
Abstract
Vascular dementia (VaD) is caused by chronic decreases in brain blood flow and accounts for 15–20% of dementia cases worldwide. In contrast to Alzheimer’s disease (AD), no effective drug treatments are currently available for VaD. Previous studies have suggested that oxidative stress and neuroinflammation in the brain play important roles in the pathogenesis of VaD. Honokiol (HKL) is a well-known bioactive and nutraceutical compound that can act as an antioxidant and anti-inflammatory molecule. HKL can protect against memory impairments in AD mouse models. In this study, we explored whether the application of HKL was also protective against the insult of chronic cerebral hypoperfusion (CCH) in rats. We found that HKL supplementation prevented the memory impairments in the inhibitory avoidance step-down and Morris water maze tasks in CCH rats. HKL also suppressed the levels of oxidative stress and inflammation in CCH rats. Moreover, HKL prevented dendritic spines abnormalities in CCH rats. We also found that HKL inhibited the activity of GSK-3β, which may be critical for the neuroprotective activity of HKL. Thus, our study demonstrated the protective role of HKL in VaD.
INTRODUCTION
Vascular dementia (VaD) is the second most common form of dementia that results in considerable morbidity and mortality [1]. The clinical manifestations of VaD are similar to other dementias, which are characterized by progressive cognitive impairment, memory loss, and thinking or speech problems [2]. Unlike other causes of dementia, such as Alzheimer’s disease (AD), Parkinson’s disease, and frontotemporal lobar degeneration, effective and approved pharmacological treatments for VaD are not available. Currently, the widely used medications for AD, such as donepezil (Aricept), galantamine (Reminyl), rivastigmine (Exelon), or memantine, are not used to treat VaD. Thus, it is urgent to develop new therapeutic drugs for VaD.
Although an effective therapy for VaD has not been developed, the potential underlying mechanisms of VaD have been extensively studied. For example, interactions between aberrant oxidative stress and inflammatory processes were found in VaD, and these two hazard factors resulted in endothelial damage and blood-brain barrier (BBB) failure [3]. Markers of oxidative stress (isoprostanes) and inflammation (cytokines and adhesion molecules) in the damaged white matter were highly correlated with the progression of VaD [4, 5]. In rodent VaD models, indicators of oxidative stress and inflammation can be found in the white matter and have been associated with cerebral hypoperfusion [6]. Hypertension, insulin resistance, and diabetes can lead to vascular oxidative stress and inflammation, both in animal models and in humans [7, 8]. Based on these findings, numerous studies have developed different treatments that target reactive oxygen species (ROS) production and inflammation for the therapy of VaD [9–11]. Honokiol is a polyphenolic compound that can cross the BBB and plays a neuroprotective role in many diseases, such as anxiety, pain, cerebrovascular injury, and epilepsy [12]. The neuroprotective roles of HKL have also been found in many animal models with memory impairments [13, 14]. However, whether HKL can be used to prevent oxidative stress, inflammation and memory impairments in VaD is not clear.
We designed this study to examine whether HKL administration is able to restore memory impairments in VaD rats. We also wanted to determine whether HKL treatment could prevent oxidative stress, neuroinflammation, and synaptic degeneration in the brain and to explore the possible mechanisms.
MATERIALS AND METHODS
Animals
Wistar rats (age, 3 months; weight, 220–250 g) were obtained from Zhengzhou University Laboratory Animal Center. They were housed in polypropylene cages in a vivarium with food and water available ad libitum under a 12:12 h light/dark cycle (lights on at 8 a.m.). All animal experiments were approved by the Care and Use of Laboratory Animals of HeNan Province. The animals were randomly divided into four groups: a sham-operated control group (sham; n = 12), a cerebral hypoperfusion group (CCH; n = 12), a cerebral hypoperfusion group with HKL treatment 4 weeks (5 mg/kg/day, CCH+HKL, n = 12) and an HKL single administration group (5 mg/kg/day, HKL, n = 12). We administered HKL (5 mg/kg; Sigma-Aldrich Inc., St. Louis, MO, USA) dissolved in DMSO once daily by intraperitoneal injection beginning the first day after surgery (Fig. 1).
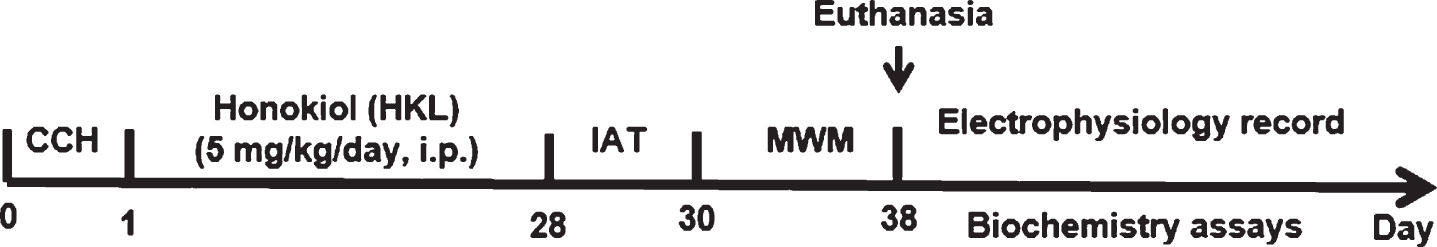
Timeline of the experimental procedures in the study. After chronic cerebral hypoperfusion (CCH), the animals were administered HKL (5 mg/kg/d·rat per os) for four weeks. Then, the learning and memory abilities of rats were assessed in an inhibitory avoidance task (IAT) and a Morris water maze (MWM). After the behavioral tasks were completed, some rats (n = 6 each group) were subjected to in vivo electrophysiology and Golgi staining, and the other rats were prepared for providing hippocampal samples to detect biochemical biomarkers.
Chronic cerebral hypoperfusion
Chronic cerebral hypoperfusion (CCH) was performed as previously described by bilateral common carotid artery stenosis [15]. Briefly, rats were anesthetized with 10% chloral hydrate (350 mg/kg, i.p.), then the bilateral common carotid arteries were exposed and freed from the surrounding connective tissues through a midline cervical incision. For hypoperfusion, metal coils (180μm inner diameter, Sawane Spring Company, Hamamatsu, Japan) were positioned to encircle the common carotid arteries, reducing blood flow to approximately 70%. Laser Doppler flowmetry was used to analyze cerebral blood flow (CBF) [16]. During surgery, the body temperature of the rats was monitored and maintained at 37.5±0.5°C by means of a heating lamp until the rats had recovered to thermal homeostasis. Four weeks after surgery, the percentage of mortality is about 35%. Then the rats with successful recovery performed the behavioral tasks at 9:00 every day.
