
Abstract
Alzheimer’s disease (AD) and other tauopathies are characterized by intracellular accumulation of microtubule-associated tau protein leading to neurodegeneration. Calpastatin is the endogenous inhibitor of calpain, a calcium-dependent cysteine protease that has been increasingly implicated in tauopathies. In this study, we generated a neuron specific calpastatin overexpressing knock-in transgenic mouse model and crossed it with the PS19 tauopathy mouse model expressing human P301S mutant tau protein. The forced expression of calpastatin in neurons significantly alleviated tau hyperphosphorylation measured by immunocytochemistry and immunoblot. The genetic inhibition of calpain by calpastatin also greatly suppressed characteristic hippocampal neuron loss and widespread astrogliosis and microgliosis in PS19 mice. Consistently, PS19 mice with neuronal calpastatin overexpression exhibited remarkably alleviated cognitive deficits, muscle weakness, skeletal muscle atrophy, and neuromuscular denervation, together implying the neuroprotective effects of neuronal calpastatin in PS19 mice of tauopathy. In sum, this study provides additional evidence supporting the pathological role of calpain in neurodegenerative diseases associated with tau pathology, and suggests that targeting calpain is likely a promising therapeutic approach for these devastating diseases.
Keywords
INTRODUCTION
Intracellular accumulation of microtubule-associated protein tau (MAPT) is a characteristic pathological feature of a wide range of neurodegenerative diseases known as tauopathies, which include Alzheimer’s disease (AD), frontotemporal dementia (FTD), parkinsonism linked to chromosome 17 (FTDP-17), Pick’s disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), and chronic traumatic encephalopathy (CTE) [1, 2]. AD is the most prevalent form of dementia characterized by progressive neurodegeneration in cerebral cortex and hippocampus, while FTD is the second most common form of early-onset dementia caused by neuronal loss in the frontal and temporal cortex [3]. Although no mutation in the tau gene, MAPT, has been identified in AD, mutations in MAPT account for the vast majority of FTDP-17 [4], establishing the direct link between tau dysfunction and dementia. However, although considerable efforts have been devoted to tau-based drug development [5], the mechanism underlying tau toxicity remains elusive.
Calpains are ubiquitous cysteine proteases composed of a large 80 kDa catalytic subunit and a small 30 kDa regulatory subunit, the activity of which is strictly regulated by calcium, the endogenous inhibitor named calpastatin, and other regulators such as CAPNS1[30 K]), CAPN3[p94], CAPN8[nCL-2], and CAPN9[nCL-4] [6]. Pathological activation of calcium-sensitive calpain through intracellular calcium influx has been regarded as the major cause of neuronal death in neurodegenerative conditions [7]. Not surprisingly, increased calpain activity or reduced expression of its endogenous inhibitor calpastatin have been repeatedly reported in brain samples obtained from patients with AD or other tauopathies [8–11]. Interestingly, a previous study reported the association of activated calpains with tau aggregates in AD and other tauopathies but not inclusions containing other pathogenic proteins [12], further indicating the likely specific role of calpains in tauopathies. Noteworthily, it has been shown that calpain aberrant activation can precede tau phosphorylation and even synaptic loss in AD patients [13], implying the possible involvement of calpain in disease progression.
Transgenic mice expressing FTDP-associated tau exhibit common pathological hallmarks of tauopathies such as tau aggregation, tau hyperphosphorylation, neurofibrillary tangles (NFTs) formation, synapse loss, inflammation, robust neurodegeneration, and behavioral deficits [14]. Abnormally high calpain activation was noted in the JNPL3 tauopathy mouse model expressing human P301L mutant tau protein [15]. The same study used conventional transgenic mice overexpressing human calpastatin to cross with JNPL3 mice, and found that the inhibition of calpain was able to prevent mutant tau induced motor neuron degeneration and motor dysfunction [15], indicating the protective effects of calpain inhibition against tau toxicity in motor neurons. However, it remains largely unknown whether the suppression of calpain by calpastatin can also alleviate brain neuron loss and associated pathological changes and cognitive deficits in transgenic mice for tauopathies. Here, we established a novel triple-transgenic tauopathy mouse model with neuronal calpastatin overexpression, and examined the impact of calpain genetic inhibition on total tau, hyperphosphorylated tau, brain neurons, neuroinflammation, skeletal muscles, neuromuscular junction, and cognitive function.
