
Abstract
General anesthesia increases the risk for cognitive impairment and Alzheimer’s disease (AD) in vulnerable individuals such as the elderly. We previously reported that prior administration of insulin through intranasal delivery can prevent the anesthesia-induced cognitive impairment and biochemical changes in the brain. However, little is known about the underlying molecular mechanisms. Here, we report that general anesthesia resulted in downregulation of mammalian/mechanistic target of rapamycin (mTOR) and eukaryotic elongation factor 2 (eEF2) in the brain along with reduction of presynaptic proteins and brain-derived neurotrophic factor and cognitive impairment in aged mice. Prior administration of intranasal insulin prevented these anesthesia-induced changes. These results suggest the involvement of the mTOR-eEF2 signaling pathway in the anesthesia-induced brain changes and cognitive impairment and in the prevention of these changes with insulin. Correlation analyses and the use of eEF2 kinase inhibitor further support our conclusions. These studies shed light on the molecular mechanism by which anesthesia and insulin could act on synaptic proteins and cognitive function.
INTRODUCTION
Clinical studies have demonstrated that general anesthesia increases the risk for cognitive decline in elderly individuals and probably also in individuals with Alzheimer’s disease (AD) [1–4]. Some animal studies also support the risk of cognitive dysfunction with general anesthesia. General anesthesia in rodents with various types of anesthetics can cause detectable cognitive impairment [5–8]. Anesthesia also causes lasting learning and memory deficits that are detectable even two months later in mice [7]. Our recent studies found that aging and some preconditions make mice more vulnerable to neurotoxicity and cognitive impairment induced by general anesthesia [7]. However, little is known about the underlying mechanisms by which exposure to general anesthesia causes cognitive impairment. To date, there is no effective treatment that can prevent the anesthesia-induced risk for cognitive decline and AD.
Insulin has been recently recognized to play an important role in the central nervous system, including neural plasticity, learning, and memory [9]. In previous studies, we treated mice with insulin through intranasal administration, which can bypass the blood-brain barrier and deliver insulin directly into the brain [10], and found that intranasal insulin administration can prevent anesthesia-induced cognitive impairment and other long-term neurobehavioral changes [6, 7]. Intranasal insulin administration can also promote brain insulin signaling and the expression of synaptic proteins and attenuate anesthesia-induced hyperphosphorylation of tau in the mouse brain [6, 11]. It appears that insulin administered into the brain might benefit cognitive function through several pathways.
In the present study, we aimed to investigate the potential mechanism leading to the beneficial role of insulin in preventing anesthesia-induced memory deficit. We found that general anesthesia induced reduction of brain synaptic proteins and brain-derived neurotrophic factor (BDNF) and that intranasal insulin administration prevented this reduction, both likely through the mTOR-eEF2 (mammalian/mechanistic target of rapamycin – eukaryotic elongation factor 2) signaling pathway.
MATERIALS AND METHODS
Antibodies and reagents
Primary antibodies used in this study are listed in Table 1. Peroxidase-conjugated anti-mouse and anti-rabbit IgG were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). The enhanced chemiluminescence (ECL) kit was from Pierce (Rockford, IL, USA). Propofol was purchased from MP Biomedicals (Solon, OH, USA). Insulin (Humulin R U-100) was from Eli Lily (Indianapolis, IN, USA). Other chemicals were from Sigma-Aldrich (St. Louis, MO, USA).
Primary antibodies used in this study
Animals and animal treatments
The C57BL6/129 mice, which were originally obtained from Jackson Laboratory (New Harbor, 124 ME, USA) and bred in our institutional animal facility, were housed (4∼5 animals per cage) with a 12/12 h light/dark cycle and with ad libitum access to food and water. The housing, breeding, and animal experiments were in accordance with the approved protocol from the Institutional Animal Care and Use Committee of New York State Institute for Basic Research in Developmental Disabilities, according to the PHS Policy on Human Care and Use of Laboratory animals (revised March 15, 2010). Female mice at the ages of 17, 18 months were used for this study. Old mice were chosen because we previously found the aged mice are more vulnerable to anesthesia-induced cognitive impairment [7].
Intranasal administration was performed manually by using a 10μl Eppendorf pipette while mouse head was restrained in a supine position in the experimenter’s hand with the neck in extension, as described [12]. The mouse was held for an additional 5–10 s to ensure the fluid was taken into the nose. A total of 1.75 U/17.5μl insulin or 0.9% saline was delivered over both nares alternatively. All mice were treated with insulin or, as a control, saline daily for 7 consecutive days. The insulin dose and the times of administration was selected because such treatment was previously found to prevent anesthesia-induced deficit in spatial learning and memory, attenuate anesthesia-induced hyperphosphorylation of tau, and promote the expression of synaptic proteins and insulin signaling in the mouse brains [6].
One day after the last intranasal administration, anesthesia of the mice was induced by intraperitoneal (ip) injection of propofol (150 mg/kg body weight) and maintained by inhalation of 2.5% sevoflurane for 1 h. Mice in the control group received the equivalent amount of intralipid, the solvent for propofol. Some mice (n = 8/group) were sacrificed immediately post anesthesia by cervical dislocation. Other mice (n = 14/group) were returned to their home cages for Morris water maze test.
Separate experiments were performed to investigate the impact of eEF2K inhibition. For these experiments, mice were administered with the eEF2K inhibitor A484954 (10 mg/kg dissolved in 50% DMSO) or, as control groups, 50% DMSO alone through ip injection 30 min before induction of anesthesia with propofol.
After sacrifice of the mice, the forebrains were divided into rostral and caudal halves (separated coronally at the bregma level), flash frozen in dry ice, and stored at –80°C for biochemical analyses at a later date.