Step-down inhibitory avoidance task
The step-down inhibitory avoidance task (IAT) was used to assess short-term memory and long-term memory. The experimental device (STT-2, Chinese Academy of Medical Sciences, Beijing, China) comprised two chambers of equal size (30 cm×25 cm×25 cm), made of Plexiglas on three sides and black plastic on the other side, with a bottom that was an electrified grid made of a series of parallel caliber bronze bars spaced 1.0 cm apart. A platform (height 5 cm, diameter 5 cm, rubber) was fixed on the bottom of the chamber in one corner to provide a shelter from the electric shock. Before the training period, the animals were given 5 min of free exploration and were then placed on the platform; immediately after stepping down onto the grid, a 0.4-mA, 1.0-s scrambled foot shock was administered. The test sessions without foot shock were performed 1.5 h and 24 h after training. The step-down latency (maximum 180 s) was used as a measure of memory retention of the animals [17].
Morris water maze test
The Morris water maze was used to assess the spatial learning and memory of rats [18, 19]. The animals were trained to find a hidden platform submerged 1-2 cm below the water surface in a pool (180 cm diameter) using a stationary array of cues in the room. Acquisition training with the hidden platform was performed over five consecutive days and included 20 trials. In each trial, the rat was placed into the pool at one of four possible locations (randomly ordered). The escape latency was defined as the time spent to reach the escape platform. If the rat did not find the platform within 60 s, it was gently guided by the observer to the platform where the rat remained for 30 s. The day after training, a 60-s probe test was conducted in which the platform was removed. During the test period, the swimming paths, percentage of time spent in the target quadrant, platform area crossings, and swimming speed were recorded and calculated by computer software.
Golgi staining
Golgi staining was performed as previously described [19, 20]. Briefly, tissue slices (∼3 mm thick) were placed in mordant (5% chloral hydrate, 5% potassium dichromate, 10% formalin in ddH2O) for 3 days. Then, the tissue was placed in 1% silver nitrate under continuous vacuum for 4 days. Finally, the brain tissue was cut into 50-μm thick sections with a vibratome and analyzed using a light microscope (Olympus BX60, Tokyo, Japan) with identical settings (×100 objective lens). The number of dendritic spines and the percent of mushroom-like spines on hippocampal dentate gyrus (DG) pyramidal neurons were analyzed. For each experimental group, a minimum of 40 cells per animal (n = 4) were analyzed by Image-Pro Plus 6.0 software.
Western blotting
Western blotting was performed as described previously [21]. The rats were sacrificed 24 h after the behavioral tasks. The cerebral hemisphere was carefully removed under a dissection microscope. The right hippocampi were removed and homogenized in lysis buffer (50 mM Tris (pH 8.0), 150 mM NaCl, 0.02% sodium azide, 0.2% SDS, 100μg/mL phenylmethylsulfonyl fluoride, 100 mM NaF, 0.5% sodium deoxycholate, 0.5 mM EDTA, 0.1 mM EGTA). The homogenates were centrifuged at 10000×g for 30 min at 4°C, and the supernatant fractions were collected for western blotting. After measurement of protein concentration using a BCA kit (Pierce, Rockford, IL, USA), a final concentration of 10% β-mercaptoethanol and 0.05% bromophenol blue was added, and the samples were boiled in a water bath for 10 min. An equal amount (20μg) of protein from each sample was resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat milk for 1 h and then incubated with antibodies GSK-3β (anti-total GSK-3β, 1:1000) and pGSK-3β (anti-phospho-GSK-3β at Ser9, 1:1000). After 24 h, the membranes were rinsed three times, incubated with secondary antibodies for 2 h at room temperature, and visualized using the Odyssey Infrared Imaging System. Protein bands were quantitatively analyzed by Kodak Digital Science 1D software (Eastman Kodak Company, New Haven, CT, USA).
Enzyme activity assay
Activities of GSK-3β and Akt (Protein kinase B, PKB) were determined by the GENMED kit (Genmed, MA, USA) according to manufacturer’s instructions respectively. Briefly, the hippocampi of different group rats were rinsed twice by reagent A and then homogenized with the extraction buffer reagent B. After incubation, the homogenized mixture was centrifuged twice at 10 000×g for 10 min at 4°C. For GSK-3β activity assay, by mixing 65 ml reagent C, 10 ml reagent D, 10 ml reagent E and 10 ml reagent F and incubation for 3 min at 30°C. Immediately after the addition of 5 ml supernatant to the reagent mixture, the optical density was measured at 340 nm every 30 s for 2 min. The activity was measured as the difference between the absorbance value at 0 and 120 s and repeated twice. For Akt activity assay, 10μg protein was incubated with antibody-HRP 60 min at 37°C, then 50μl substrate A and 50μl substrate B were added to the reaction system. After 15 min at 37°C, an absorbance of 340 nm at 0 min and 5 min was measured. The difference between the absorbance at the fifth minute and the immediate absorbance represented the activity.
Real-time PCR
The total RNA from the hippocampus was extracted by TRIzol reagent (Invitrogen) according to the manufacturer’s instructions and 1 lg RNA was reversely transcripted. The cycle conditions were set as follows: initial template denaturation at 95°C for 1 min, followed by 40 cycles of denaturation at 95°C for 5 s, and combined primer annealing/at 60°C for 30 s, and elongation at 72°C for 30 s. This cycle was followed by a melting curve analysis, ranging from 60 to 95°C, with temperature increasing by steps of 0.5°C every 10 s. All reactions were performed in triplicate. For the evaluation of mRNA, synthesis of cDNA was performed using a RNA PCR kit (Takara, Otsu, Shiga, Japan) and quantitative real-time PCR was carried out with the SYBR premix Ex TaqII kit (Takara) according to the manufacturer’s instructions. The primer sequences were as followed: GSK-3β forward, 5’- ATAGATGTATGGTCTGCAGGCTGTG-3’ and reverse, 5’-AAGACCTTAGTCCAAGGATGTGCCT-3’; β-actin forward, 5’-CACGAAACTACCTTCAACTCC-3’ and reverse, 5’-CATACTCCTGCTTGCTGATC-3’. The 2-ΔΔCt method was adopted and applied to calculate the relative quantities of each gene [22]. β-actin was used as an endogenous control.
