
Abstract
Advanced glycation end products (AGEs) have been implicated in the disease process of diabetes mellitus. They have also been found in senile plaques and neurofibrillary tangles in the brains of Alzheimer’s disease patients. Furthermore, abnormally high levels of D-ribose and D-glucose were found in the urine of patients with type 2 diabetes mellitus, suggesting that diabetic patients suffer from dysmetabolism of not only D-glucose but also D-ribose. In the present study, intravenous tail injections of ribosylated rat serum albumin (RRSA) were found to impair memory in rats, but they did not markedly impair learning, as measured by the Morris water maze test. Injections of RRSA were found to trigger tau hyperphosphorylation in the rat hippocampus via GSK-3β activation. Tau hyperphosphorylation and GSK-3β activation were also observed in N2a cells in the presence of ribosylation-derived AGEs. Furthermore, the administration of ribosylation-derived AGEs induced the suppression of brain-derived neurotrophic factor (BDNF) and tropomyosin-related kinase B (TrkB). Both GSK-3β inhibition and BDNF treatment decreased the levels of phosphorylated tau in N2a cells. In particular, the administration of BDNF could rescue memory failure in ribosylated AGE-injected rats. Ribosylation-derived AGEs downregulated the BDNF-TrkB pathway in rat brains and N2a cells, leading to GSK-3β activation-mediated tau hyperphosphorylation, which was involved in the observed rat memory loss. Targeting ribosylation may be a promising therapeutic strategy to prevent Alzheimer’s disease and diabetic encephalopathies.
Keywords
INTRODUCTION
Advanced glycation end products (AGEs) are the final derivatives of the nonenzymatic glycation of proteins by reducing saccharides, such as D-glucose (Glc) and D-ribose (Rib) [1, 2]. AGEs have been implicated as being involved in many human diseases, such as diabetes [3], cataracts [4], renal failure [5], and sarcopenia [6]. More recently, it has become evident that AGEs are also involved in neurodegenerative diseases, including Alzheimer’s disease (AD) [7] and Parkinson’s disease [8].
With respect to glycation and AGEs, Glc is the most commonly investigated sugar [9]. However, it has recently been found that urine Rib levels are higher in patients suffering from type 2 diabetes mellitus (T2DM) than in age-matched controls [10]. Rib is a major contributor to glycated human serum protein and glycated hemoglobin and has been identified by basic and clinical investigations [11, 12]. Urine Rib levels of AD patients were markedly higher than those of cognitively normal subjects [13]. Administration of Rib induces elevation of formaldehyde levels in mouse brain [14, 15] and hepatic triglyceride [16]. It is reasonable to assume that T2DM patients may be suffering from a dysmetabolism of not only Glc but also Rib [17]. Therefore, the dysmetabolism of Rib and the resultant complications should be further investigated.
Rib is a naturally occurring pentose monosaccharide that is essential for the production of energy in cells [18, 19]. It is obtained from food or ingested as a supplement intended to support cardiac energy metabolism [20–22]. Rib is a metabolite of the pentose phosphate pathway [23, 24]. The concentrations of Rib are approximately 0.02 and 0.1 mM in human serum [25] and the human brain [10], while the concentrations of Glc are about 5 mM and 2 mM in human serum and brain [26, 27], respectively. Although the role of Rib in the glycation and cross-linking of proteins such as collagen has been investigated [28, 29], the mechanisms underlying the effects of ribosylated proteins on brain functions are still unclear.
Abnormally phosphorylated tau has been discovered to be the major proteinaceous constituent of paired helical filaments that constitute the neurofibrillary tangles [28] that are a histopathological hallmark of AD. Tau hyperphosphorylation is usually considered to be an early event in the neurodegenerative process in AD [30]. It has been shown that hyperphosphorylated tau is also present in the brain of a T2DM animal model [31]. Li and colleagues have shown that the AGEs of Glc can induce tau hyperphosphorylation in rats through the activation of glycogen synthase kinase 3β (GSK-3β) [32]. GSK-3β is an important tau kinase that phosphorylates tau at epitopes such as Thr181 and Ser396 [33, 34]. Tau proteins from patients with AD were found to be glycated and able to induce oxidative stress [35], while the level of glyoxalase I was reported to be higher in the brains of patients with AD than in the brains of healthy controls [36]. Although these data provide further evidence supporting the existence of a relationship between tau hyperphosphorylation and AGE formation, the role of ribosylation in this process has not been addressed.
Brain-derived neurotrophic factor (BDNF) has been shown to play an important role in cognitive function during normal neuronal development [37]. When released, BDNF binds to and activates the receptor tropomyosin-related kinase B (TrkB) [38]. The activation of TrkB by BDNF initiates downstream signaling pathways that modulate synaptic plasticity. In the brain, BDNF is active in the hippocampus, cortex, and other areas [39] and plays an important role in the formation of long-term memory [40]. In support of this hypothesis, BDNF (+/–) mice show impairments in spatial memory acquisition during the Morris water maze test [41]. Navaratna and colleagues found that treatment of brain microvascular endothelial cells with AGEs caused reduced expression and secretion of BDNF in an extracellular signal-related kinase (ERK)-dependent manner [42]. BDNF-TrkB-CREB pathway was also reported to involve in the cognitive impairment after treatment with propofol [43]. As previously shown by us [44], Rib induces cognitive impairment and an accumulation of AGEs in the brains of mice. However, the effect of ribosylation on the BDNF-TrkB pathway and the related tau hyperphosphorylation has not been investigated.
In the present study, we employed both animal and cellular models to demonstrate that ribosylation-derived AGEs can trigger tau hyperphosphorylation through the reduction of BDNF. Furthermore, we show that the activation of GSK-3β plays a role in mediating this effect.
MATERIALS AND METHODS
Antibodies
The antibodies utilized in this study were directed against tau phosphorylated at the following epitopes: pThr181 (1:1,000 dilution, Signalway Antibody, USA); pSer214, pSer396 and AT8 (1:2,000, Invitrogen, USA); and Tau-1 (tau not phosphorylated at Ser199/202, 1:5,000, Millipore, USA). The total expression level of tau was detected using Tau-5 (1:2,000, Millipore, USA). The levels of kinase involved in tau phosphorylation were examined using antibodies against GSK-3β (1:2,000) and phospho-GSK-3β (Ser9, 1:2,000; Tyr216, 1:2,000) from Cell Signaling Technology (USA). Antibodies against BDNF (1:2,000, Abcam, USA), its receptor TrkB (1:1,000, Cell Signaling Technology, USA), AGEs (6D12, monoclonal antibody, Wako, Japan) and β-actin (1:5,000, Santa Cruz Biotechnology, USA) were also employed.