Morris water maze test
The spatial learning and memory of mice were evaluated using Morris water maze (MWM) test on the next day post anesthesia. The test was done in a circular pool (180 cm in diameter, 60 cm in height) filled with white dye-tinted water that was maintained at room temperature (20±1°C). The water maze was designated into four equal quadrants, named north (N), south (S), east (E), and west (W) quadrant. A platform was placed in the middle of one quadrant submerged 1 cm below water surface. Each mouse was trained in the water maze for four trials per day for four consecutive days from semi-random start positions to find the hidden platform. When the mouse climbed onto the hidden platform, the training trial was terminated. If a mouse failed to find the platform within 90 s, it was gently guided to the platform. At the end of each trial, the mouse was left on the platform for ∼20 s, then dried and returned to its home cage. Approximately 24 h after the last training trial, probe test was performed for 60 s without platform in the water maze. The swim path, swim distance (cm), escape latency (s), swim speed (cm/s), time spent in each quadrant (s), distance traveled in each quadrant (cm), latency to enter the platform site zone (s), and the number of platform site zone crossings were recorded by using an automated tracking system (Smart video tracking system, Panlab; Harvard Apparatus).
Western blot analysis
The rostral halves of the mouse brains were homogenized in pre-chilled buffer containing 50 mM Tris-HCl (pH7.4), 50 mM GlcNAc, 20μM UDP, 1.0 mM EGTA, 2 mM Na3VO4, 100 mM NaF, 0.5 mM AEBSF, 1.0μg/ml aprotinin, 10μg/ml leupeptin, and 2.0μg/ml pepstatin A. Protein concentrations of the homogenates were determined by the Pierce 660 nm Protein Assay. The samples were resolved in 10% SDS-PAGE, and the protein bands of the gel were electrotransferred onto Immobilon-P membrane (Millipore, Bedford, MA, USA). The blots were then probed with primary antibody (see Table 1) and developed with the corresponding horseradish peroxidase-conjugated secondary antibody and ECL kit.
Statistical analysis
The MWM probe test data and the quantitative data of western blots were analyzed by using one-way ANOVA, followed by Tukey’s post hoc tests using GraphPad Prism 5. The MWM training trial data were analyzed by using repeated measures ANOVA, followed by Tukey’s post hoc tests. All data are presented as means±SEM, and p < 0.05 was considered statistically significant. Linear correlation analysis was done by using MS Excel.
RESULTS
Prior administration of intranasal insulin prevents spatial memory deficit induced by anesthesia in mice
To investigate the effect of general anesthesia on cognitive function in mice, we tested spatial memory of the mice one day after anesthesia in MWM. We found that all the mice were able to learn the platform location during the training phase, as evidenced by the reduction in latency to reach the submerged platform from day 1 to day 4 (Fig. 1A). However, the anesthesia-treated mice (red) took significantly more time (p < 0.001) to locate the platform than the control mice (green), indicating impaired learning in mice after exposure to anesthesia as compared to the control mice. The learning curve of mice after prior treatment with intranasal insulin (blue) was found to be indistinguishable from those of the control mice and significantly different from those of the anesthesia group without insulin treatment, suggesting that the prior treatment of insulin prevented the anesthesia-induced learning impairment.

Effects of anesthesia and intranasal insulin on spatial learning and memory in mice. Mice were tested in Morris water maze one day after anesthesia with and without prior treatment with intranasal insulin administration. A) The latency to reach the platform during 16 training trials (4 trials/day for 4 days). The first latency to reach the platform location (B), the swimming distance within the target quadrant (C), the number of the platform location crossings (D), and the swim speed (E) of mice during the 60 s. probe test are shown. Data are presented as mean±SEM (n = 12–14 per group).
Probe trial was performed one day after the last training trial. As expected, we found increased latency to locate the platform position (Fig. 1B), decreased swimming distance within the target quadrant (Fig. 1C), and decreased number of platform position crossing (Fig. 1D) in mice after exposure to anesthesia, as compared to the control mice. These results suggest impairment in spatial memory in mice after exposure to anesthesia. The anesthesia-induced impairment in special memory was found to be largely prevented in mice with prior treatment with intranasal insulin, as these parameters between the control mice and the Anes/Ins mice were not significantly different (Fig. 1B–D). Neither anesthesia nor insulin affected the swim speed of the mice (Fig. 1E), which otherwise could affect the interpretation of the water maze data. Taking together, these observations suggest that prior administration of intranasal insulin can prevent anesthesia-induced spatial learning and memory in mice.
Intranasal insulin prevents anesthesia-induced downregulation of synaptic proteins and BDNF in the mouse brain
To investigate the possible underlying mechanism through which anesthesia and insulin affect learning and memory, we determined the levels of synaptic proteins and found that the levels of both synapsin and synaptophysin were decreased after anesthesia and that prior treatment with intranasal insulin prevented these decreases in the mouse brains (Fig. 2). Because BDNF is a neurotrophic factor important to cognitive function [13] and may also regulate synaptic protein expression [14, 15], we determined its level in these mouse brains. We found that the brain level of both BDNF precursor and mature BDNF were changed in the same way as synapsin and synaptophysin with anesthesia and intranasal insulin (Fig. 2). These results suggest a possibility that anesthesia may downregulate synaptic proteins and BDNF and that insulin may prevent these changes.