Assays of oxidative damage
The left hippocampi were dissected on ice and ground into 10% tissue homogenates with 0.1 M PBS buffer solution (pH 7.4). The tissue homogenates were centrifuged at 10000×g at 4°C for 10 min, and the supernatants were collected and prepared for the measurement of superoxide dismutase (SOD) activity, malondialdehyde (MDA) content, and lactate dehydrogenase (LDH) according to the manufacturer’s instructions and protocols for the detection of inflammatory cytokines. The ROS content was measured using the dichlorofluorescein method as previously described [23].
Detection of inflammatory cytokines
The inflammatory cytokine levels in the hippocampus were assessed by using TNF-α, IL-1β, IL-6 and IL-10 assay kits (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. The concentrations of these inflammatory cytokines were calculated according to the spectrometrically determined optical density using a micro enzyme-linked immunosorbent assay (ELISA) reader.
Statistical analysis
Statistical analysis was performed using SPSS 19.0. The differences between the groups were assayed by ANOVA followed by LSD’s post hoc test. For a single comparison, the significance of differences was determined by the t-test. A value of p < 0.05 is considered to be statistically significant.
RESULTS
Effects of HKL on CCH-induced memory deficits
We first examined the effect of HKL treatment on the memory retention of CCH rats. Four weeks after surgery, the rats were subjected to the step-down IAT. When the training period ended, the short-term memory (STM) and long-term memory (LTM) were measured at 1.5 h and 24 h. Two-way repeated measures ANOVA showed a significant time-course effect (F(6, 22) = 18.36, p < 0.001) and different treatment effects (F(3, 36) = 6.31, p < 0.05) in STM, while the interaction between time-course and treatment was not significant (F(18, 36) = 1.67, p > 0.05). CCH did not significantly decrease the latency during training and in the STM test. In the LTM test, however, CCH significantly decreased the escape latency of rats (F(1, 20) = 12.66, p < 0.01), and treatment with HKL not only prolonged the reduced latency produced by CCH (F(1, 20) = 7.65, p < 0.05) but also resulted in better performance (F(1, 20) = 8.18, p < 0.05) than that in the controls. These results suggest that HKL reverses long-term fear memory deficits induced by CCH.