Preparation of ribosylated RSA/BSA
Rat serum albumin (RSA, Sigma, USA) and bovine serum albumin (BSA, AMRESCO, USA) were mixed with Rib or Glc prepared in 0.02 mM Tris-HCl (pH 8.0) to final concentrations of 10 mg/ml protein and 1 M sugar (named RRSA, GRSA, RBSA, and GBSA). Separate preparations of RSA and BSA without sugar were used as controls. The reaction mixtures were incubated at 37°C for 7 days. After incubation, the preparations were extensively dialyzed against Tris-HCl to remove free Rib/Glc before being used in cell cultures. RRSA and GRSA were not dialyzed before being administered to rats, and Rib and Glc alone were used as controls. All solutions were filtered with 0.22μm membranes (Millipore, USA). In the in vivo study, RRSA was used to reduce autoimmunity in rats. For the in vitro experiments involving N2a cells, because both RRSA and RBSA behaved the same in mouse-derived cells, inducing somewhat heterogeneous reactions, we chose BSA, which is much less expensive than RSA.
Ethics statement
The protocols for the handling of the rats and the experimental procedures were approved by the Animal Welfare and Research Ethics Committee of the Institute of Biophysics, Chinese Academy of Sciences (permit number: SYXK2013-77), and the methods were performed in accordance with the approved guidelines.
Animals and the administration of RRSA/GRSA
Male Sprague Dawley rats (8 weeks) were obtained from Vital River Laboratory Animal Technology Co. Ltd. (China). The rats were randomly divided into six groups and allowed to acclimatize to the cages for four days. Then, the rats received a daily tail intravenous injection of RRSA or GRSA for 30 days, and RSA, Rib, Glc, and saline were used as the controls. The administration dosage was 25 mg/kg·W. All rats were maintained under pathogen-free conditions.
Measurement of body weight and grip strength
Body weight and grip strength were recorded before and after tail intravenous injections as described previously [45, 46].
Open field test
Rats were subjected to an open field test in an apparatus that consisted of a 200×200 cm open arena with walls that were 40 cm high (Huaibeizhenghua Biological Instrument Equipment Co., LTD, China). The procedure was described previously [45].
Elevated plus maze
The elevated plus maze (EPM) was composed of four arms (50×10 cm, Huaibeizhenghua Biological Instrument Equipment Co., LTD, China) that were arranged in the shape of a ‘+’ sign, with two open arms across from each other that were perpendicular to two closed arms with a center platform (10×10 cm). The open arms had neither side nor end walls, whereas the closed arms had side and end walls (40 cm in height) but were open on the top. The procedure was described previously [45].
Morris water maze
The Morris water maze test was performed as described previously [45, 47]. Rats were placed on a visible platform before they were exposed to a hidden platform. All data were recorded with a computerized video system.
Sample collection from animals
After the behavioral tests, the rats were sacrificed, and the hippocampi and cortexes were quickly dissected for subsequent western blotting or fixed in 4% paraformaldehyde for immunohistochemistry.
Cell culture
Mouse neuroblastoma Neuro 2a (N2a) cells (obtained from Cell Resource Center, China) were cultured in Dulbecco’s modified Eagle’s medium: Nutrient Mixture F-12 (DMEM/F12, Corning, USA) supplemented with 100 IU/ml penicillin and 100μg/ml streptomycin at 37°C in a humidified 5% CO2 incubator. The medium contained 10% fetal bovine serum. Cells were grown to 70–80% confluence in 100 mm diameter dishes and provided with nutrients every three days. For all experiments, the culture medium was replaced with serum-free medium before the addition of RBSA/GBSA.
CCK-8 assay
N2a cells were seeded on a 96-well plate at a concentration of 104 cells per well and exposed to RBSA/GBSA (0, 0.1, 0.3, 0.5, 0.8, 1.0, 1.5, and 2 mg/ml) for 24 h, except for the control. The CCK-8 (Beyotime, China) reagent was added after 24 h. The plates were incubated at 37°C for 1 h, and the absorbance was read at 450 nm.
Lactate dehydrogenase cytotoxicity detection
Lactate dehydrogenase (LDH) cytotoxicity assays were performed according to the manufacturer’s protocol (Beyotime, China). This colorimetric assay quantifies the activity of LDH released from the cytosol of damaged cells into the supernatant and thus serves to quantify cell death.
Flow cytometric analysis
Cells undergoing apoptosis were detected by double staining with Annexin V-FITC/PI in the dark according to the manufacturer’s protocol (Beyotime, China). Cells were stained with Annexin V-FITC and PI and immediately analyzed by flow cytometry (BD FACS Calibur, USA).
Cell lysate protein extract preparation
After treatment, N2a cells were harvested in ice-cold WIP (Beijing Cellchip Biotechnology Co., Ltd., China) containing phosphatase and protease inhibitor (1 mM PMSF). The samples were then centrifuged at 20,000× g at 4°C for 15 min, and the supernatants were collected and analyzed for protein content using a bicinchoninate acid (BCA) assay kit (ThermoFisher Scientific, USA). The samples were stored at –80°C until further immunoblotting analysis.
Western blotting
Protein samples from brain tissues and cell lysates were mixed with 5× SDS-PAGE Sample Buffer (Genestar, China) and boiled for 10 min. Cell or brain lysate extracts containing ∼30μg of protein were separated on 12% SDS-PAGE gels and then transferred onto PVDF membranes (Millipore, USA). The nonspecific binding sites of the membranes were blocked with 5% nonfat milk in PBS containing 0.1% Tween-20 (PBST, pH 7.4) for 1 h at room temperature. The membranes were then incubated overnight at 4°C with antibodies. The following day, the membranes were washed three times and then incubated for 1 h at room temperature with a horseradish peroxidase-linked secondary anti-mouse or anti-rabbit antibody (1:4000 dilution; ZSGB-BIO, China). The membranes were then washed three times with PBST. The immunoreactive bands were reacted with enhanced chemiluminescence detection reagents (Applygen, China), visualized after exposure of the membranes to Kodak X-ray film and quantified by Quantity One 1D analysis software 4.5.2 (Bio-Rad, USA).
Immunohistochemical analysis
Rat brains were processed for immunohistochemistry using standard protocols, as previously reported [45]. The primary antibodies used were rabbit anti-pS396 and rabbit anti-AT8 (both 1:500). The secondary antibody used was biotin-labeled goat anti-rabbit antibody. The proteins were detected using horseradish peroxidase-labeled antibodies. The staining was visualized with an AEC system (ZSGB-BIO, China), and light microscopy (Nikon Optical, Japan) was used to observe the staining.