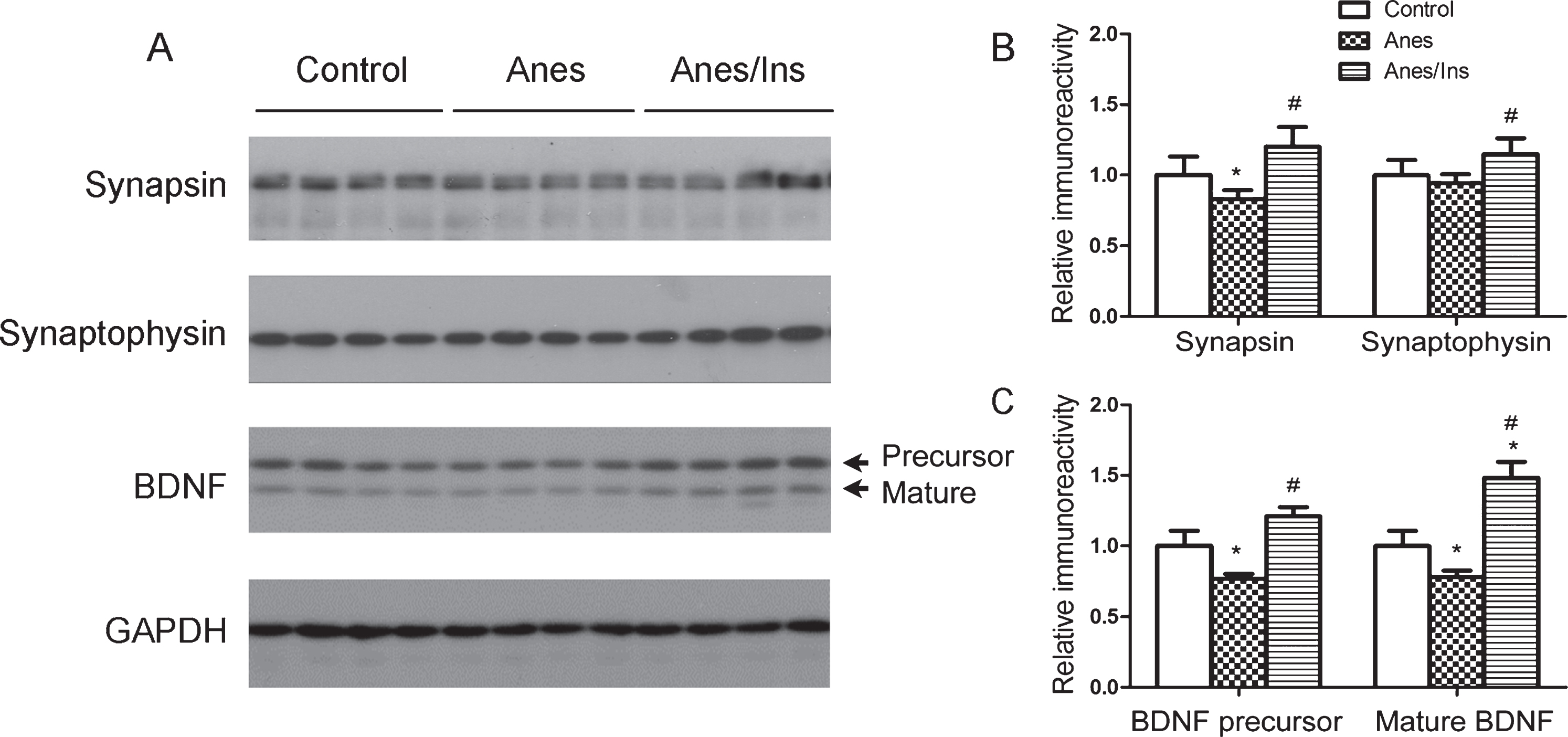
Levels of brain synaptic proteins and BDNF in mice after anesthesia and intranasal insulin administration. A) Brain homogenates of mice sacrificed at the end of 1 h anesthesia with or without prior administration of intranasal insulin for 7 days were analyzed by western blots developed with antibodies indicated at the left side of the blots. B, C) Densitometrical quantifications (mean±SEM, n = 8 per group) of the blots. *p < 0.05 versus control. #p < 0.05 versus Anes group.
Intranasal insulin promotes neural protein synthesis through the mTOR-eEF2 pathway in the mouse brain
The mTOR-eEF2 signaling pathway is known to regulate brain protein synthesis [16]. The activated mTOR complex 1 after its phosphorylation at Ser2448 and Ser2481 can inhibit eEF2 kinase (eEF2K) that phosphorylates and inactivates eEF2 and thus inhibits protein biosynthesis [17]. We thus investigated whether this pathway is involved in the downregulation of synaptic proteins with anesthesia. We found that anesthesia resulted in decreased mTOR phosphorylation/activation and increased eEF2 phosphorylation (Fig. 3). These results suggest that anesthesia inhibits mTOR signaling, resulting in activation of eEF2K and thus inactivation of eEF2 and consequently inhibition of protein biosynthesis (Fig. 4A). Intranasal insulin treatment was found to increase the phosphorylation/activation of mTOR signaling and thus counteract the anesthesia-induced effects (Fig. 3).