Effect of HKL on CCH-induced spatial memory deficits
We also assessed the spatial cognitive abilities of rats using the Morris water maze. After CCH, the rats spent more time finding the hidden platform on the 5th day of the training period than the controls (p < 0.05, Fig. 3A), while the HKL-treated rats showed a shorter latency to find the hidden platform than the CCH animals (p < 0.05). In the spatial probe test, the CCH rats had a longer latency to reach the original platform zone (F(1,18) = 9.16, p < 0.01, Fig. 3B) and spent less time in the target quadrant (F(1,18) = 8.32, p < 0.01, Fig. 3C) than the controls, implying that memory retention was impaired after CCH. Compared to the CCH rats, HKL rats remembered the previous platform location. HKL rats had a shorter latency to reach the original platform zone (F(1,18) = 5.96, p < 0.05) and spent more time in the target quadrant (F(1,18) = 6.19, p < 0.05). Throughout the tasks, the swimming speed of the rats showed no significant differences among the groups, implying that the effect of the drugs was on learning and memory rather than on swimming ability (Fig. 3D).
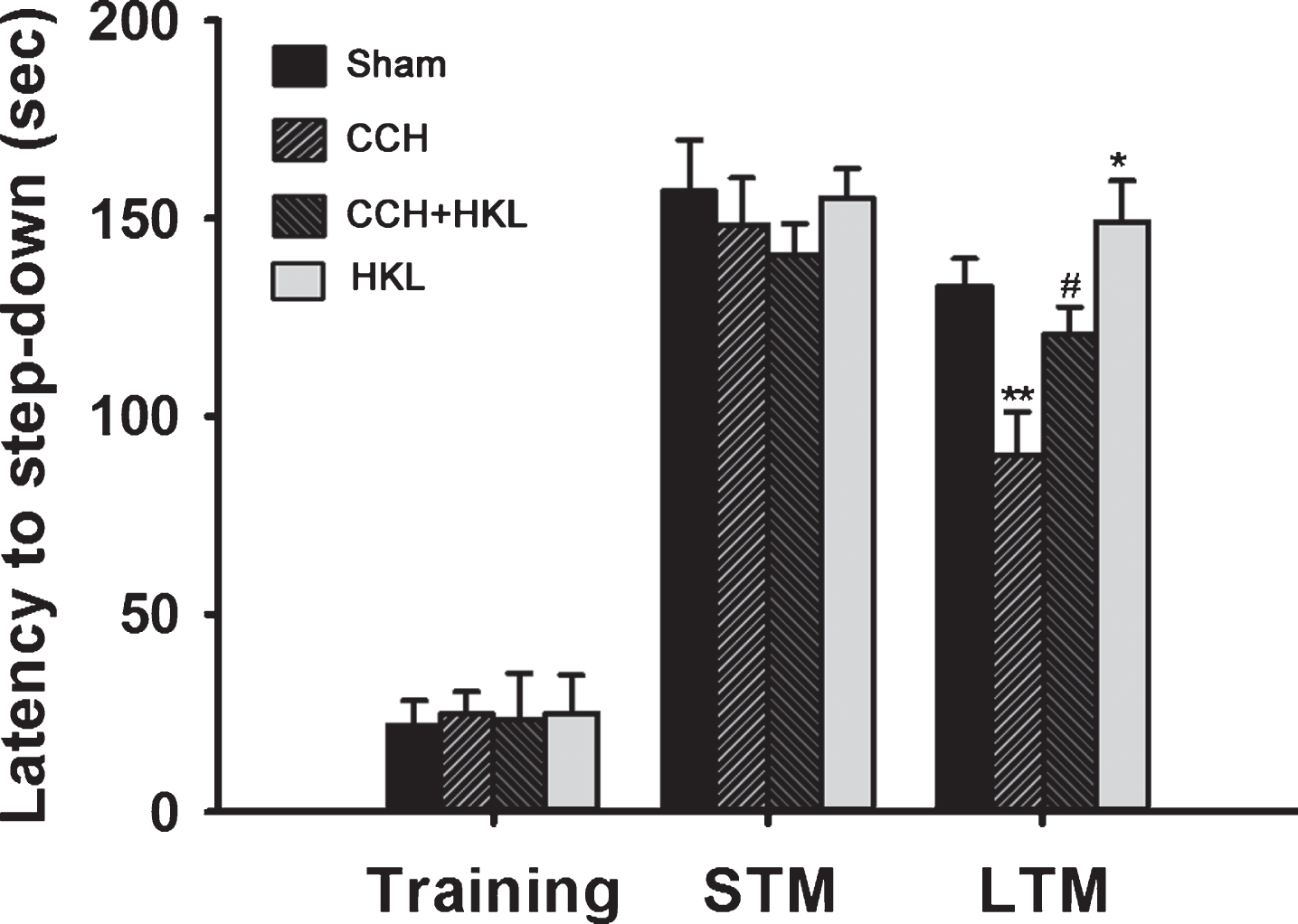
Effects of HKL on CCH-induced memory deficits. The animals were randomly divided into four groups: a sham-operated control group (sham), a cerebral hypoperfusion group (CCH), a cerebral hypoperfusion group with Honokiol treatment (CCH+HKL), and a Honokiol single administration group (HKL) as described in the Methods section. After training, rats were tested for memory retention after 1.5 and 24 h for short-term memory (STM) and long-term memory (LTM), respectively. Step-down latencies were measured up to a ceiling of 180 sec, and no foot shock was administered during test sessions. All values are expressed as the mean±SEM (n = 11). *p < 0.05, **p < 0.01 versus sham group; #p < 0.05 versus CCH group.
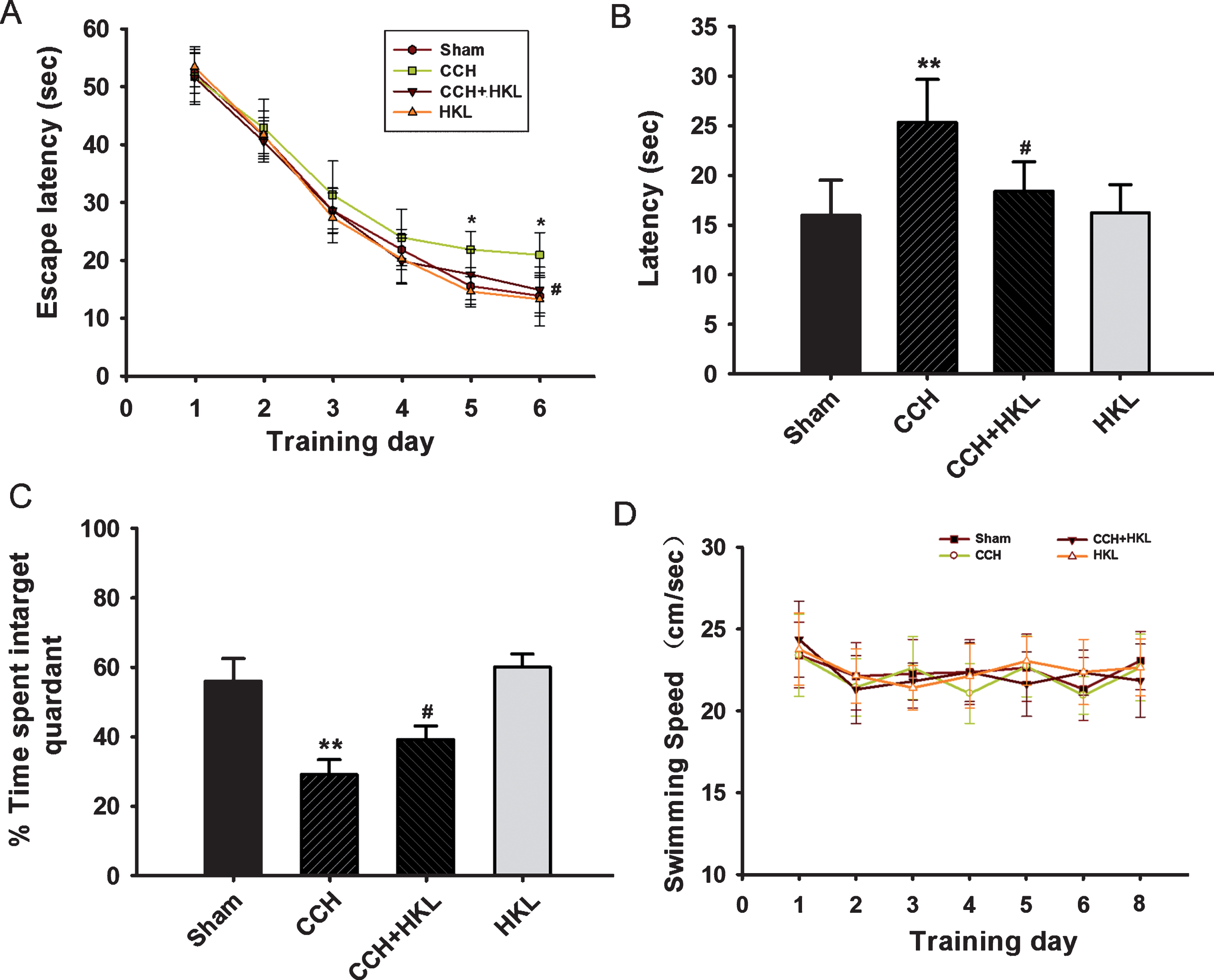
Effects of HKL on CCH-induced spatial memory impairments. A) The escape latency to find the platform during the training period. B) The latency to first cross the platform zone during the probe test. C) The time spent in the target quadrant. D) The average swimming speed of the rats each day. All values are expressed as the mean±SEM (n = 10). *p < 0.05, **p < 0.01 versus sham group; #p < 0.05 versus CCH group.