Determination of reactive oxygen species in rat brain
Reactive oxygen species (ROS) concentrations in rat brains were measured with a rat ROS ELISA kit (Shanghaijining Co. Ltd., China). Lysates of hippocampus and cortex were used and experimental procedure was performed according to the manufacturer’s protocol.
Stereotactic BDNF injection
The hippocampi of Sprague Dawley rats (10 in each group) were stereotactically injected (–3.30; ±2.0; –4.0) with BDNF-expressing viruses (packaged by AAV9, 5μl, Vigene Biosciences, China). The titer of the purified BDNF-containing viral vector was >1×1013 vg/ml. Two weeks later, the rats were intravenously injected through the tail with RBSA or saline for 30 days.
Data analysis
All data were analyzed using SPSS software; one-way ANOVA or two-way ANOVA with a post hoc test were used. Differences with a probability level of 95% (p < 0.05) were considered significant.
RESULTS
Ribosylated products impair the cognitive ability of Sprague Dawley rats
To clarify whether ribosylated proteins affect cognitive functions in vivo, we incubated Rib with rat serum albumin (RSA) for 7 days to obtain ribosylated RSA (RRSA) [2]. Next, we treated Sprague Dawley rats (n = 10) with intravenous tail injections of RRSA (25 mg/kg·W, once daily) for 30 days. Glc and its modified RSA (GRSA) were employed for the purposes of comparison. No differences in weight or muscle strength were found among the groups (Supplementary Figure 1, p > 0.05). When the rats were tested in the EPM (Supplementary Figure 2) and the open field test (Supplementary Figure 3), we did not find that RRSA administration led to anxiety-related behavior in rats, as indicated by the absence of differences among the groups in the time spent in the open arm and open arm entries in the EPM or in the time spent in the center of the open field arena.
In this study, the Morris water maze was employed to assess spatial learning and memory. Tensile test has shown no difference in animal muscle strength among rats of these groups, which excluded the interference of motor abilities on the behavioral tests. During the training session, all rats improved their performance as indicated by a shortened escape latency over successive days (Fig. 1a). There were no significant differences in the performance of the rats among the groups after a 5-day training period; however, on day 3, the escape latency of rats injected with RRSA was higher than that of control rats (p = 0.0228), and the escape latencies of rats injected with RSA (p = 0.0103) and rats injected with Rib (p = 0.0092) were shorter than those of control rats on day 2 and day 4. No significant differences were found between the Glc- and GRSA-injected groups and the control in the 5-day training period. Together, these data showed that glycated products (regardless of whether they were ribosylated or glucosylated) did not impair spatial learning in Sprague Dawley rats under our experimental conditions.
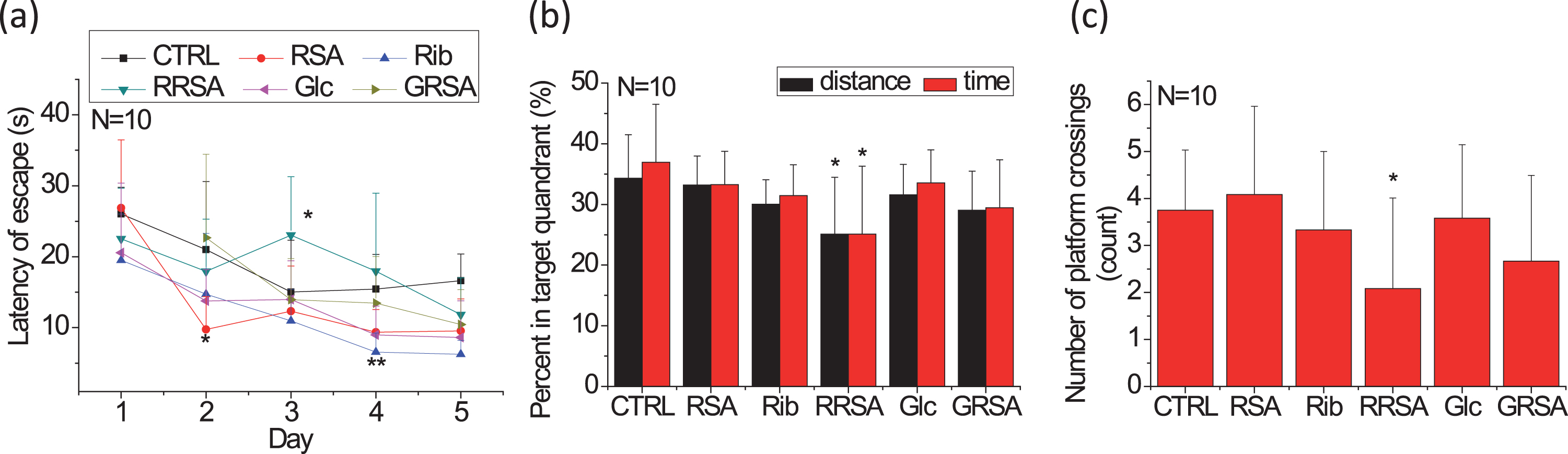
Impaired spatial memory in rats intravenously injected with ribosylated proteins demonstrated by the Morris water maze test. Rats were injected with RRSA (25 mg/kg·W, once daily) for 30 days. GRSA, RSA, Rib, Glc, and saline were used as controls. The length of time needed to find the hidden platform was recorded as the escape latency on each of the five training days (a). The percentages of the search time and the distance traveled spent in the quadrant from which the platform was removed during the probe trial are shown (b), as is the number of platform crossings in the probe trial (c). Rib, D-ribose; Glc, D-glucose; RSA, rat serum albumin; RRSA, ribosylated RSA; GRSA, glucosylated RSA. All values are expressed as the mean±SD. *p < 0.05, **p < 0.01 versu control.
After withdrawing the platform (probe trial), the time and distance the RRSA-injected rats spent swimming in the target quadrant were significantly shorter than those of the control group (p = 0.0247 and p = 0.0302, respectively) (Fig. 1b). No significant differences were observed between any other treatment group and the control group. The number of platform crossings was also lower in the RRSA-treated group (p = 0.0463) than in the other groups (Fig. 1c). This finding indicates that it is memory, but not learning ability, that is impaired in the rats treated with ribosylated protein products.
Ribosylated products induce tau hyperphosphorylation in the rat brain
It has been shown that the presence of hyperphosphorylated tau is closely related to cognitive impairment. Therefore, we determined the level of tau phosphorylation in the brain by western blotting and immunohistochemistry.