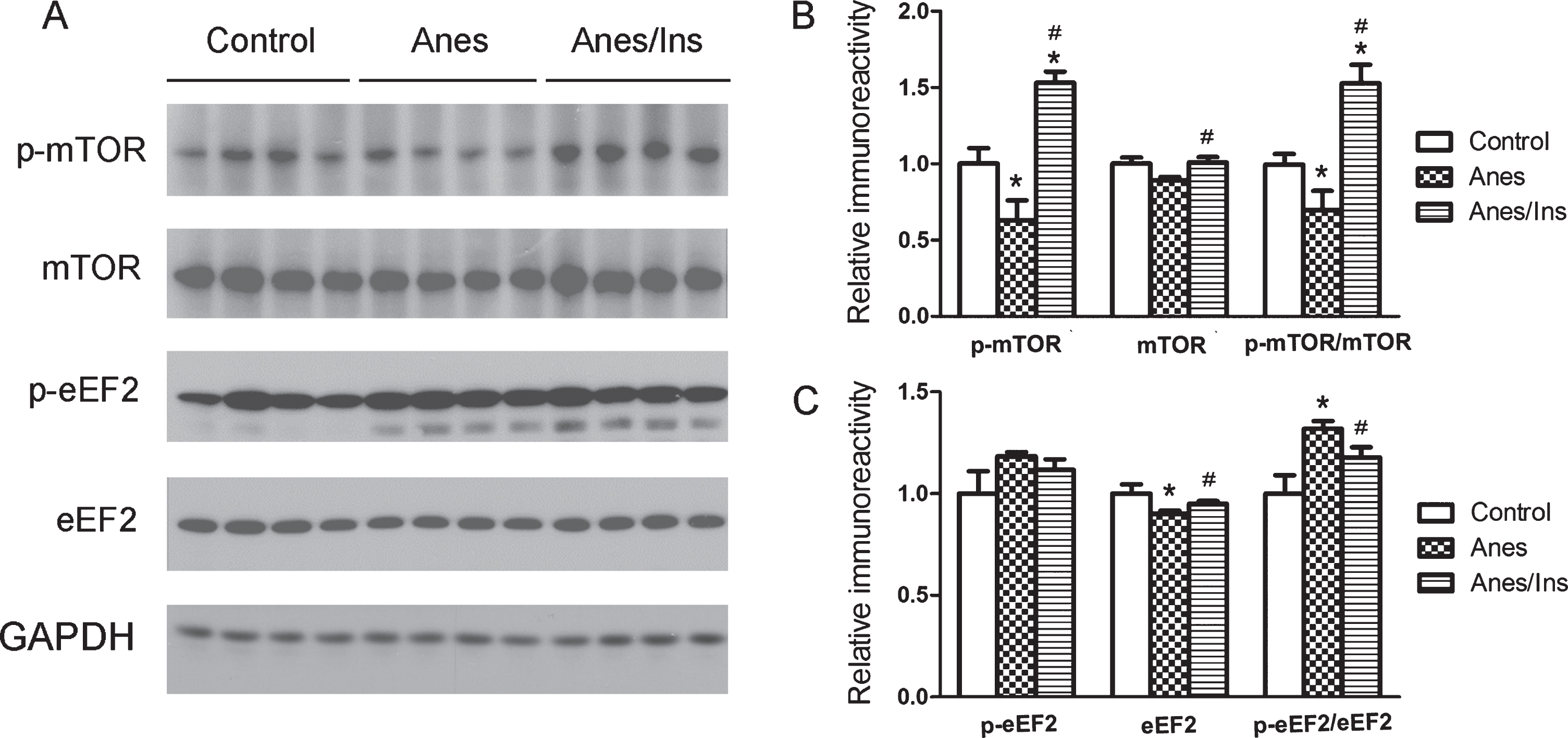
Levels and phosphorylation/activation of brain mTOR and eEF2 in mice after anesthesia and intranasal insulin. A) Brain homogenates of mice sacrificed at the end of anesthesia for one hour with or without prior administration of intranasal insulin for 7 days were analyzed by western blots developed with antibodies indicated at the left side of the blots. B,C) Densitometrical quantifications (mean±SEM, n = 8 per group) of the blots. *p < 0.05 versus control. #p < 0.05 versus Anes group.
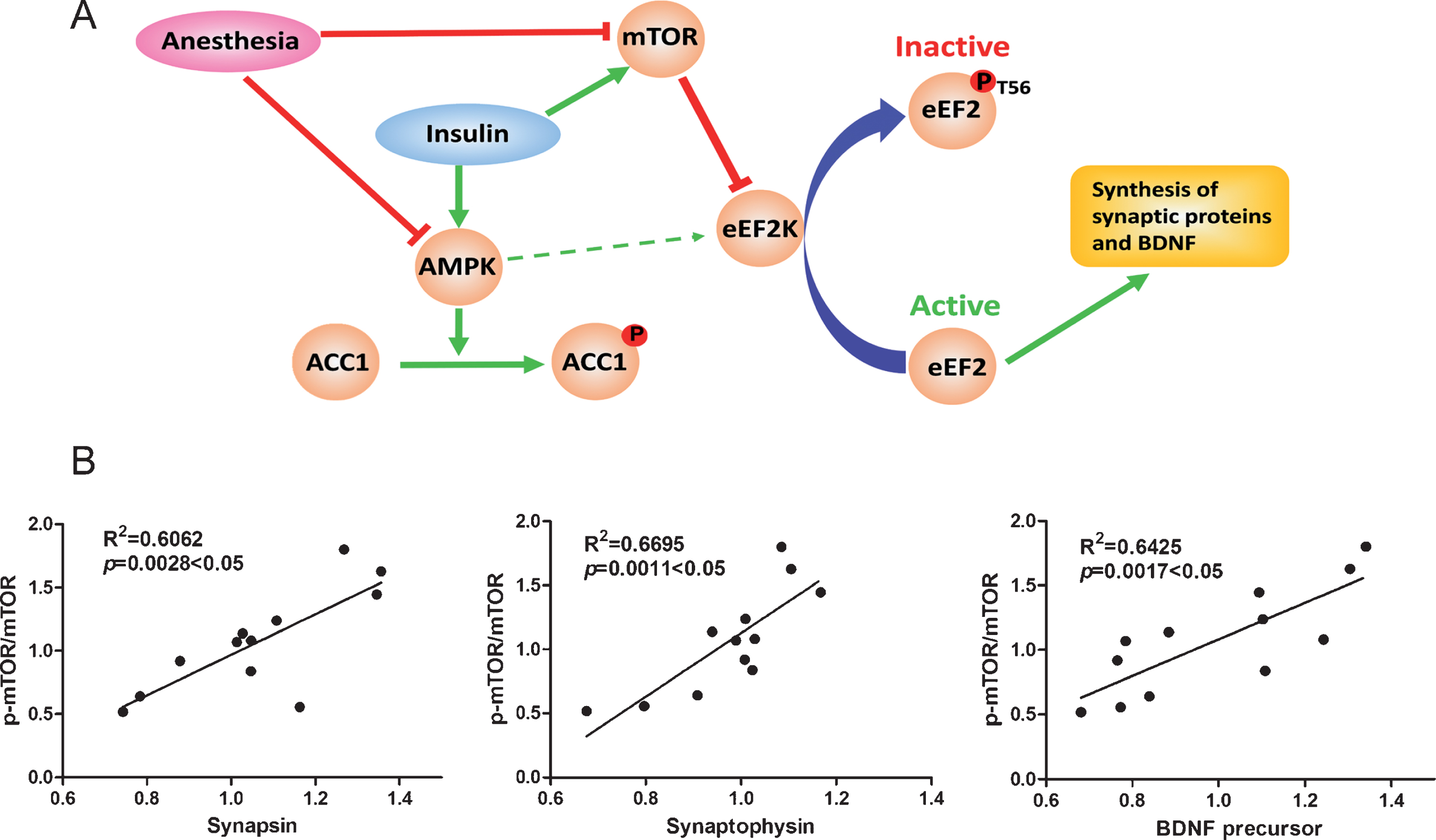
The mTOR-eEF2 signaling pathway and correlation analysis between ratios of p-mTOR/mTOR and levels of synaptic proteins and BDNF precursor in brains of mice treated with intranasal insulin and anesthesia. A) Possible mTOR-eEF2 signaling pathway by which anesthesia and insulin modulate brain protein biosynthesis. The red color indicates the consequence of general anesthesia, and the green color represents the action of insulin in the brain, which were observed in the present study. The thin, green arrowed line between AMKP and eEF2K indicates a theoretical action but it was not seen in the present study. B) The mouse brain mTOR activation, as represented by the ratio of p-mTOR/mTOR levels determined using western blots, was plotted against the levels of brain synapsin, synaptophysin, and BDNF precursor, followed by linear correlation and regression analysis (n = 12).