HKL reduces oxidative stress in the hippocampus
CCH and/or HKL can interrupt the balance of oxidation and antioxidation in the brain [24], so we examined the oxidative status, including SOD and glutathione peroxidase (GSH-Px) activity levels as well as MDA and ROS content, in the hippocampus of rats (Fig. 4). We found that compared to the control condition, CCH caused SOD (control, 4.33±0.42 U/mg; CCH, 2.46±0.54 U/mg, p < 0.01) and GSH-Px (control, 0.45±0.08 U/mg; CCH, 0.28±0.05 U/mg, p < 0.01) activity levels to decrease significantly, while compared with the CCH treatment, HKL could ameliorate the lower activity levels of SOD (3.48±0.34 U/mg) and GSH-Px (0.35±0.04 U/mg) (p < 0.05). We also found that the MDA and ROS levels in the hippocampus were much higher in the CCH group than in the control group (p values < 0.01), and HKL administration ameliorated these effects (p < 0.05 and p < 0.01, respectively). Taken together, these results implied that HKL could prevent CCH-induced oxidative stress in the hippocampus.
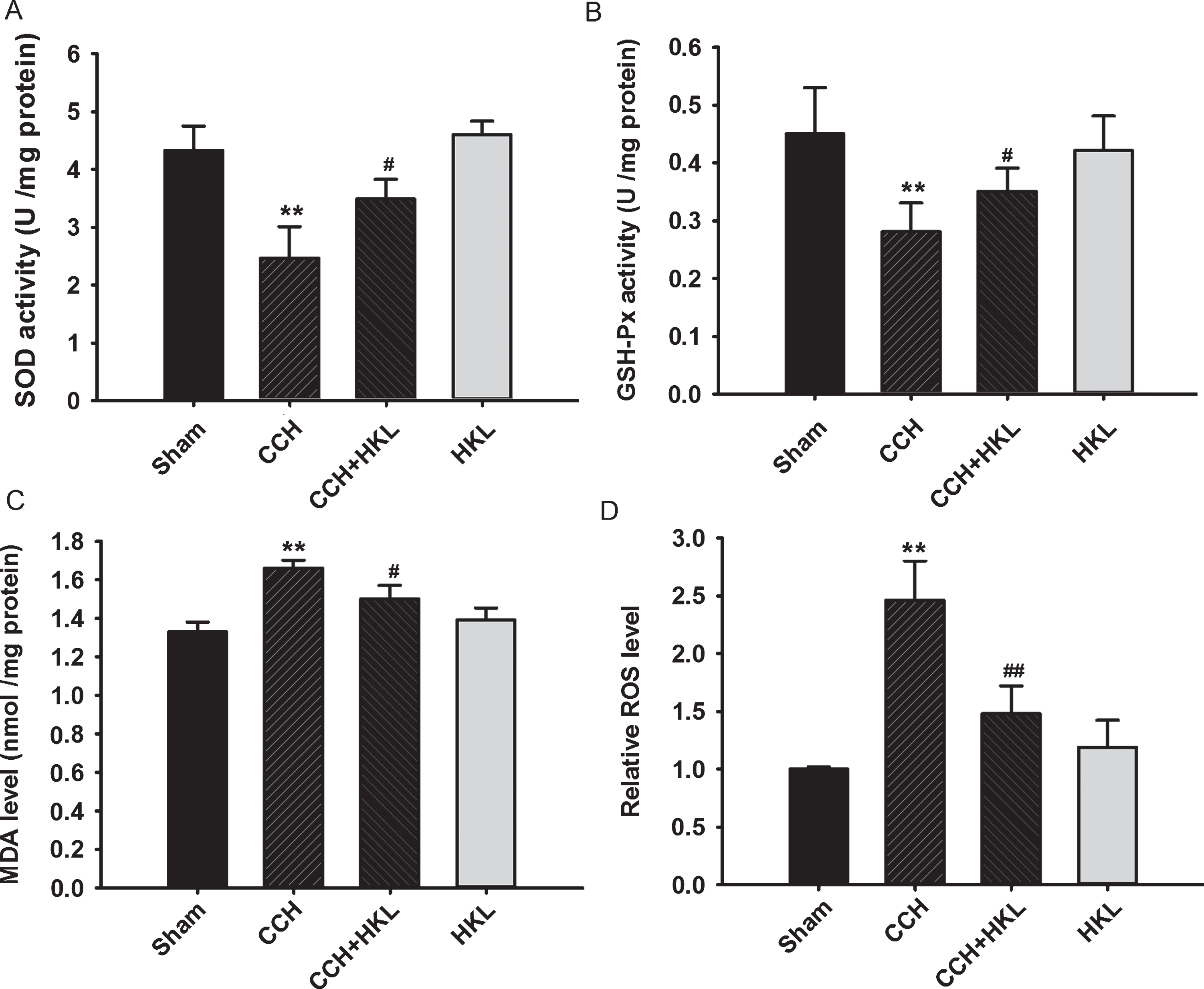
HKL prevents CCH-induced oxidative stress in the hippocampus. The activity of superoxide dismutase (SOD) (A) and glutathione peroxidase (GSH-Px) (B) and the levels of malondialdehyde (MDA) (C) and reactive oxygen species (ROS) (D) in the hippocampus were measured using enzyme-linked immunosorbent assays (ELISAs) and enzyme activity assays. Data are expressed as the mean±SD (n = 5). **p < 0.01 versus sham group; #p < 0.05, # #p < 0.01 versus CCH group.
HKL attenuates the CCH-induced inflammatory response in the hippocampus
CCH can upregulate the expression of proinflammatory factors; therefore, we detected changes in hippocampal interleukin levels using ELISA kits to investigate the anti-inflammatory effect of HKL following CCH. Compared to the control conditions, CCH significantly increased the levels of TNF-α, IL-1β and IL-6 (p values < 0.01), implying a stronger inflammatory response, which was prevented by HKL (p values < 0.05, Fig. 5A-C). In contrast, the anti-inflammatory IL-10 levels in the CCH group were lower than those in the control group (p < 0.01, Fig. 5D), and HKL partly improved IL-10 levels. These results indicated that HKL had some anti-inflammatory effect in the brain after CCH. There were no significant differences in all inflammatory factor levels after treatment with HKL alone.