The total tau level (detected with Tau-5) in the rat brain (cortex and hippocampus) did not change after RRSA treatment (Fig. 2a, c). The levels of nonphosphorylated tau in the hippocampi assayed with the monoclonal antibody Tau-1 (recognizing Ser199/202) were also not significantly different among the groups, with the exception of a lower level observed in the RRSA-treated group than in the non-RRSA-treated group (p = 0.0377) (Fig. 2a, b). The levels of phosphorylated Ser396 and AT8 (phosphorylated at epitope Ser199/202) of tau were increased in the hippocampi of RRSA-injected rats compared with the control group (p = 0.0181 and p = 0.0095, respectively), although rats in the RSA, Rib, and Glc groups also showed significantly higher AT8 levels in their hippocampi compared with the control. Among all groups, there were no significant differences in the levels of nonphosphorylated or phosphorylated tau in the cortex, with the exception of an increased level of AT8 in the RRSA group (Fig. 2c, d).
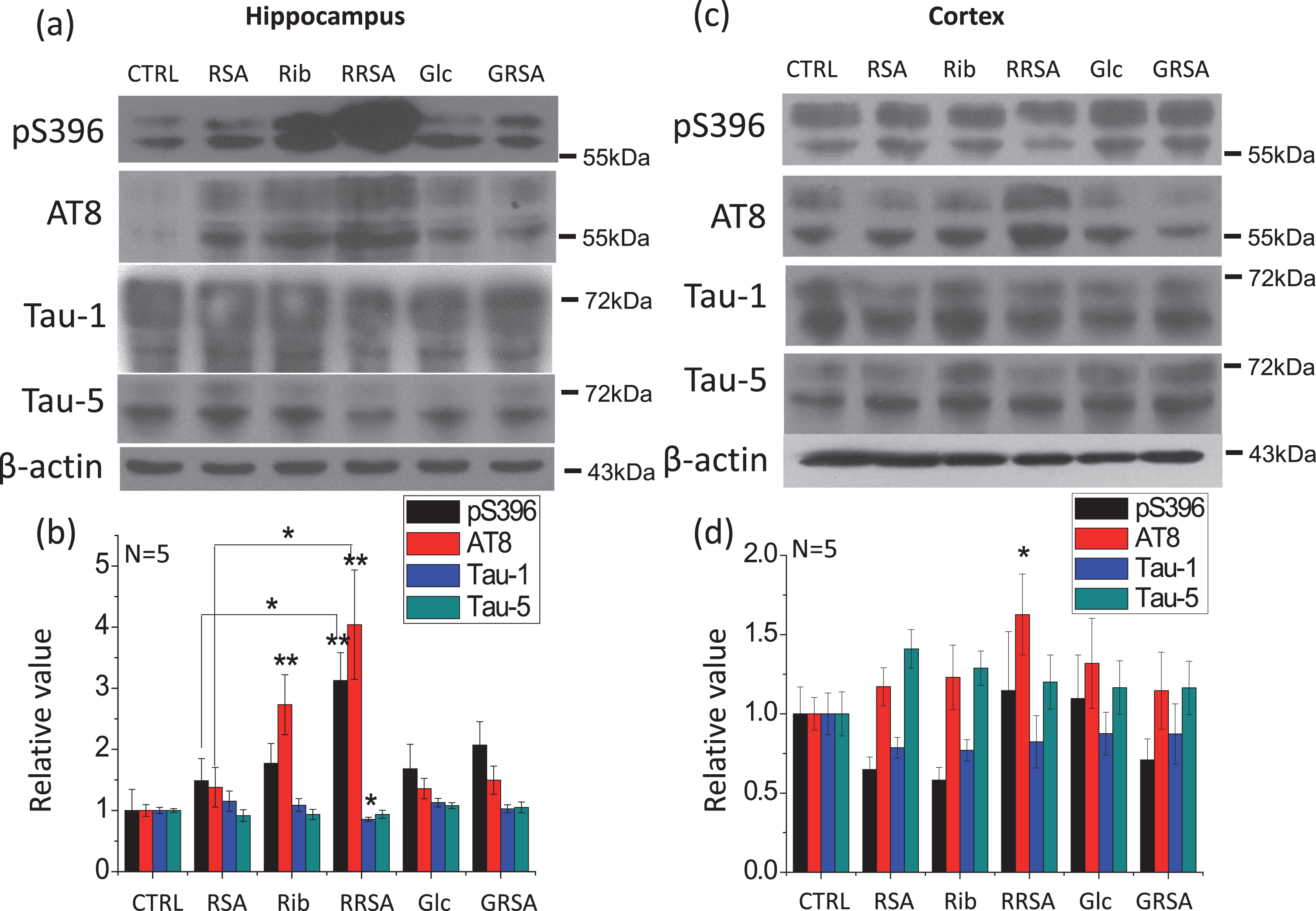
Ribosylated products induced tau hyperphosphorylation in the rat brain. The conditions for the treatment were the same as in Fig. 1. The phosphorylation levels of tau in the hippocampus and cortex were detected by western blotting using the anti-pSer396, anti-AT8, anti-Tau-1, and anti-Tau-5 antibodies a, c). β-Actin was used as a loading control. The quantification is shown in b and d. The value of the saline control was designated as 1.0. The levels of phosphorylated tau are expressed as the ratio of phospho-site to total tau. All values are expressed as the mean±SEM. *p < 0.05, **p < 0.01 versus control unless indicated otherwise in the Figure.
We further performed immunohistochemical staining of brain sections. An increase in the immunoreactivity of tau phosphorylated at Ser396 and Ser199/202 was observed in the hippocampi of rats treated with RRSA (Fig. 3). The levels of immunoreactivity were less pronounced in the other groups. No differences in immunoreactivity in the cortex were observed among all the groups (data not shown). Combining the data, this indicated that ribosylated products are able to induce tau hyperphosphorylation in the brains of rats, especially in the hippocampus.

Immunohistochemical staining of phosphorylated tau in the hippocampus. The conditions for the treatment were the same as in Fig. 1. In the hippocampus, the levels of tau phosphorylated at Ser396 and Ser199/202 were detected by immunohistochemistry using the anti-pSer396 or anti-AT8 antibodies (a). The quantification of the relative intensity is shown (b). All values are expressed as the mean±SEM. **p < 0.01 versus control.
