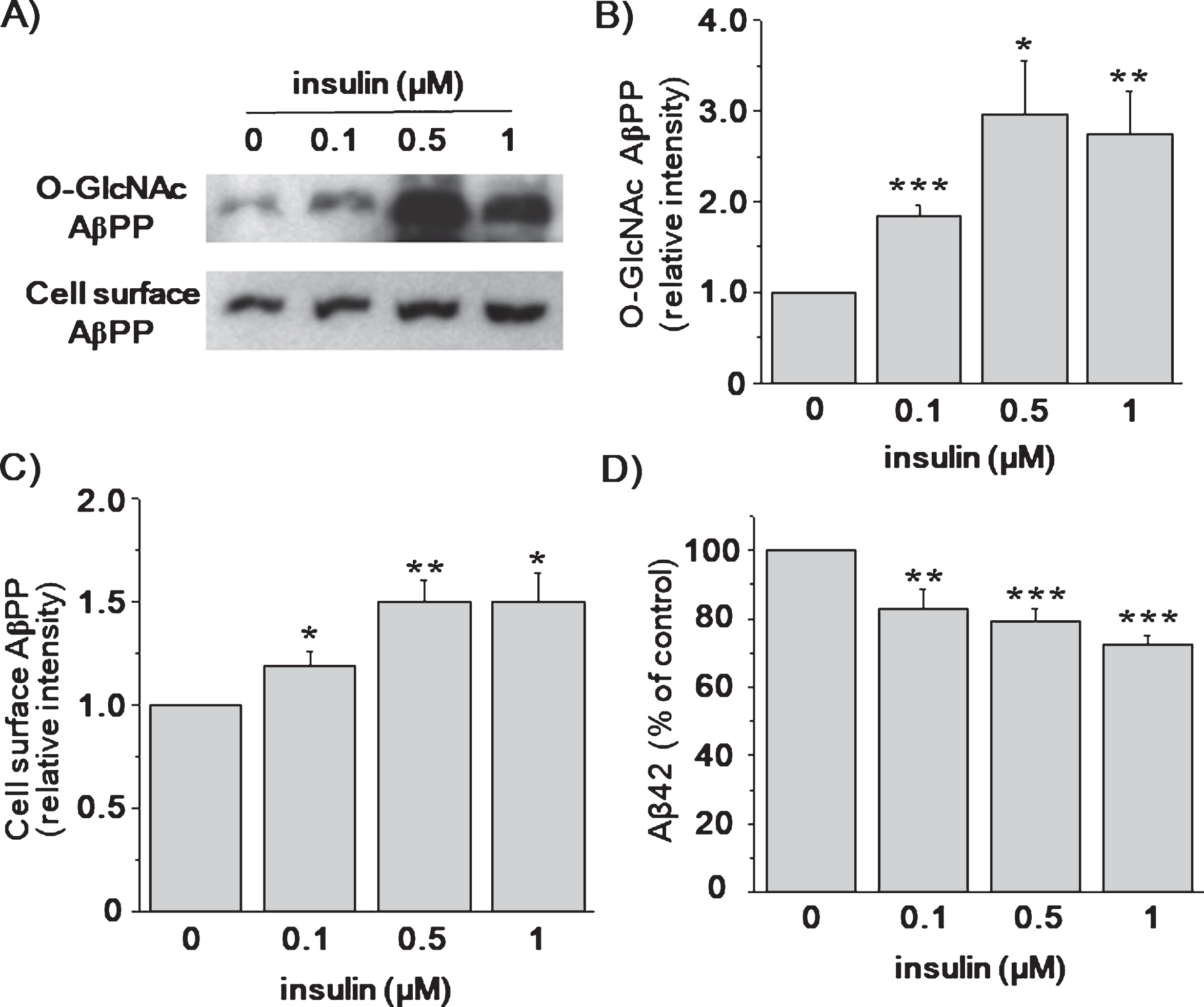
Insulin increased AβPP O-GlcNAcylation and reduced Aβ42 levels from cultured cortical neurons. Cultured cortical neurons were treated with 0.1, 0.5, and 1 μM insulin for 2 h in the neurobasal media. A) Cell lysates were prepared for the detection of the levels of O-GlcNAcylated AβPP. Levels of cell surface AβPP was measured by using cell surface biotinylation method. B) Densitometric analysis of western blot in (A) shows that the levels of O-GlcNAcylated AβPP were increased by insulin (0.1 μM insulin RD, 1.84; 95% CI, 1.08–2.24; p < 0.001; 0.5 μM insulin RD, 2.93; 95% CI, 1.83–2.97; p = 0.02; 1 μM insulin RD, 2.75; 95% CI, 2.27–3.94; p < 0.01; n = 4). C) The levels of cell surface AβPP were also increased by insulin (0.1 μM insulin RD, 1.19; 95% CI, 1.01–1.31; p = 0.04; 0.5 μM insulin RD, 1.50; 95% CI, 1.22–1.57; p < 0.01; 1 μM insulin RD, 1.50; 95% CI, 1.36–1.79; p = 0.02; n = 4). D) The levels of secreted Aβ42 were measured from the conditioned media by using ELISA methods. Insulin decreased the secreted Aβ42 levels in a dose-dependent manner (0.1 μM insulin, 82.79%; 95% CI, 81.88–98.34; p = 0.01; 0.5 μM insulin, 79.45%; 95% CI, 77.13–85.80; p < 0.001; 1 μM insulin, 72.33%; 95% CI, 62.93–78.25; p < 0.001; n = 8). One-way ANOVA: *p < 0.05; **p < 0.01; ***p < 0.001.
DISCUSSION
In this study, we demonstrated that the level of O-GlcNAcylated AβPP was increased by insulin. Consistent with our previous report [44], the increased O-GlcNAcylated AβPP by insulin enhanced sAβPPα production, while concomitantly inhibited the generation of sAβPPβ and Aβ42, without changes in the expression levels of secretases. Thus, AβPP O-GlcNAcylation by insulin promoted non-amyloidogenic pathway, and decreased the levels of β-secretase products, confirming the relationship between AβPP O-GlcNAcylation and Aβ production [43]. Our results are consistent with the previous studies showing that insulin treatment promoted sAβPPα secretion, and reduced the Aβ accumulation in neuronal cells [67, 68].