MATERIALS AND METHODS
Animals
Mouse procedures and behavior testing were performed according to the NIH guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) at Case Western Reserve University. PS19 tau transgenic mice (B6;C3-Tg:Prnp-MAPT*P301S PS19Vle/J, # 008169) were obtained from the Jackson Laboratory. CASTKI mice were generated by microinjection of the human calpastatin expression cassette, Cas9 mRNA prepared by in vitro transcription, and sgRNA, into zygotes for gene targeting into the Rosa26 locus. Microinjected zygotes were implanted into recipient C57Bl/6J followed by backcross and genotyping. The human calpastatin was assembled into mROSA KI-RR-LR-0512 vector by AIOTM Cloning (Biocytogen, Worcester, MA). The insertion of human calpastatin expression cassette was confirmed by PCR using the primers: Rosa26-L-GT-F (CCTCAGAGAGCCTCGGCTAG) and CAG-L-GT-R (AGTCCCTATTGGCGTTACTATGG) for a product size of 1331bp, or WPRE-R-GT-F (ACGAGTCGGATCTCCCTTTGG) and Rosa26-R-GT-R (CATGAGTCAAGCCA GTCCAAGAG) for a product of 1408bp (data not shown). CASTKI mice were crossed with Thy1-Cre mice (FVB/N-Tg(Thy1-cre)1Vln/J; #006143) to generate double transgenic TCAST mice. TCAST mice were further mated with PS19 mice to obtain triple transgenic tTg mice.
Barnes maze
As a well-known hippocampal-dependent task, Barnes maze was used to test spatial learning and memory. Twenty circular holes with 5 cm in diameter representing distinct spatial cues were located all around a circular table with a diameter of 122 cm. A target box, placed under one of the holes with the aid of visual cues for mouse to reach, was kept constant throughout the study. The four genotypes of mice included NTG, TCAST, PS19, and tTg, all of which were tested at 8 months of age. Each mouse was placed in the center of the maze at the start of the trial and given 2 min to find the target box. The time spent locating the target box was recorded. The mouse was led into the target box if it did not reach the target box in time. The mouse was trained twice every day. The test results were recorded on the fifth day. Trials were recorded with ANY-maze behavioral tracking software (Stoelting Co., Wood Dale, IL).
Open field
The open field test was used to assess general locomotor activity, anxiety, and aspiration to explore in mice. The four genotypes of mice included 8-month-old NTG, TCAST, PS19, and tTg. Each mouse was placed in the center of a square area with walls to prevent escaping. Trials were recorded for 10 min with ANY-maze behavioral tracking software (Stoelting Co., Wood Dale, IL).
Grip strength
Hindlimb grip strength was examined using the grip strength test meter from Bioseb (Vitroles, France). The mouse hind paws were placed on a metal pull bar or a wire grid while its tail was gently pulled backwards. The four genotypes of mice included 8-month-old NTG, TCAST, PS19, and tTg. Each mouse was tested 5 times and the maximum force was recorded for statistical analysis.