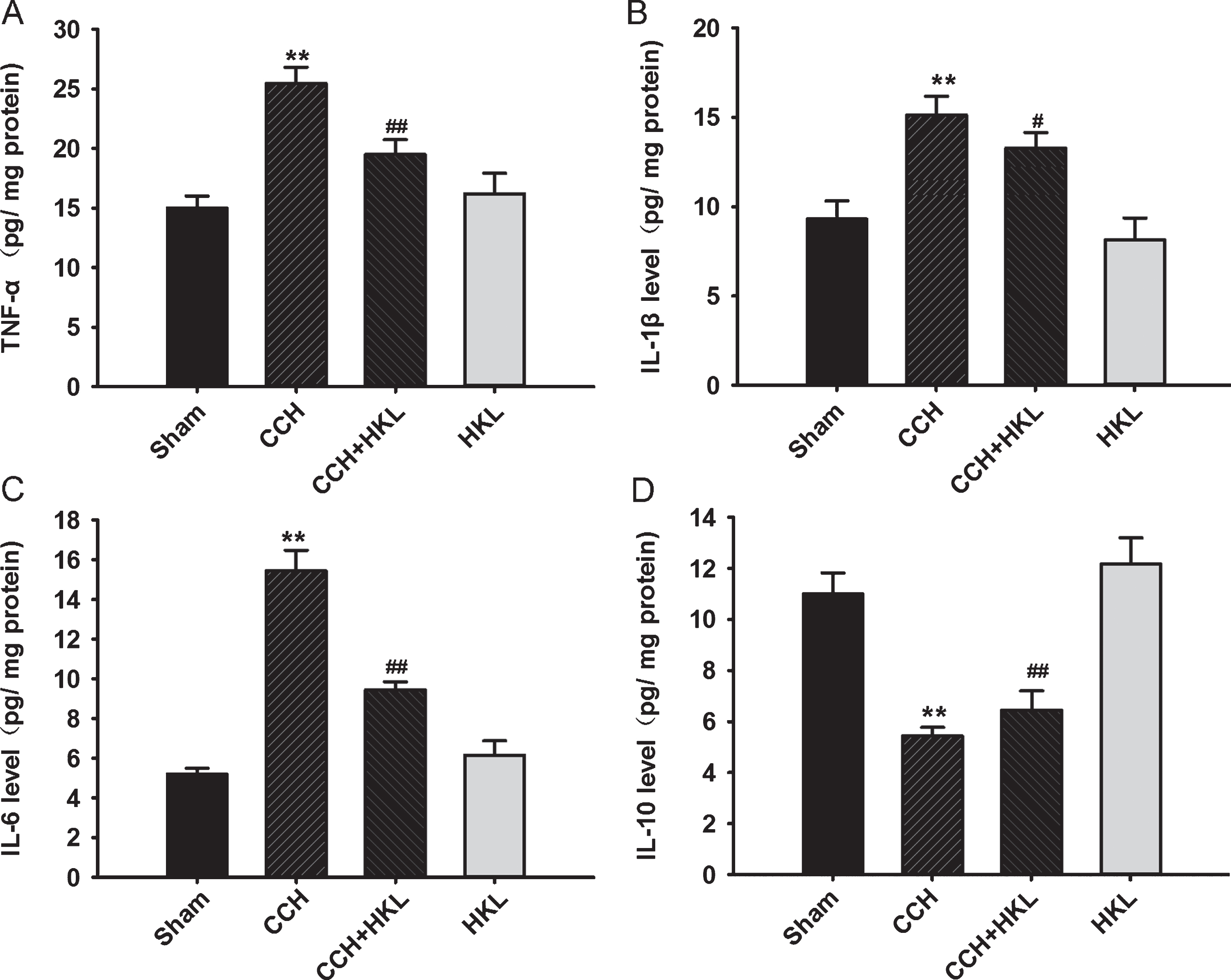
HKL prevents CCH-induced inflammation in the hippocampus. After the behavioral tasks, the levels of TNF-α (A), IL-1β (B), IL-6 (C), and IL-10 (D) in the hippocampus were detected using enzyme-linked immunosorbent assay (ELISA). Data are expressed as the mean±SD (n = 5). **p < 0.01 versus sham group; #p < 0.05, # #p < 0.01 versus CCH group.
HKL protects the dendritic spines in the DG region
Because the alterations in synaptic connections are considered essential for learning and memory formation [25], we also examined the morphological changes of synapses by assessing dendritic spines in the DG region by Golgi staining. The analysis used a well-known and previously reported method and found that CCH significantly decreased not only the density of the dendritic spines (Fig. 6A, 6B) but also the percentage of mushroom-type spines (Fig. 6A, 6C); pretreatment with HKL strongly improved the spinogenesis that was inhibited by CCH. These data confirmed that pretreatment with HKL may alter the morphology of spines.