We also showed that the effects of insulin on AβPP O-GlcNAcylation were mediated by OGT through Akt signaling. However, Akt inhibitr alone reduced the level of AβPP O-GlcNAcylation, without changes in Aβ42 level, suggesting that Akt inhibitor might inhibit the basal OGT activity toward AβPP O-GlcNAcylation without affecting Aβ42 level. We speculate that this discrepancy may result from the combination of various effects of Akt signaling [51]. Akt influences many cellular processes such as cell survival, growth, proliferation, cell migration, and angiogenesis by phosphorylating a wide range of intracellular proteins, which may affect Aβ42 level. Additional studies are needed to elucidate the roles of Akt signaling in AβPP metabolism. More importantly, however, Akti completely inhibited the effects of insulin on AβPP O-GlcNAcylation and Aβ42 level. Thus, our findings may provide the underlying molecular mechanisms of beneficial insulin treatment for AD. Lately, it is known that not only O-GlcNAcylation plays an essential role in regulating numerous cellular processes, but also may protect brain and neurons against Aβ- or tau-induced impairments [69–71]. We tried to estimate the proportions of O-GlcNAcylated AβPP out of total AβPP in our experimental conditions. Quantitation of the western blot results revealed that about half of AβPP was O-GlcNAcylated in control condition, and most of AβPP was O-GlcNAcylated by insulin treatment (Supplementary Figure 10). This might explain the significant effects of insulin on Aβ42 and AβPP endocytosis.
The role of insulin in the brain has been less studied, compared with its role in peripheral tissues. However, there is evidence that insulin in the brain has important metabolic, neurotrophic, and neuromodulatory functions. Recent studies have demonstrated that both insulin and insulin receptors are ubiquitously expressed in the brain [28, 72]. The physiological concentration of insulin in peripheral tissues is known at 1∼10 nM [73, 74], while brain insulin levels can reach 10 to 100-fold higher than in plasma, especially in hippocampus, hypothalamus, cortex, and olfactory bulb [72]. Since we observed the insulin effects on AβPP O-GlcNAcylation at 0.5 μM and on Aβ42 production as low as 0.1 μM, insulin concentrations we used were within the physiological range in the brain.