Immunoblot
Mice were perfused with cold phosphate-buffered saline (PBS) under deep avertin anesthesia. Brains were collected and homogenized in Cell Lysis Buffer (Cell Signaling, Danvers, MA) with 1 mM PMSF (Sigma-Aldrich, St. Louis, MO), Protease Inhibitor Cocktail (Roche, Indianapolis, IN), and Phosphatase Inhibitor Cocktail 3 (Sigma-Aldrich, St. Louis, MO). Protein extracts were separated by SDS-PAGE, followed by immunoblot analysis as described [16]. The blots were developed using Immobilon Western Chemiluminescent HRP Substrate (WBKLS0500, EMD Millipore, Burlington, MA) and imaged by the ChemiDoc Imaging System (BioRad, Hercules, CA). Primary antibodies used for immunoblot included GAPDH (Cell Signaling Technology, Danvers, MA), Calpastatin (Santa Cruz Biotechnology, Dallas, TX), and Tau46 (Cell Signaling Technology, Danvers, MA).
Hematoxylin and eosin (H&E) stain
Brain tissues were embedded in paraffin and then cut into 6- μm thick sections. After rehydration, sections were incubated in hematoxylin stain solution (RICCA, Arlington, TX) for 10 min. Sections were incubated in water for 10 min followed by differentiating with 1% acid alcohol. Then sections were counterstained in eosin solution for 5 min. Finally, sections were dehydrated using graded ethanol and xylene, and mounted with Permount mounting medium (Hatfield, PA).
Immunochemistry and immunofluorescence
Immunocytochemistry was performed by the peroxidase anti-peroxidase protocol as we described [16]. Brain tissues were embedded in paraffin and then cut into 6- μm thick sections. After rehydration, sections were treated with 1X ImmunoDNA Retriever Citrate (BioSB, Goleta, CA) for 3 min at high pressure to inactivate endogenous peroxidases. The sections were incubated with 10% normal goat serum (NGS) in tris buffered saline (TBS buffer 50 mM Tris-HCl and 150 mM NaCl, pH = 7.6) followed by rinsed with distilled H2O. After incubating the blocking serum for 30 min, sections were incubated overnight at 4°C with primary antibodies in TBS containing 1% NGS. Primary antibodies include anti-NeuN (1:1000; Millipore), anti-AT8 (Phospho-Tau Ser202/Thr205, 1:1000; Invitrogen), anti-PHF1 (Phospho-Tau Ser396/Ser404, 1:1000; PHF-161 (gift of Dr. Sharon Greenberg and Dr. Peter Davies)), anti-GFAP (1:1000; Invitrogen), and anti-Iba1 (1:1000; Wako). The sections were rinsed and incubated with 1% and 10% NGS in TBS respectively, followed by immunostaining via the peroxidase-antiperoxidase method, and developed using DAB chromogen (BioCare Medical, Pacheco, CA). After rinsed with distilled water, sections were dehydrated using graded ethanol and xylene, and mounted with Permount mounting medium (Hatfield, PA). Immunofluorescent staining of neuromuscular junction was performed as we described [16]. Gastrocnemius muscles were immersed into 30% (w/v) sucrose in phosphate buffered saline (PBS buffer 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 17.6 mM KH2PO4, pH = 7.4) in 4°C, followed by fixation in 10% formalin (Sigma-Aldrich, St. Louis, MO) for 30 min. After embedding in Tissue-Plus O.C.T compound (Fisher, Pittsburgh, PA), the GAST muscle was sectioned into 25- μm thick sections. Sections were dried in 37°C for 30 min. Then sections were incubated in the blocking buffer (2% bovine serum albumin (BSA), 0.2% non-fat dry milk, 5% NGS, and 0.5% Triton X-100 in PBS) at 4°C overnight followed by washing with PBS. The next day, sections were incubated with primary antibody in antibody buffer (2% BSA and 5% NGS in PBS) for 48 h at 4°C. Primary antibody used include anti-Neurofilament (1:100, Developmental Studies Hybridoma Bank, University of Iowa), and anti-Synaptic vesicle glycoprotein 2A (SV2) (1:50, Developmental Studies Hybridoma Bank, University of Iowa). After washing with PBS, sections were incubated with secondary antibody (Alexa Fluor 488 and α-bungarotoxin conjugated to Alexa Fluor 555, Invitrogen, Carlsbad, CA) for 2 h in room temperature in dark. After washing with PBS, sections were mounted with Fluoromount-G mounting medium (SouthernBiotech, Birmingham, AL).