High cytotoxicity and tau hyperphosphorylation induced by ribosylated proteins in cells
To investigate the effects of ribosylated products on tau phosphorylation in cells, we used the mouse neuroblastoma neuro-2a cell line (N2a) for in vitro experiments. To this end, N2a cells were incubated with RBSA for 24 h. First, cell viability was determined using the Cell Counting Kit-8 (CCK-8) assay (Fig. 4a). Only RBSA was found to induce decreased cell viability. Activity assays measuring the release of LDH also showed that RBSA was highly cytotoxic to N2a cells (Fig. 4b). Furthermore, a flow cytometric analysis using Annexin V-FITC/propidium iodide (PI) staining also showed a high level of cytotoxicity (Fig. 4c and Supplementary Figure 5). RBSA treatments resulted in a clear increase in both the upper right and lower right (indicating early and late apoptosis, respectively) populations after 24 h. These data demonstrate that ribosylated protein products exert a highly cytotoxicity effect on N2a cells.

Cell viability was measured using the CCK-8 assay, the LDH cytotoxicity assay and the detection of apoptosis in N2a cells cultured with RBSA. RBSA (0, 0.1, 0.3, 0.5, 0.8, 1.0, 1.5, and 2 mg/ml) was added to cells on a 96-well plate. The CCK-8 assay was performed 24 h later (a). The LDH assay was also used to measure the cytotoxicity of RBSA (0.5 and 1.0 mg/ml) after incubation for 24 h. GBSA and BSA were used as controls. Cells incubated without proteins were used as a negative control (b). Apoptosis was measured by flow cytometry using Annexin V-FITC/PI staining, and the statistical results from Supplementary Figure 4 are shown here (c). All values are expressed as the mean±SEM. **p < 0.01, ***p < 0.001 versus control unless indicated otherwise in the Figure. LR, lower right, UR, upper right.
To determine whether ribosylated products also trigger tau hyperphosphorylation in N2a cells, we determined the phosphorylation of tau at Thr181, Ser214, Ser396, and Ser199/202 (AT8) epitopes in N2a cells. Incubation with RBSA (1 mg/ml) resulted in a significant increase in tau phosphorylation at Thr181, Ser214, Ser396, and Ser199/202, along with a decrease in tau nonphosphorylation (Tau-1) (Fig. 5a, b). Incubation with RBSA at a lower concentration (0.5 mg/ml) also induced tau hyperphosphorylation at Thr181, Ser396, and Ser199/202, but not at Ser214. GBSA, in contrast, did not exert the same effect, indicating that ribosylated but not glucosylated products induced tau hyperphosphorylation in N2a cells under our experimental conditions.
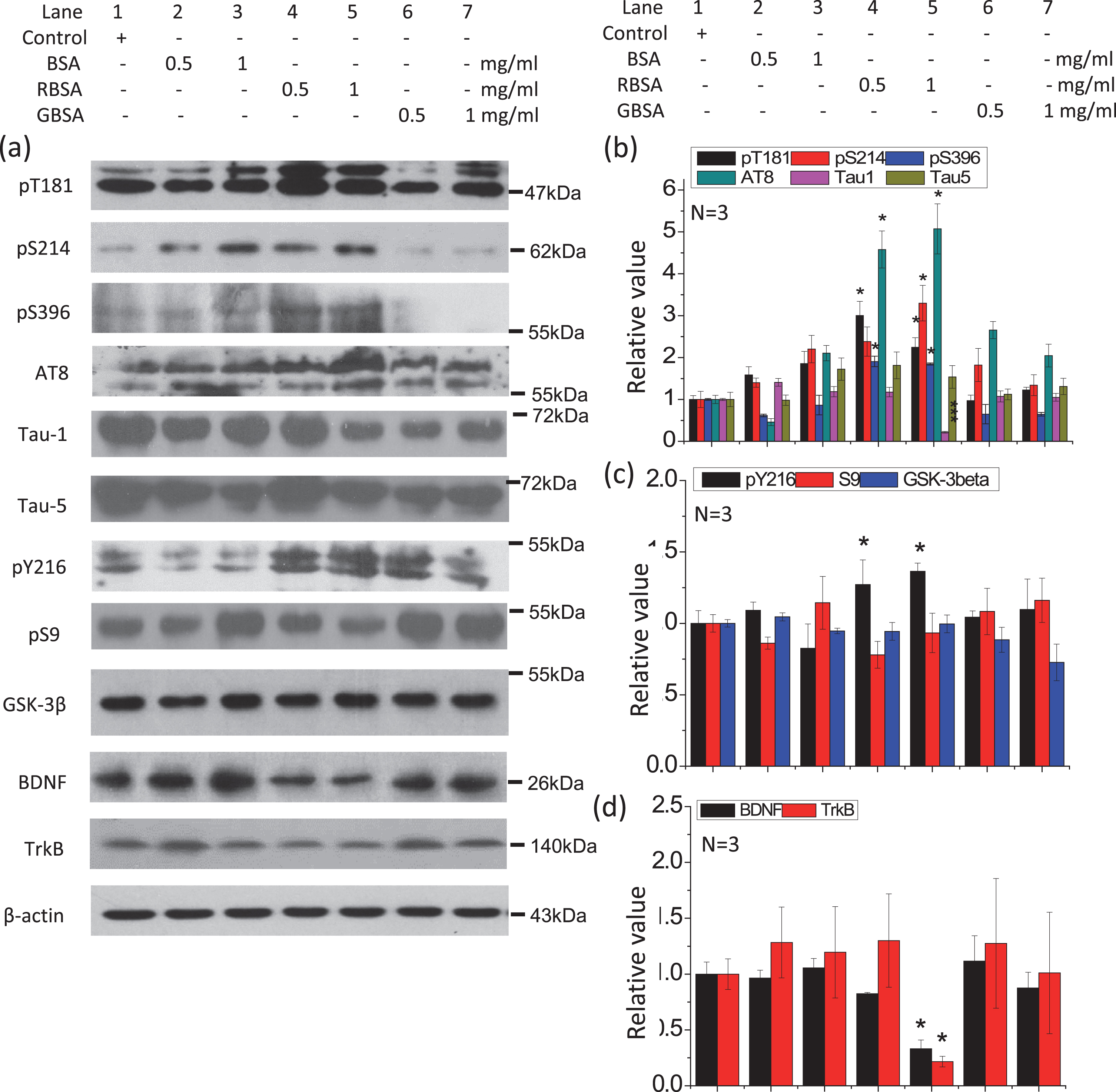
RBSA activated GSK-3β, promoted tau hyperphosphorylation, and reduced BDNF and TrkB levels in N2a cells. Cells were incubated with different concentrations of RBSA (0.5 and 1.0 mg/ml) for 24 h on 6-well plates. GBSA and BSA were employed as controls. The levels of phosphorylated tau, tau kinase, BDNF, and TrkB in cells were determined by western blotting (a). β-Actin was used as a loading control. The quantification is shown in b, c, and d. The value of the saline control value was designated as 1.0. All values are expressed as the mean±SEM. *p < 0.05, ***p < 0.001 versus control.