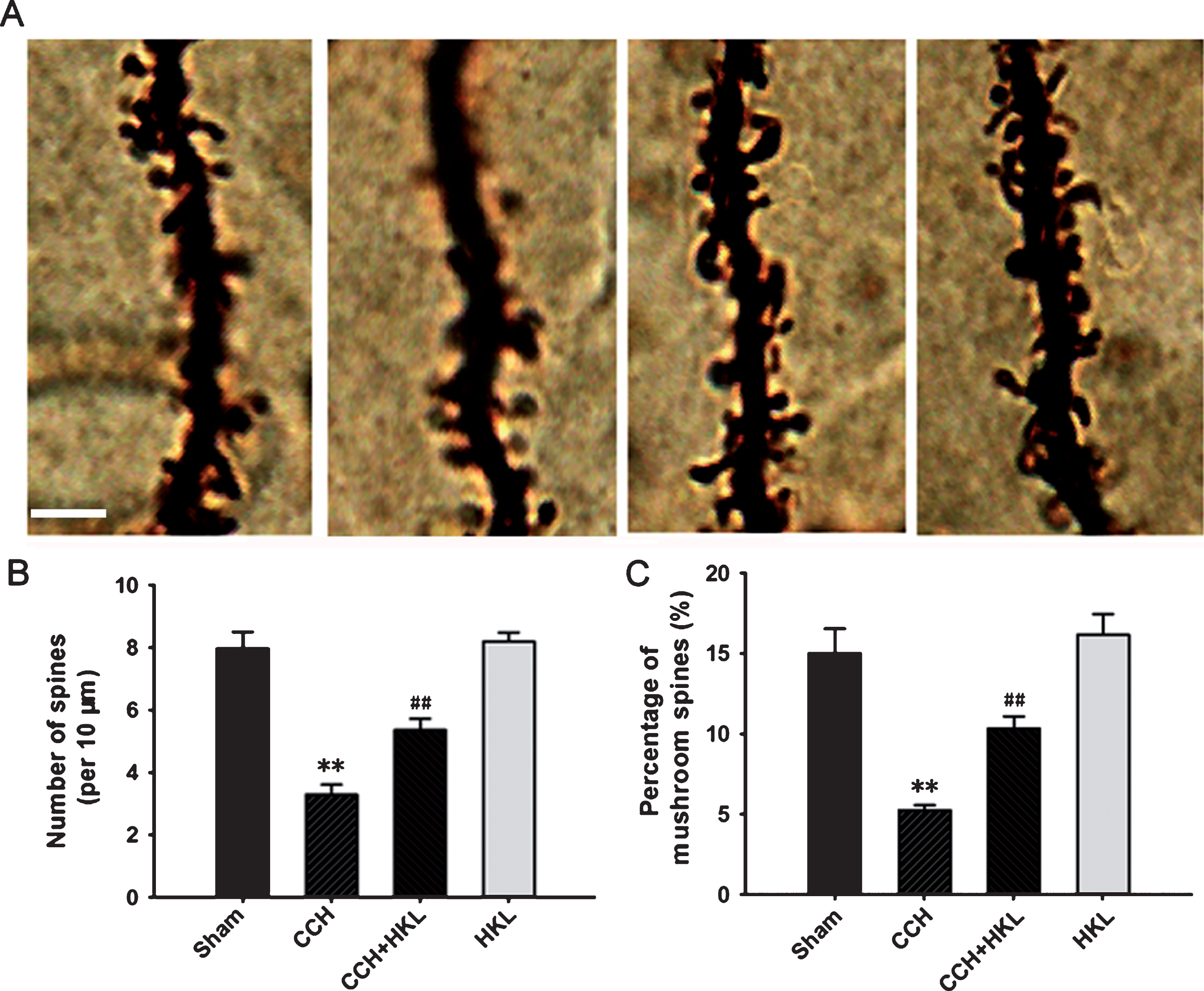
Changes in the dendritic spines. Representative images of dendritic spines (A) were captured from Golgi staining in the hippocampal dentate gyrus (DG) region, and the quantitative analysis of the density of spines (B) and the percentage of mushroom types (C) were conducted. Bar = 5μm. **p < 0.01 versus sham group; # #p < 0.05 versus CCH group.
HKL reverses CCH-induced GSK-3β activation in the hippocampus
It is known that the activation of GSK-3β plays important role in mediating the synaptic disorder, memory loss, and neurodegeneration in the dementia [26] and both the in silico and in vivo evidences suggested that GSK-3 inhibition can be used as glycogen a potential pharmacological target for vascular dementia [27]. We finally detected the levels and activity of GSK-3β of pGSK-3β and total GSK-3β in the hippocampus of rats by western blotting. We found no significant differences in the total GSK-3β, while the immunostaining of phosphorylated GSK-3β at the Ser9 site (inactivated form of the kinase) was significantly decreased in the CCH group compared to the control group (Fig. 7A). Quantitative analysis of the relative pGSK-3β/GSK-3β ratio confirmed these differences (p < 0.01, Fig. 7B), which indicated that GSK-3β in the hippocampus was activated after CCH treatment. Compared to the CCH condition, HKL pretreatment significantly enhanced GSK-3β phosphorylation (p < 0.01). To verify the activity of GSK-3β, we measured the activity of GSK-3β by a commercial assay kit. We can see that the results were consistent with western blot. CCH increased the activity of GSK-3 about 2.5-fold of the control level, and the activation of GSK-3β was partially antagonized by pretreatment with HKL. We also detected the expression of GSK-3β at mRNA level, there was no significance in each group (Fig. 7D). GSK-3β can be inactivated through the phosphorylation of the Ser9 site by Akt [28], So we measured the activity of Akt (Fig. 7E). The results showed that CCH improved the activity of Akt, while HKL pretreatment significantly this activation. HKL treatment alone did not change the activity of Akt.
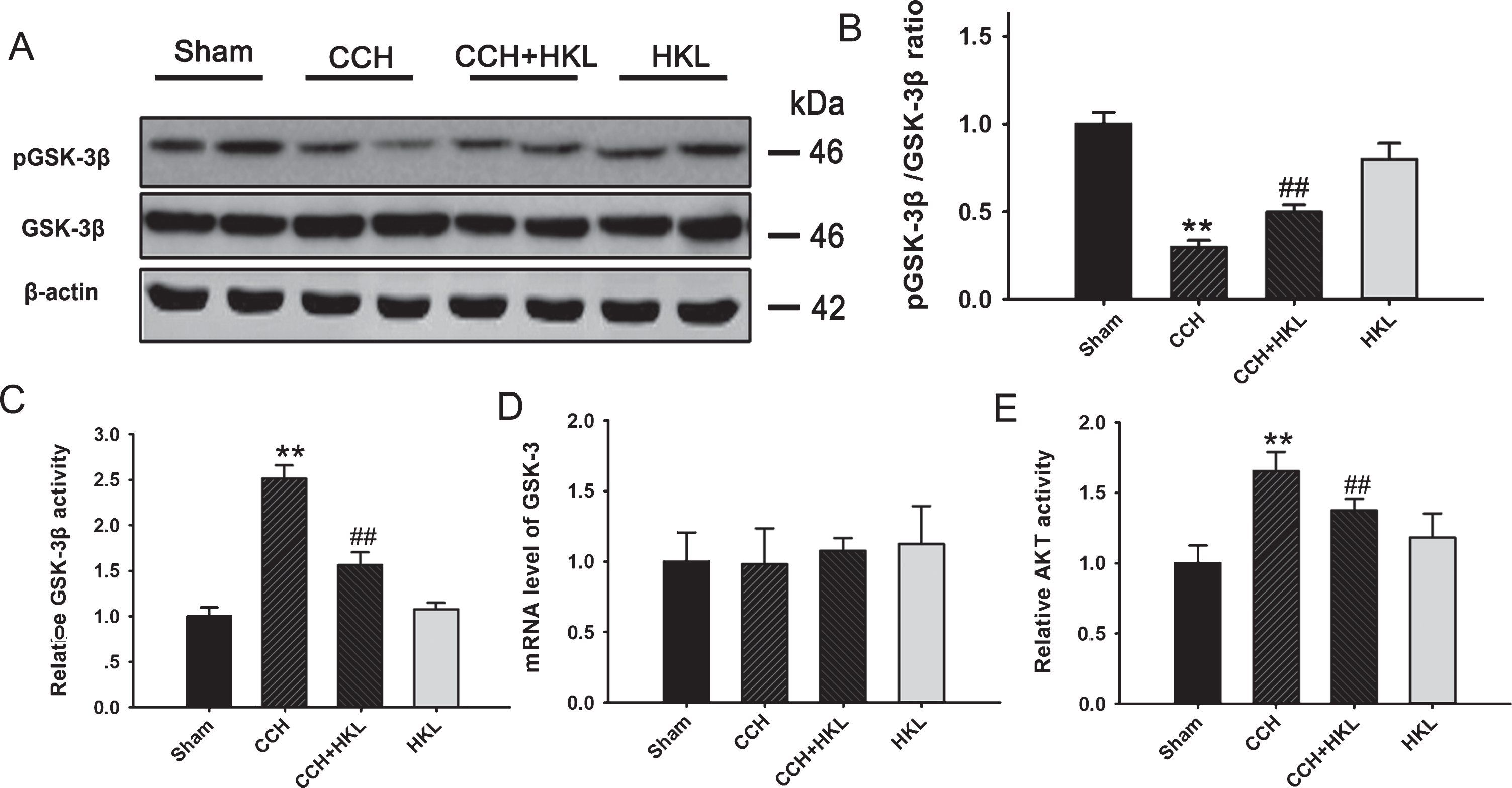
HKL reverses CCH-induced GSK-3β activation in the hippocampus. A) The levels of phosphorylated GSK-3β at serine-9 (pGSK-3β) and total GSK-3β (GSK-3β) were measured by western blotting. B) The ratio of pGSK-3β/GSK-3β was calculated. C) The activity of GSK-3β detected by the GENMED kit. D) The mRNA level in different group. E) The activity of PKA activity. **p < 0.01 versus sham group; # #p < 0.05 versus CCH group.
DISCUSSION
Due to its multiple facets of neuroprotection, HKL has been used as a therapeutic drug for cognitive deficits in many rodent models [13, 29–32]. In VaD animals, cerebral hypoperfusion results in the depletion of ATP and mitochondrial dysfunction, which in turn leads to increased ROS production and an imbalance of ROS and antioxidases, also referred to as oxidative stress, causing the further production of ROS and more severe oxidative damage. Oxidative stress characterized by elevated levels of protein oxidation, lipid peroxidation, DNA oxidation, and ROS formation with significantly decreased levels of antioxidant enzymes plays an important role in the pathogenesis of neurodegeneration, as seen in the VaD brain [33]. Furthermore, increased levels of oxidative alterations promote Aβ production to facilitate protein misfolding and induce the production of H2O2, which ultimately causes neuronal damage with cognitive decline [34]. Several studies have suggested that the inhibition of oxidative stress has a beneficial effect in CCH therapy [35]. In our study, HKL treatment ameliorated the overproduction of oxidants and restored the levels of antioxidant enzymes, which is consistent with a previous study [13].