Statistical analysis
Statistical analysis was performed using one-way analysis of variance (ANOVA) or Student’s t-tests. Data are means±SEM. All experiments were independently performed at least three times. A value of p < 0.05 was considered significant, and n represents number of mice per experiment.
RESULTS
Generation of triple-transgenic tauopathy mice with neuronal calpastatin overexpression
Recently, based on a conditional transgenic mouse model (CASTKI mice) with targeted ROSA26 locus insertion of an expression cassette for tissue-specific overexpression of human calpastatin, we established neuron specific calpastatin overexpression double transgenic mice (TCAST mice) by crossing CASTKI mice with transgenic mice expressing Cre under the neuron-specific promoter thymus cell antigen 1 (Thy1-Cre mice) [16]. PS19 transgenic mice expressing the P301 S mutant form of human T34 tau with one N-terminal insert and four microtubule binding repeats (1N4 R) driven by the mouse prion protein promoter are commonly used as a mouse model of tauopathy [17]. To study the role of neuronal calpain in tauopathy, we mated TCAST mice with PS19 mice to obtain triple-transgenic tau mouse (tTg mice) with neuronal calpastatin overexpression (Fig. 1A). TCAST, PS19, or tTg mice are viable and phenotypically normal at birth, and demonstrate no difference, compared to control mice (CASTKI mice or non-transgenic mice), in feeding and survival until 8 months of age (data not shown). Using an antibody specific to human calpastatin, immunoblot analysis of mouse brain tissues confirmed the similar expression of human calpastatin in both TCAST and tTg mice (Fig. 1B, C). As expected, compared with age-matched control or TCAST mice, TCAST and tTg mice exhibited greatly increased expression of total tau recognized by the Tau46 antibody (Fig. 1B). Further quantification demonstrated that human calpastatin overexpression in neurons had no significant impact on total tau expression in PS19 mice (Fig. 1D).

Generation of mutant tau transgenic mice with neuronal calpastatin overexpression. A) The flowchart of the generation of triple transgenic tTg mice. B–D) Representative immunoblot (B) and quantification (C and D) of human calpastatin (hCAST, C) and total tau (Tau46, D) in total brain lysates from NTG, TCAST, PS19, and tTg mice at 8-month old (n = 3–6 mice per group). Data are means±SEM. Experiments were independently performed at least three times. Student’s t-test or one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. *p < 0.05; ns, non-significant.
Improved cognitive function and muscle strength in tTg mice
Although motor dysfunction and cognitive deficits have consistently been reported in young or aged PS19 mice, PS19 mice begin to exhibit hind limb paralysis beginning around 9–10 months associated with a hunched-back posture [18–20], making it difficult to assess cognitive function after 9 months. Therefore, we conducted behavioral tests to investigate the cognitive and motor performances in 8-month-old PS19 or tTg mice before the appearance of paralysis. All control, TCAST, PS19, or tTg mice showed similar traveling distance and moving speed in open field tests, indicating that the locomotor activity and anxiety-related behavior were not significantly altered by mutant tau or human calpastatin expression (data not shown). The Barnes maze task consisting of 5 blocks was first used to assess the cognitive function. Unlike control or TCAST mice showing a decreased time to enter the escape hole during the course of training, PS19 mice showed no apparent decrease in time for escape in all trials (Fig. 2A), indicating impaired performance over the trials. In contrast, tTg mice displayed greatly improved ability to find the escape hole in training and testing days (Fig. 2A), suggesting that the suppression of calpain activity by calpastatin in neurons enhanced spatial learning of PS19 mice. Furthermore, despite unchanged locomotor activity, 8-month-old PS19 mice showed significant weakness and atrophy of hindlimb skeletal muscles, both of which were completely blocked in tTg mice (Fig. 2B, C). Consistently, loss of neuromuscular synapses or denervation of neuromuscular junction noted in PS19 mice was also abolished in tTg mice (Fig. 2D, E), together indicating the strong protective effect of neuronal calpastatin against mutant tau.