Correlation analysis between the ratios of p-mTOR/mTOR, which represent its activity, and the levels of synaptic proteins and BDNF precursor was performed to assess if mTOR-eEF2 signaling is involved in anesthesia-induced reduction of synaptic proteins and BDNF and the insulin’s effect. We found that the p-mTOR/mTOR ratio was positively correlated to the levels of synapsin, synaptophysin, and BDNF (Fig. 4B). These results support that the mTOR signaling pathway is involved in the regulation of expression of synaptic proteins and BDNF under anesthesia and after intranasal insulin treatment.
Inhibition of eEF2K mimics insulin in preventing anesthesia-induced downregulation of synaptic proteins and BDNF in the mouse brain
To confirm the involvement of the mTOR-eEF2 signaling pathways in the modulation of brain protein biosynthesis after anesthesia and intranasal insulin treatment, we treated mice with the eEF2K inhibitor A484954 by intraperitoneal injection before anesthesia. Quantitative western blots of the brain homogenates of these mice revealed an increased ratio of p-eEF2/eEF2 and decreased levels of BDNF precursor and synaptophysin in mice after exposure to anesthesia, though some of these changes did not reach statistical significance (Fig. 5). As expected, pretreatment with A484954 prevented anesthesia-induced increase in eEF2 phosphorylation, as shown in the p-eEF2/sEF2 ratio, and also prevented anesthesia-induced reduction of BDNF precursor and synaptophysin levels (Fig. 5). Thus, the effects of the eEF2K inhibitor on BDNF and synaptophysin were similar to those of intranasal insulin, supporting that anesthesia and intranasal insulin may modulate BDNF and synaptic protein synthesis through the mTOR-eEF2 signaling pathways (Fig. 4A).