Our findings show that AβPP O-GlcNAcylation by insulin decreased AβPP endocytosis rate. Cell surface levels of AβPP were increased, and the rate of AβPP internalization was reduced by insulin, which might regulate Aβ generation. Consistent with these results, the inhibitory effect of insulin on Aβ42 level was blocked by using inhibitors for endocytosis. In addition, the inhibitory effect of insulin on AβPP endocytosis was selective, because insulin did not affect the endocytosis of transferrin, which undergoes clathrin-dependent endocytosis. The YENPTY endocytic motif of AβPP C-terminal residue is important for its internalization [54]. It interacts with many intracellular adaptor proteins, such as Fe65, Mint, and Dab1, to regulate not only the cell surface level of AβPP, but also modulation of Aβ production [75]. For example, Fe65 interacting AβPP binds to several low-density lipoprotein receptors. This complex is rapidly internalized, resulting in decrease of sAβPPα secretion, and increase of Aβ generation [76]. Also, loss of Mint proteins reduces Aβ production [77]. Thus, AβPP endocytosis may play an important role in AβPP metabolism. Recently, the trafficking and localization of AβPP are known as the critical determinant for undergoing amyloidogenic or non-amyloidogenic pathway [14–17]. Several studies have already shown that the endocytic pathway in neurons is disrupted in AD [78–80]. Also, the endocytic pathway pathology was observed in non-human primate brain as the abnormally enlarged endosomes accumulated [81]. In addition, recent genome-wide association studies confirmed several AD-associated variants in endocytosis-related genes [82–84]. These studies strongly suggest that the disturbance of endocytosis could be involved in AD pathogenesis, and that regulating AβPP endocytosis is critical for Aβ production.
Alterations in insulin signaling have been reported in the brain of AD patients [20–26]. In the AD brain, the levels of insulin related proteins and its activity are changed [25, 85]. The levels of brain insulin, insulin receptor, insulin receptor substrate 1, phosphorylated insulin receptor, and its activity are decreased [25, 86]. In AD patients, not only the downstream of insulin signaling, Akt and GSK3-β, but other insulin signaling molecules, such as phosphoinositide-dependent kinase-1 (PDK1) and protein kinase C (PKC), are reduced [20–23, 87]. Lately, it is reported that the global levels of O-GlcNAcylation in post mortem brain cortical and hippocampal tissue of AD subjects are decreased [88]. Although the levels of Akt and O-GlcNAcylation are reduced in AD patients, the direct relationship between Akt and AβPP O-GlcNAcylation has never been reported. Thus, additional studies are needed to elucidate the mechanism of the effects of insulin signaling and AβPP O-GlcNAcylation in the brain of AD patients.
Recently, it is reported that AβPP undergoes a post-translational lipid modification called palmitoylation [89]. The primary function of palmitoylation is to enhance hydrophobicity, and facilitate association with membrane components of the cells and hydrophobic domains of other proteins [90]. Protein palmitoylation consists of a thioester linkage between palmitic acid and a cysteine residue, often resulting in lipid raft localization of the protein [91, 92]. The authors showed that palmitoylation targets AβPP to lipid rafts, and palmitoylated AβPP is a preferential substrate for β-secretase and Aβ generation. Lipid rafts are plasma membrane microdomains that are enriched in cholesterol and sphingolipids. Recent studies have suggested that lipid raft proteins are trafficked through the endocytic pathway, resulting from invagination of the plasma membrane rafts into the endocytic vesicles [93]. Also, lipid raft endocytosis is characterized as a general mechanism for pathogen entry [94], recycling of extracellular ligands [95], and cell surface trafficking [96]. Recently, we reported that a significant amount of AβPP is localized in lipid rafts, and that increasing cholesterol level increases the localization of AβPP in lipid rafts [97]. Since our findings show that AβPP O-GlcNAcylation by insulin decreases the AβPP endocytosis rate, it is possible that the effects of insulin on AβPP O-GlcNAcylation and the inhibition of AβPP internalization may occur via the change of lipid raft-mediated endocytosis of AβPP. Further studies are required to elucidate whether AβPP O-GlcNAcylation induces changes in AβPP localization in the lipid raft. Also, further studies are required to fully understand the interaction between AβPP and diverse adaptor proteins associated with endocytic pathways.