Neuronal calpastatin improves cognitive function and alleviates muscle wasting and denervation in PS19 mice. A) Barnes maze performance. All mice are at 8 months old. n = 10 for NTG (7 male/3 female), 8 for TCAST (4 male/4 female), 8 for PS19 (4 male/4 female), and 10 for tTg (5 male/5 female). B) The hind-limb strength of 8-month-old mice. n = 7 for NTG (4 male/3 female), 8 for TCAST (4 male/4 female), 8 for PS19 (4 male/4 female), and 9 for tTg (5 male/4 female). C) Quantification of gastrocnemius (Gast) muscle weight of 8-month-old mice. n = 7 for NTG (4 male/3 female), 8 for TCAST (4 male/4 female), 8 for PS19 (4 male/4 female), and 9 for tTg (5 male/4 female). D, E) Representative images (D) and quantification of NMJ innervation (E) in Gast muscles of 8-month-old mice. n = 7 for NTG (4 male/3 female), 8 for TCAST (4 male/4 female), 8 for PS19 (4 male/4 female), and 9 for tTg (5 male/4 female). Data are means±SEM. Experiments were independently performed at least three times. One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. *p < 0.05, **p < 0.01; ns, non-significant.
Alleviation of tau pathology in tTg mice
To understand how cognitive function and muscle strength were improved in tTg mice, we further examined the impact of neuronal calpastatin overexpression on tau pathology using two widely used specific antibodies (AT8 and PHF1) recognizing tau protein phosphorylated at serine 202/threonine 205 or serine 396/serine 404, respectively. Immunocytochemical analysis revealed that 8-month-old PS19 mice developed remarkable AT8 and PHF1-positive intraneuronal accumulation of phosphorylated tau pathology in brain neurons mainly in the hippocampus and neocortex, whereas control or TCAST brains only showed background AT8 and PHF1 staining without association with any large granules (Fig. 3A, B). Extensive extracellular PHF1 staining was also noted in PS19 mice (Fig. 3A, B). Notably, AT8/PHF1-positive intraneuronal phosphorylated tau and widespread PHF1 immunoreactivity were all greatly suppressed in tTg mice (Fig. 3A, B). Further immunoblot analysis confirmed that the levels of phosphorylated tau recognized by both AT8 and PHF1 were greatly increased in total brain lysate from PS19 mice (Fig. 3C, D). Consistent with immunostaining findings, tau hyperphosphorylation was significantly reduced in tTg mice (Fig. 3C, D), suggesting that the suppression of calpain activity by neuronal calpastatin is sufficient to prevent tau hyperphosphorylation in PS19 mice.