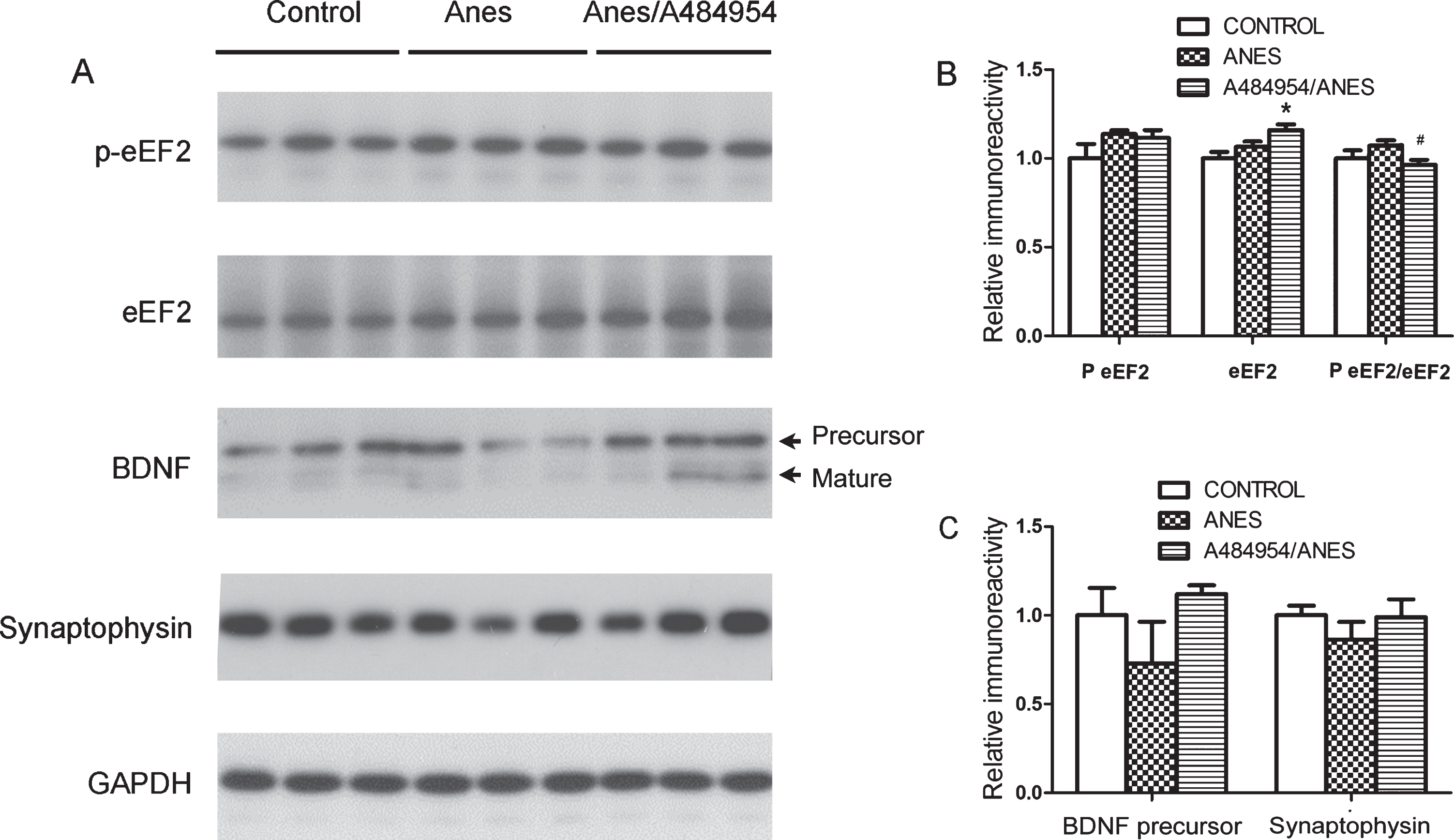
Phosphorylation of eEF2 and level of synaptophysin and BDNF in the mouse brain. A) Brain homogenates of mice sacrificed at the end of anesthesia with or without prior injection (i.p.) of A484954 were analyzed by western blots developed with antibodies indicated at the left side of the blots. B,C) Densitometrical quantifications (mean±SEM, n = 6 per group) of the blots. *p < 0.05 versus control. #p < 0.05 versus Anes group.
AMPK is activated by intranasal insulin but the activation fails to counteract the promotion of protein synthesis in the mouse brain
AMP kinase (AMPK) is a potential target of insulin and its activation involves in age-related synaptic remodeling [18]. It was also reported that AMPK can activate eEF2K [19, 20], which could in turn phosphorylate and inactivate eEF2 and inhibit protein biosynthesis (see Fig. 4A). We, therefore, investigated if AMPK is altered after anesthesia and intranasal insulin treatment. We found that anesthesia led to marked reduction of brain AMPK phosphorylation at Thr172 and that insulin increased AMPK phosphorylation remarkably (Fig. 6A, B), indicating the impact of anesthesia and insulin on brain AMPK activity. To confirm the inhibition and activation of AMPK by anesthesia and insulin, respectively, we determined the phosphorylation of a known AMPK substrate, acetyl-CoA carboxylase 1 (ACC1), at Serine 79. As expected, the phosphorylation of ACC1 was found to be increased with intranasal insulin treatment (Fig. 6A, B), confirming the activation of AMPK by insulin treatment.