In VaD patients, cognitive deficits were also highly correlated with elevated neuroinflammation in the central nervous system [36]. The levels of inflammatory biomarkers, such as α1-anti-chymotrypsin, interleukin 6, and C-reactive protein, were increased in both plasma and cerebrospinal fluid [24]. The ischemic events in VaD are recognized as the main initiating cause of inflammatory cascades because the debris of necrotic neurons are strong stimulators for immune cells [37], including astrocytes and microglia. Subsequently, many cytokines and chemokines will be released, and the inflammatory pathways will be activated. In an animal model of VaD, neuroinflammation apparently occurs in the corpus callosum and hippocampus, the two important brain regions involved in learning and memory [38]. Although anti-inflammatory drugs do not improve outcomes in clinical settings of stroke [37], they are effective in rescuing memory deficits in VaD animal models [38]. We provide evidence here that HKL supplementation suppresses the levels of TNF-α, IL-1β, and IL-6 but restores the level of IL-10, suggesting that HKL could restore the inflammatory status in the hippocampus of CCH rats. These data are consistent with a previous study showing that treatment with 10μM HKL inhibited IFNγ±LPS-induced inflammatory cytokine release from microglia [39].
We also found that HKL treatment could suppress the activity of GSK-3β, an important kinase mediating tau hyperphosphorylation, synaptic degeneration, and memory impairments in neurodegenerative disease [40, 41]. A previous study revealed that HKL treatment protected against amyloid-β-induced neurotoxicity via the GSK-3β and β-catenin signaling pathways in PC12 cells [42]. HKL treatment increased the phosphorylation of Akt (PKB) [43], the upstream regulator of GSK-3β. Thus, it is possible that HKL inhibited GSK-3β by elevating the activity of Akt and promoting the inhibitory phosphorylation of GSK-3β. As reported previously, activation of GSK-3β induced a reduction in dendritic branches and the density of dendritic spines both in vivo [44] and in vitro [45]. Activation of GSK-3 has also been shown to increase the expression of proinflammatory cytokines such as IL-1β and TNF-α by inhibition of CREB [45]. Our study suggested that HKL can restore dendritic spine density and suppress neuroinflammation in CCH rat brains. We propose that the inhibition of GSK-3β plays an important role in the neuroprotective effects of HKL. Interestingly, in neuronal cells, the activation of GSK-3β could be induced by oxidative stress [46], and the inhibition of GSK-3β was found to be important for controlling oxidative stress [47]. Moreover, application of the inhibitors of GSK3β were shown to be beneficial in many neurological diseases with neuroinflammation, including AD, multiple sclerosis, and AIDS dementia complex [48]. Considering the antioxidant, anti-neuroinflammation and GSK-3β inhibition effects of HKL, we believe that HKL administration is a promising treatment for VaD.