Ribosylated products induce tau hyperphosphorylation through BDNF-mediated activation of GSK-3β
To clarify the mechanism by which ribosylated products induce tau hyperphosphorylation, we examined changes in the activation of the specific tau kinase GSK-3β, which is involved in the regulation of major phospho-epitopes of tau phosphorylation [48]. In the rat hippocampus (but not the cortex) and N2a cells, treatment with ribosylated products (either RRSA or RBSA) resulted in a significant increase in the active form (pY216) of GSK-3β but not the inhibitory form (pS9) compared with the levels in the other groups (Figs. 5a, c and 6a, b, d, e), indicating that GSK-3β might play a role in tau hyperphosphorylation. Since RSA and BSA have highly conserved protein sequences, it is reasonable to expect that ribosylated RSA and BSA would have similar effects on tau phosphorylation and GSK-3β activation.

RRSA activates GSK-3β and reduces BDNF and TrkB in the rat brain. Conditions for the treatment were the same as in Fig. 2, except that the levels of tau kinase, BDNF, and TrkB in the hippocampus and cortex were detected by western blotting (a, d). β-Actin was used as a loading control. The quantification is shown in b, c, e, and f. The value of the saline control value was designated as 1.0. The levels of phosphorylated GSK-3β are expressed as the ratio of phospho-site to total GSK-3β. All values are expressed as the mean±SEM. *p < 0.05 versus control unless specific indication in the Figure.
We next inhibited GSK-3β using a specific inhibitor, CHIR99021 [49] (Fig. 7). This markedly rescued tau phosphorylation (Thr181, Ser214, Ser396, AT8, and Tau-1) in a concentration-dependent manner, whereas there were no significant changes in the total level of tau (Tau-5). These data confirm that GSK-3β is involved in tau hyperphosphorylation in the presence of ribosylated products.
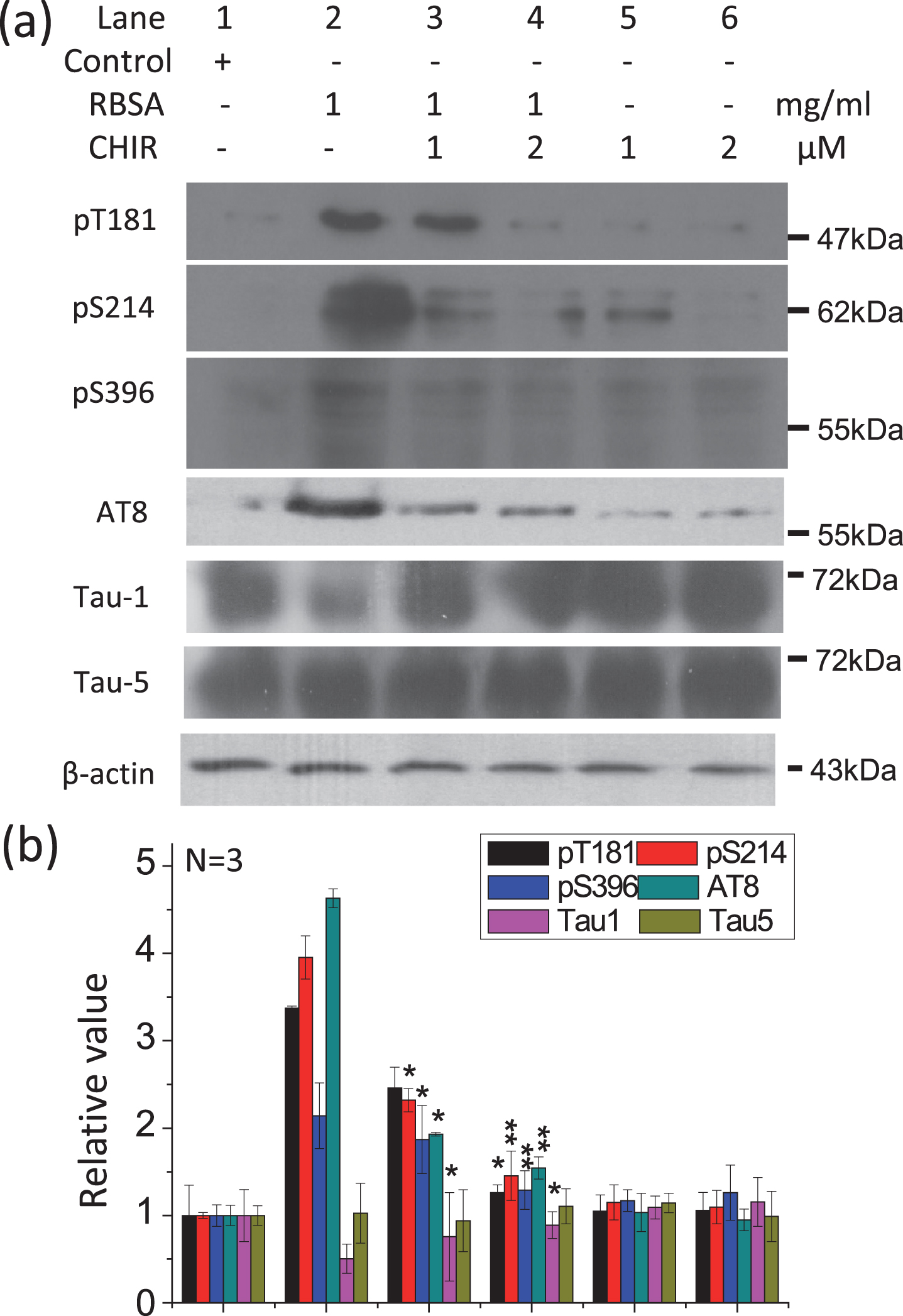
GSK-3β catalyzed the hyperphosphorylation of tau in the presence of RBSA. The GSK-3β-specific inhibitor CHIR99021 (1μM and 2μM) was added to the cells in the presence of 1.0 mg/ml RBSA and incubated for 24 h on 6-well plates. The levels of phosphorylated tau were determined by western blotting (a). β-Actin was used as a loading control. The quantification is shown in b. The value of the saline control value was designated as 1.0. All values are expressed as the mean±SEM. *p < 0.05, **p < 0.01, ***p < 0.001 versus control.