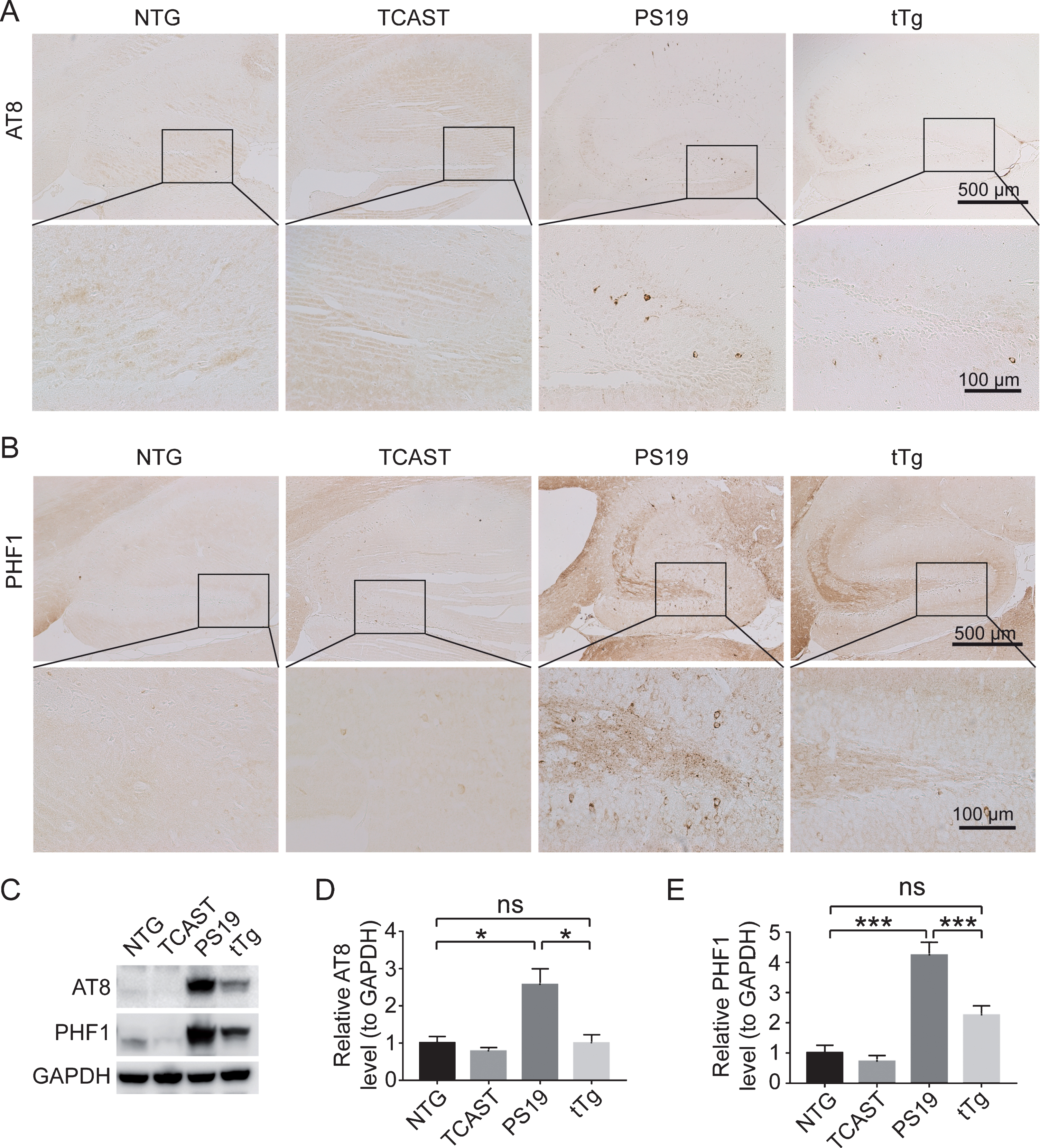
Neuronal calpastatin suppresses tau hyperphosphorylation in PS19 mice. A, B) Representative immunocytochemistry of phosphorylated tau at serine 202/threonine 205 (recognized by AT8, A) or serine 396/serine 404 (recognized by PHF1, B) in the hippocampal regions of NTG, TCAST, PS19, and tTg mice at 8 months old. C, D) Representative immunoblot (C) and quantification (D and E) of phosphorylated tau recognized by AT8 (D) or PHF1 (E) in total brain lysates from NTG, TCAST, PS19, and tTg mice at 8-month old (n = 3–8 mice per group). Data are means±SEM. Experiments were independently performed at least three times. One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. *p < 0.05; ***p < 0.001; ns, non-significant.