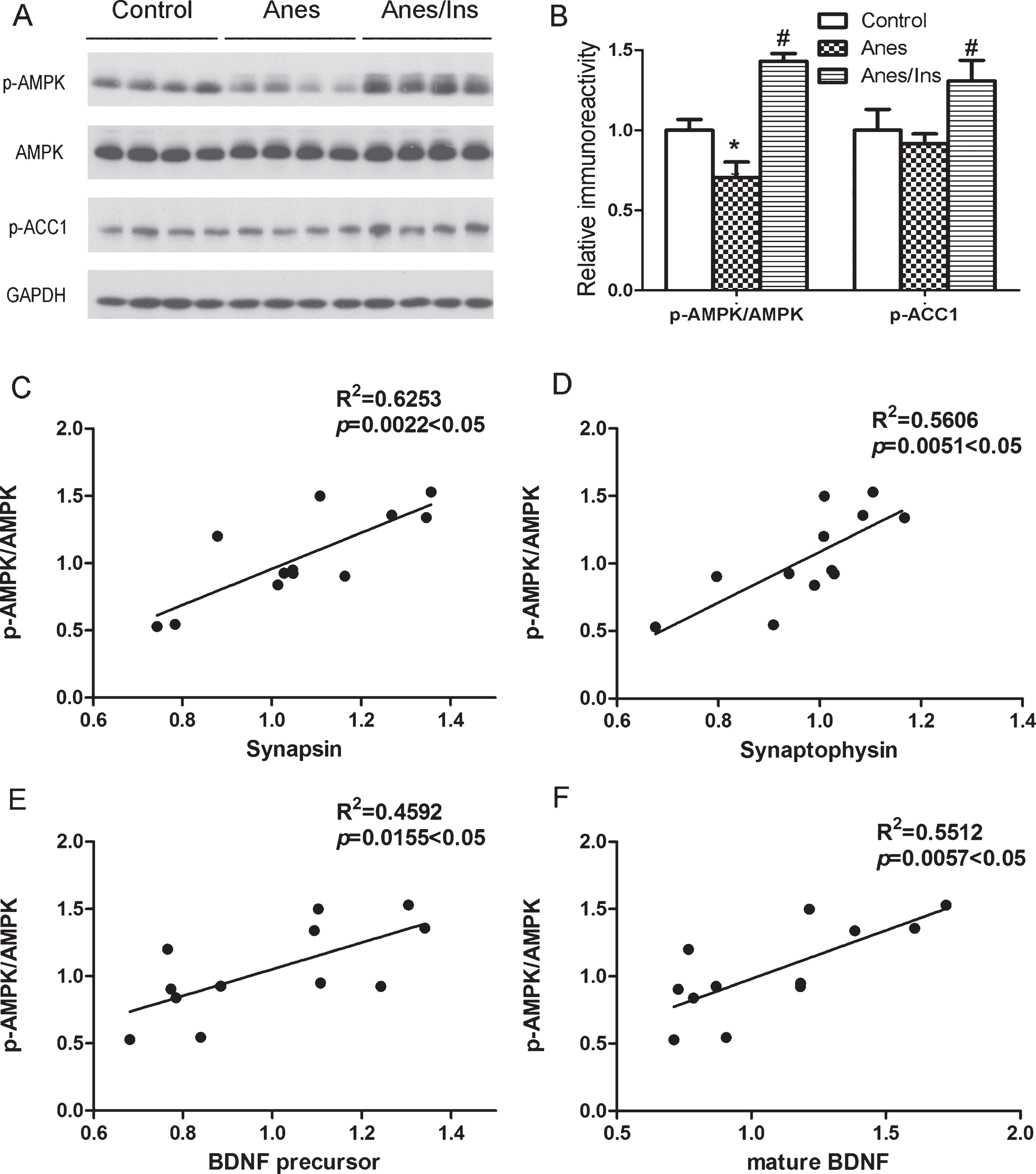
Western blots of AMPK and ACC1 and correlation analysis between p-AMPK/AMPK level and levels of synaptic proteins and BDNF. A) Brain homogenates of mice sacrificed at the end of anesthesia with or without prior administration of intranasal insulin were analyzed by western blots developed with antibodies indicated at the left side of the blots. B) Densitometrical quantifications (mean±SEM, n = 8 per group) of the blots as shown in panel A. *p < 0.05 versus control. #p < 0.05 versus Anes group. Correlation analysis between AMPK activation, as represented by the ratio of p-AMPK/AMPK levels, and the levels of synapsin (C), synaptophysin (D), BDNF precursor (E), and mature BDNF (F). A line is drawn when the correlation is statistically significant (p < 0.05).
Insulin-induced activation of AMPK could theoretically counteract mTOR-mediated activation of neuronal protein biosynthesis through activating eEF2K (see Fig. 4A). Our results that eEF2 was actually more phosphorylated after anesthesia and less phosphorylated with insulin treatment (see Fig. 3) suggest that the anesthesia- and insulin-induced changes of AMPK activity failed to counteract the action of mTOR on eEF2K and the consequent protein biosynthesis. If AMPK activity dominates the regulation of synaptic proteins and BDNF under our conditions, we would expect negative correlations between AMPK activity, as represented by the p-AMPK/AMPK ratio, and the levels of these brain proteins. However, our correlation analysis indicated positive correlations instead (Fig. 6C–F). These results further support that the anesthesia- and insulin-induced changes in AMPK activity failed to counteract the action of mTOR on eEF2K and the consequent protein biosynthesis.
DISCUSSION
Animal studies have shown that general anesthesia can cause cognitive impairment [5–8]. Both in vitro and in vivo studies suggest that anesthetics accelerate AD pathology [11, 21–24]. However, little is known how anesthesia promote cognitive impairment and AD-related brain pathology. Here, we found that the anesthesia-induced cognitive impairment was accompanied with the reduced brain levels of BDNF and presynaptic proteins synapsin and synaptophysin, likely through inhibition of the mTOR-eEF2 pathway. More importantly, we found that intranasal administration of insulin can promote mTOR-eEF2 signaling and prevent anesthesia-induced cognitive impairment and reduction of brain levels of BDNF and presynaptic proteins. Our observations provide initial evidence for the molecular mechanism explaining the impact of anesthesia and brain insulin on cognitive function. The negative impact of general anesthesia on cognitive function does not appear to be specific to anesthetic agents since various anesthetics, including injection agents and inhalational agents, can all cause cognitive impairment in animals [5–8]. Postoperative cognitive dysfunction is also seen after anesthesia with different general anesthetics [1–4]. It is a common clinical practice to induce general anesthesia by an injection anesthetic, such as propofol, and to maintain anesthesia by an inhalational agent, such as sevoflurane. We, therefore, used this combination of anesthetics in the present study for more clinical relevance. Among the possible mechanisms through which general anesthesia induces cognitive impairment, tau hyperphosphorylation [7, 25], amyloid-β protein accumulation and oligomerization [26], neural apoptosis [26], and neuroinflammation [27] have been suggested in animal studies. There are significant inconsistencies among various studies, probably due to different anesthetics, anesthesia regimens, and animal species/ages used for individual studies. Nevertheless, we found in previous studies that intranasal insulin administration prior to anesthesia, which can bypass the blood-brain barrier and deliver insulin directly into the brain [10], can prevent anesthesia-induced tau hyperphosphorylation and cognitive impairment and promote the level of synaptic proteins in the brain [6, 11]. We are particularly interested in insulin-induced brain synaptic protein levels, as they likely associate with neural plasticity and cognitive function.