It is known that the BDNF-TrkB pathway regulates GSK-3β activity, as described previously [32]. Therefore, we assessed the levels of BDNF and TrkB in both the rat brain and N2a cells (Figs. 5, 6). In the hippocampus of the rat brain, there were significant decreases in BDNF and TrkB levels in the RRSA-treated group compared with the other groups, whereas no differences were detected in the cortex among all groups (Fig. 6a, c, d, f). An analysis of the N2a cell experiments yielded the same result (Fig. 5a, d). The BDNF and TrkB levels in N2a cells were significantly decreased under RBSA treatment in a concentration-dependent manner. This suggests that the BDNF-TrkB pathway is inactivated by ribosylated products and could therefore be involved in tau hyperphosphorylation.
To determine whether the BDNF-TrkB pathway is involved in the hyperphosphorylation of tau induced by ribosylation-derived AGEs, we added extracellular BDNF to the cultured cells to determine whether there was a rescue effect on tau phosphorylation. The addition of BDNF 2 h before RBSA treatment significantly suppressed tau phosphorylation at Thr181, Ser214, Ser396, and Ser199/202 compared with RBSA treatment alone (Fig. 8). The effect was concentration-dependent. Additionally, BDNF preincubation decreased the active form of GSK-3β, indicating that BDNF acts upstream of GSK-3β. Furthermore, the rescue effect of BDNF treatment was also observed in an in vivo study. When RBSA-treated rats were given BDNF by stereotactic injection of the AAV9 virus containing the BDNF sequence, their spatial memory ability increased significantly compared with that of RBSA-treated rats in the Morris water maze test. They spent more time swimming in the target quadrant than control or RBSA-treated rats (Supplementary Figure 5). These data together demonstrated that ribosylated products induced tau hyperphosphorylation through BDNF-mediated GSK-3β activation. BDNF treatment can rescue the memory impairment caused by ribosylated products.
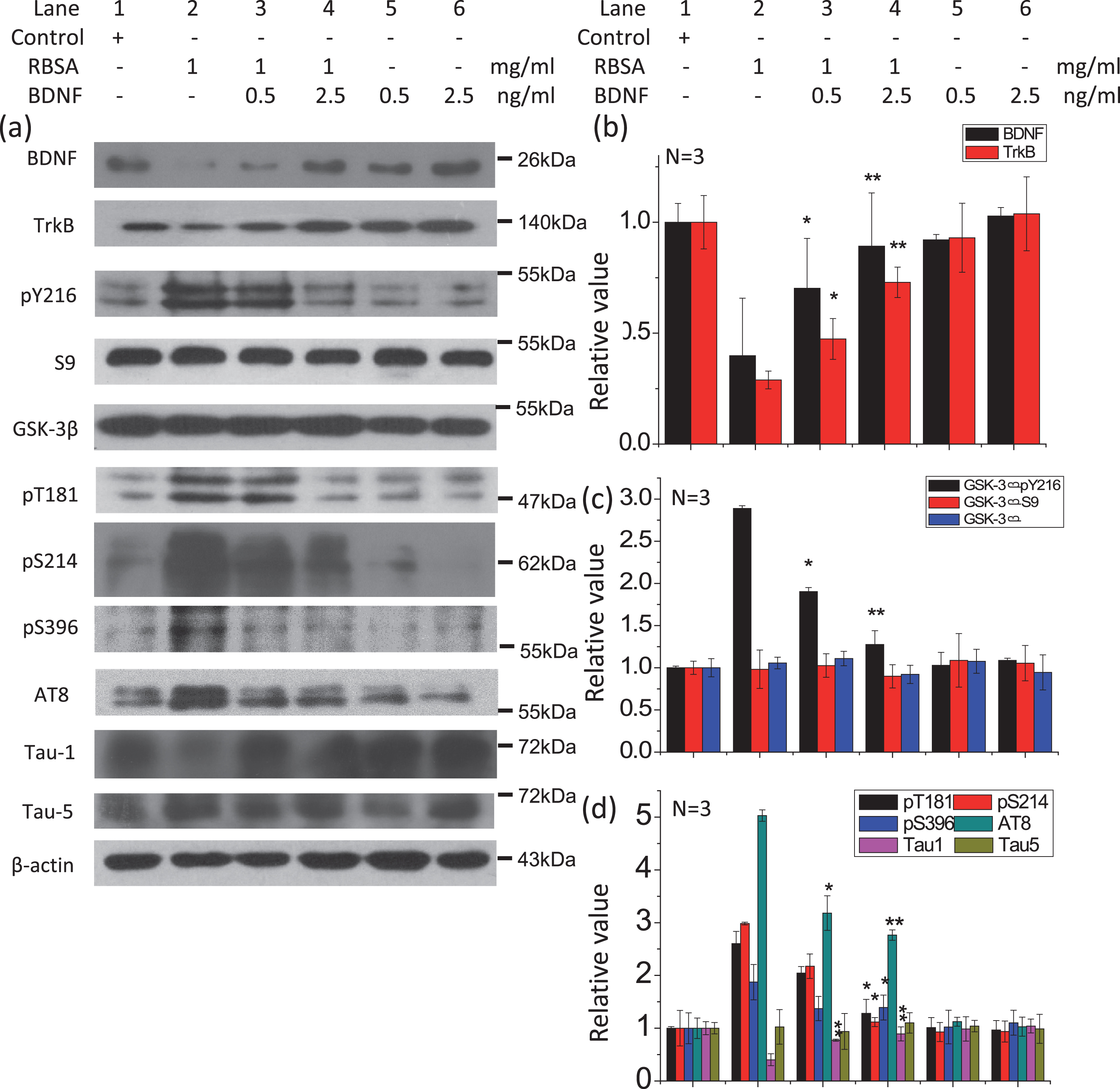
The effect of preincubation with BDNF on the level of tau phosphorylation in N2a cells. Cells were cultured with BDNF (0.5 ng/ml and 2.5 ng/ml) for 2 h before RBSA (1.0 mg/ml) was added and then incubated for 24 h on 6-well plates. The levels of GSK-3β, phosphorylated tau, BDNF, and TrkB were determined by western blotting (a). β-Actin was used as a loading control. The quantification is shown in b, c, and d. The value of the saline control was designated as 1.0. All values are expressed as the mean±SEM. *p < 0.05, **p < 0.01 versus control.