Protection against neuron loss and gliosis in tTg mice by neuronal calpastatin
Given our findings that behavioral deficits and tau hyperphosphorylation were alleviated in tTg mice, we also investigated the possible impact of neuronal calpastatin overexpression on tauopathy-associated neuronal loss by performing histological analysis of brains with hematoxylin and eosin (H&E) staining and immunocytochemistry using neuronal marker NeuN. Consistent with the previous study [17], 8-month-old PS19 mice showed obvious loss of neurons specifically in the CA3 region of the hippocampus (Fig. 4A, B), not necessarily associated with intraneuronal accumulation of phosphorylated tau. In contrast, neuronal density in the hippocampal CA3 of the tTg mice at 8 months of age was similar to that seen in control or TCAST mice (Fig. 4A, B), indicating that suppressing the calpain activity prevented further neuronal loss. As a prominent pathological feature of AD, neuroinflammation characterized by the proliferation and activation of microglia and astrocytes (i.e., microgliosis and astrogliosis) have also been reported in PS19 mice [17]. To examine the extent of microgliosis and astrogliosis in 8-month-old PS19 and tTg mice, we immunostained brain sections with antibodies specific against ionizing calcium binding adaptor molecule 1 (Iba1) and glial fibrillary acidic protein (GFAP) for activated microglia and astrocytes, respectively. Likely corresponding to degenerating neurons in the hippocampus, PS19 mice exhibited greatly increased microgliosis and astrogliosis in this specific brain area (Fig. 5A, B). Interestingly, age-matched tTg mice exhibited remarkably reduced both microgliosis and astrogliosis (Fig. 5B), suggesting the inhibition of tauopathy-associated gliosis by overexpressing calpastatin in neurons.

Neuronal calpastatin prevented neuronal loss in PS19 mice. A) Representative H&E staining of hippocampal neurons in 8-month old NTG, TCAST, PS19, and tTg mice. B) Representative immunocytochemistry of the pan-neuronal marker NeuN in the hippocampus of 8-month old NTG, TCAST, PS19, and tTg mice. The regional loss of neurons in the hippocampal CA3 of PS19 mice was alleviated in tTg mice.

Neuronal calpastatin inhibits astrogliosis and microgliosis in PS19 mice. A) Representative immunocytochemistry of astrocyte marker GFAP in the hippocampus of 8-month-old NTG, TCAST, PS19, and tTg mice. B) Representative immunocytochemistry of microglia marker Iba1 in the hippocampus of 8-month-old NTG, TCAST, PS19, and tTg mice. Compared with aged matched NTG or TCAST, PS19 mice showed enhanced astrogliosis and microgliosis, both of which were alleviated in tTg mice.
DISCUSSION
Here, we used our newly generated knock-in mouse model with neuronal calpastatin overexpression to test whether the genetic suppression of calpain activity is able to alleviate tauopathy-associated brain neuron loss and cognitive dysfunction. We found that the widely used PS19 tau transgenic mice demonstrated impaired spatial learning, weakness, atrophy, and denervation of skeletal muscle, tau hyperphosphorylation, hippocampal neuron loss, and gliosis at 8 months, all of which were greatly alleviated by the overexpression of calpastatin in neurons. Therefore, this study provides additional evidence supporting the pathological role of the calpain-calpastatin system in tauopathies, and implying targeting calpain as a promising therapeutic approach.
Using the Barnes maze task, we assessed the cognitive function in the PS19 mice expressing human tau bearing the P301S mutation. At 8 month of age before progressive paralysis, PS19 mice showed significant impairments of spatial learning. As significant neuronal loss and gliosis was readily seen in the hippocampus, cognitive deficits in PS19 mice should be due to dysfunction or loss of brain neurons. Although PS19 mice on the mixed B6C3F1 and C57BL/6J genetic background were unable to stay on the rotating rod, making it difficult to assess the motor coordination and balance by the rotarod performance test, they exhibited greatly diminished skeletal muscle strength, weight, and innervation, indicating the widespread toxicity of mutant tau in the central nervous system (CNS). Consistent with the previous study reporting the protective effect of calpastatin on motor neurons and motor function in the JNPL3 mice expressing tau P301L mutant [15], PS19 mice with calpastatin overexpression also showed greatly alleviated skeletal muscle weakness, atrophy, and innervation, in addition to the improved cognitive function. Although the prion protein promoter is expected to express mutant tau in the brain, spinal cord, and possibly other tissues such as heart, kidneys, and lung, calpastatin is overexpressed specifically in neurons under the neuron specific Thy1 gene promoter. Thus, the suppression of brain neuron loss and behavioral deficits in tTg mice should be a result of inhibited calpain activity in neurons.