The integrity of synapses is critical to neural plasticity and cognitive function. Any changes of synaptic proteins would likely alter the synaptic integrity and consequently affect neural plasticity and cognitive function. The observations of decreased levels of synaptic proteins post general anesthesia observed in the present study, as well as in previous studies [8, 29], suggest damages of synaptic integrity with anesthesia, which could underlie the cognitive impairment observed in animal studies and post-operative cognitive decline seen in patients after exposure to general anesthesia. It is known that eEF2 is an important elongation factor for neuronal protein biosynthesis [16]. Our findings of the inhibition/phosphorylation of eEF2 post anesthesia and its activation/dephosphorylation suggest that both anesthesia and insulin might impact cognitive function through the regulation of eEF2 and in turn the synaptic integrity. The activity of eEF2 is mainly regulated negatively by eEF2K that in turn is regulated by mTOR [30]. Our findings of the inhibition/dephosphorylation of mTOR and the inhibition/dephosphorylation of eEF2 in the brain post anesthesia suggest that the mTOR-eEF2 pathway is involved in the anesthesia-induced alteration of synaptic integrity and cognitive impairment (see Fig. 4A). The prevention of these changes with intranasal insulin treatment and with eEF2K inhibitor further supports our conclusions. Regulation of eEF2K by mTOR and of mTOR by insulin have been well known under other conditions [31–33].
AMPK is also known to regulate eEF2K by phosphorylating it at Ser398 and thus activate it [19, 35], and insulin can regulate AMPK [36, 37]. In the present study, we indeed observed inhibition of AMPK with anesthesia and activation of AMPK with insulin. Theoretically, AMPK could counteract mTOR’s action on eEF2K (see Fig. 4A). However, while AMPK was found to be inhibited/dephosphorylated with anesthesia, the inactivation/phosphorylation of eEF2 was not found and the opposite was observed instead. Similar scenario was seen with insulin treatment. These observations suggest that in the mouse brain, eEF2K is dominantly regulated by mTOR and AMPK fails to counteract mTOR’s action on eEF2K (see Fig. 4A). Lack of regulation of eEF2K by AMPK was also reported in skeletal muscles [38].
BDNF is an important neurotrophic factor critical to neurodevelopment and maintaining neural plasticity and cognitive function [39]. Its expression in the brain is also regulated by eEF2 [40]. Our present observations of the reduction of brain BDNF level post anesthesia and its elevation with insulin treatment, as well as the positive correlation between p-mTOR/mTOR and BDNF precursor level in the brain suggest that its expression is regulated through the mTOR-eEF2 pathway. The alteration of BDNF level under these conditions can in turn affect the cognitive outcomes as observed in the present study. The anesthesia-induced changes of BDNF level and mTOR signaling are likely to be dynamic and transient, since only transient changes in phosphorylation of tau and CREB in the mouse brains are seen after anesthesia [6, 11]. Nevertheless, the transient alterations of mTOR and BDNF signaling could have long-term impact because the downstream of the signaling and neurotrophic changes can last long.
Recent studies have demonstrated that insulin actually plays important roles in regulating neural development, neuronal activities, and learning and memory [41, 42]. Intranasal administration of insulin, which bypasses the blood-brain barrier and reaches the brain through several pathways including olfactory- and trigeminal-associated extracellular pathways and perivascular pathway, can avoid the adverse effects of peripheral administration of insulin [10]. Improved memory in both healthy adults and in AD patients have been observed after intranasal insulin treatment [43–47]. Treatment of 3×Tg-AD mice, a commonly used mouse model for AD, with intranasal insulin in our previous study led to increased expression of synaptic proteins [48]. The present observation that intranasal insulin treatment elevate synaptic protein in the brain is consistent with our previous study. In addition, the present study also provides further mechanistic insight that insulin’s action on synaptic protein expression and cognitive function are likely through the mTOR-eEF2 signaling pathway.
It should be noted that the impact of anesthesia on cognitive function was mild and the alterations of the brain proteins studied were small. These small changes indicate the complex regulation of cognitive function and the mild effect of general anesthesia only on a small part of the complex regulation. Though all the observed changes were small and some of them even did not reach statistical significance, the overall trend was consistent and clearly supports the involvement of the mTOR-eEF2 signaling pathway in the anesthesia-induced cognitive impairment. The correlation analyses further support this conclusion. More importantly, our findings suggest that insulin can act on the same pathway and prevent anesthesia-induced cognitive impairment.
In conclusion, we found that general anesthesia induced reduction of brain BDNF and synaptic proteins in addition to mild cognitive deficit and that intranasal insulin administration prevented these alterations. More importantly, we found that the mTOR-eEF2 signaling pathway is likely involved in both the anesthesia’s effect and the insulin’s action on synaptic protein expression and cognitive impairment. These studies shed light on the possible molecular mechanism through which anesthesia and insulin impact on cognitive function post general anesthesia.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the New York State Office for People with Developmental Disabilities, Albany, NY. We thank Jeffrey Goodman, PhD, of New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY, USA, for the use of his gas anesthesia equipment.