DISCUSSION
Impaired energy metabolism in the brain has been implicated in age-related cognitive impairment [50]. More specifically, AGEs have been implicated in T2DM and its related complications, such as diabetic encephalopathy [51]. As AD has been called type 3 diabetes [52], the relationship between AGEs and typical pathological features of AD (such as the hyperphosphorylation of tau) is gaining more attention. In patients with neurodegenerative diseases, AGEs have been found in amyloid plaques [53], neurofibrillary tangles [54], and Lewy bodies [55]. Meanwhile, increased levels of Rib and Glc have been found in the urine of patients with T2DM [10]. Rib also acts as a major contributor to the formation of HbA1c [11]. This makes the investigation of the role of Rib in glycation and the resulting AGE formation even more important. In the present study, we compared the effects of ribosylation and glucosylation on tau hyperphosphorylation and memory. We found that compared with glucosylated proteins, ribosylated proteins (RRSA and RBSA) are more active in suppressing the activity of the BDNF-TrkB pathway and triggering the hyperphosphorylation of the tau protein. These results may provide a new path for studying the relationship between Rib and age-related cognitive impairment.
To measure spatial cognitive functions in rats, the Morris water maze paradigm was used. Rats injected with RRSA did not show impaired learning (in the training process). However, in the probe trial, RRSA-treated rats showed an observable impairment of spatial memory, as indicated by decreased time and distance spent in the target quadrant and fewer platform crossings compared to the control group. Under our experimental conditions, we found that memory, but not learning, was affected by treatment with ribosylated products. These data suggest that ribosylation-derived AGEs may affect brain areas that play roles in memory. In fact, the level of tau phosphorylation in the rat hippocampus (an area that plays a role in memory storage [56]) was shown to increase after treatment with RRSA, while in the cortex, there was no significant increase in tau phosphorylation (Fig. 2). Besides, RSA treated rats were shown to spend more time in the open arm compared to control rats in the EPM. They seemed to show more curiosity than the rats of other groups. However, there was no difference in the open arm entries among the rats of different groups. It is hard to consider that RSA induced more curiosity in rats.
As described previously, Rib administration by intraperitoneal injection has been shown to induce AGE accumulation in the serum and brains of mice and to lead to impaired spatial learning and memory through RAGE-dependent astrocytic inflammation [44, 57]. However, chronic oral administration of Rib for 6 months affected cognitive abilities without AGE accumulation [45]. Rib is rapidly metabolized by cells when it is absorbed in the human body. Injection of Rib leads to not only the glycation of proteins but also the activation of complex metabolic pathways such as energy metabolism pathways and the pentose phosphate pathway (’pentose shunt‘) [23, 58]. Therefore, whether Rib-derived AGEs can cause cognitive impairment was addressed in the present study by tail intravenous injection of rats with RRSA, and RRSA was found to impair memory in rats. Although RBSA and RRSA were shown to be rich in AGEs production while GBSA and GRSA were relatively low in AGEs contents (Supplementary Figure 6), we only observed that serum glycated protein levels, but not AGE levels in brain tissues, were elevated (Supplementary Figures 7 and 8). It appears that the impairment of cognitive ability occurs as an indirect effect of AGE accumulation. Ribosylated products contain some small glycated products (such as ROS). We have measured the ROS production in both cortex and hippocampus of rats (Supplementary Figure 9). Ribosylated serum albumin treated rats showed higher ROS levels in the hippocampus compared to control rats, while no difference in the ROS levels of the cortex was shown among different groups of rats. Moreover, we found that BDNF was shown to rescue the ROS increment in ribosylated AGEs rats. These data can add more evidence to our hypothesis. The administration of ribosylated products in rats results in small glycated products crossing the blood-brain barrier and affecting the hippocampus, inducing cognitive impairment.
Tau hyperphosphorylation is thought to be closely related to cognitive impairment in AD patients [59]. Under our experimental conditions, tau hyperphosphorylation was also shown in the brains of rats treated with ribosylation-derived AGEs. To clarify the cause of memory impairment, we studied the mechanism of tau hyperphosphorylation induced by ribosylated products in cultured N2a cells. Tau hyperphosphorylation was replicated in the in vitro system. We used this cell model to further investigate the regulation of tau phosphorylation by treatment with ribosylated products and found that the kinase GSK-3β partly controls tau phosphorylation. Inhibition experiments confirmed that GSK-3β is one of the major kinases involved in ribosylated AGE-induced tau phosphorylation. It should be noted that, although glucosylated AGEs were not effective to induce tau hyperphosphorylation in the present experimental condition, high concentrations of GBSA can also promote tau hyperphosphorylation in N2a cells (data not shown), indicating lower efficacy of glucosylated AGEs compared to ribosylated AGEs.
BDNF plays a role in the activity-dependent regulation of synaptic structure and function [60]. As reported by several laboratories, continuous downregulation of the hippocampal expression of BDNF by antisense oligodeoxynucleotides and acute injection of an anti-BDNF antibody into the hippocampus also disrupted the performance of rats in inhibitory avoidance learning and in the radial maze task [61–63]. The BDNF-TrkB pathway is also activated by GSK-3β [32]. The signaling cascades upstream of GSK-3β, BDNF, and TrkB were found to be involved in the activation of GSK-3β in our investigation. Treatment with ribosylation-derived AGEs decreased the levels of BDNF and TrkB in both the rat hippocampus and N2a cells. Treatment with BDNF can rescue the destructive effect of treatment with ribosylated proteins. Thus, we propose that ribosylation-derived AGEs induce tau hyperphosphorylation through BDNF reduction and GSK-3β activation, leading to cellular dysfunction and memory loss (Supplmentary Figure 10). Other factors, such as ROS, may also play roles in the effect of ribosylated AGEs on memory. Besides western blotting, more investigation should be carried out to confirm this conclusion.
In conclusion, using animal and cell models, the present study demonstrates that the BDNF-TrkB pathway is involved in ribosylated AGE-mediated tau hyperphosphorylation and memory loss.
Footnotes
ACKNOWLEDGMENTS
We are grateful to Mr. Yingge He (Institute of Biophysics, Chinese Academy of Sciences) for his assistance with the animal behavioral tests and to Ms. Junfeng Hao (Immunodeficiency Animal Laboratory, Institute of Biophysics, Chinese Academy of Sciences) for her processing of the brain tissue sections. Thanks to Dr. Jürgen Götz (Queensland Brain Institute, The University of Queensland, Australia) for editing the English language in this paper. This work was supported by grants from the National Key Research and Development Program of China (2016YFC1305900, 2016YFC1306300), the Beijing Municipal Science and Technology Project (Z161100000217141; Z161100000216137), the Natural Scientific Foundation of China NSFC (31670805, 81573763), the Youth Innovation Promotion Association CAS (2017132), the 973 Project (2012CB911004) and the External Cooperation Program of BIC, Chinese Academy of Sciences (GJHZ201302).