Intraneuronal tau accumulation and tau hyperphosphorylation, prominent features of tauopathies, were recapitulated in cortical neurons of PS19 mice. Interestingly, consistent with the previous study of motor neurons [15], these tau proteinopathies could also be greatly suppressed by genetic calpain inhibition, suggesting calpain activation as a likely pivotal pathological event upstream of tau pathology. The level of total tau remained unchanged in the presence of neuronal calpastatin overexpression, indicating the unlikely direct effect of calpastatin on transgene expression. Notably, protein kinases can be substrates of calpain [21]. Future studies will be interesting to investigate whether calpastatin suppresses hyperphosphorylated tau by regulating kinases responsible for tau phosphorylation. Nevertheless, since previous studies reported the generation of tau fragments by calpain-mediated tau cleavage [11, 15], the inhibition of aggregation prone tau fragments, combined with reduced levels of phosphorylated tau, may be a possible mechanism by which neuronal calpastatin alleviates tau pathology and associated neuroinflammation and neurodegeneration in tTg mice. It remains to be determined how the calpain-calpastatin system is activated in response to mutant expression. Considering the critical role of calcium for both calpain activation and tau phosphorylation [22], future studies are expected to address the interplays between calpain activation, tau phosphorylation, and calcium homeostasis in tauopathies. Notably, calpain inhibition alone has no effect on AT8 or PHF-1 recognized tau phosphorylation in TCAST mice, further suggesting that the calpain-calpastatin system may play a specific role in tau pathology rather than under generalized physiological conditions.
Neuropil vacuolization but not neuronal loss has been reported in the hippocampus of very old transgenic mice overexpressing wild type tau [23]. Hippocampal neuron loss in PS19 mice should be specifically induced by the mutation but unlikely due to human tau overexpression. In 8-month-old PS19 mice, the intraneuronal accumulation of tau or tau hyperphosphorylation is not restricted to the CA3 region of the hippocampus where neurodegeneration was observed, suggesting that tau pathology and neuronal loss may be independent pathological events during disease progression. Since calpain over-activation through ion channel mediated intracellular calcium influx has long been implicated as the major cause of neuronal death [24–26], the suppressed neuronal loss in tTg mice may at least be partially attributed to calpain-mediated death pathways not related to tau pathology. Along this line, as a prominent early feature of tauopathies, mitochondrial dysfunction has been reported in PS19 mice before the appearance of tau pathology or brain atrophy [27]. And, our most recent study has found that mitochondria can act like miniature conveyors to transiently carry calpastatin-enriched mitochondria-associated membranes to mediate the activity of calpain [16]. Thus, although beyond the scope of the present study, the potential involvement of mitochondria in calpastatin-mediated neuronal protection could not be ruled out, and future studies are needed to explore this aspect.
In summary, we show that the genetic inhibition of calpain activity by calpastatin overexpression is sufficient to greatly alleviate mutant tau-associated cognitive deficits, skeletal muscle abnormalities, tau pathology, brain neuron loss, and neuroinflammation, consolidating the pathological role of calpain in tauopathies. Mutant tau may exert its neuronal toxicity via the aberrant activation of calpain. And, inhibition of calpain is likely a promising therapeutic approach for neurodegeneration in AD and other tauopathies.