
Abstract
Apolipoprotein (apo) E4 is the major genetic risk factor for Alzheimer’s disease (AD). It is shown that apoE4 preferentially undergoes aberrant cleavage in neurons, yielding neurotoxic C-terminal-truncated apoE4 fragment. Endoplasmic reticulum (ER) stress has also been known to be involved in the pathogenesis of AD. However, little is known about the contribution of ER stress to the neurotoxicity of apoE4 fragment. In the present study, we established the neuron-specific expression human C-terminal-truncated apoE4(1–272) fragment transgenic mice and also transfected apoE4(1–272) fragment in neuroblastoma N2a cells. We found that human apoE4(1–272) fragment could trigger ER stress as evidenced by increasing the expression of ER stress markers both in vivo and in vitro. Meanwhile, the apoE4(1–272) transgenic mice presented obviously AD-like neuropathological changes, including the impairment of spatial learning and memory, prominent axonal morphological changes, and hyperphosphorylation of tau. At the same time, we also found that glycogen synthase kinase-3 activities were significantly increased. Furthermore, these neuropathological changes, especially tau hyperphosphorylation and axonal transport impairment, were significantly rescued by the ER stress protector 4-phenylbutyric acid (4-PBA) in apoE4(1–272)-transfected N2a cells. Pretreatment with 4-PBA not only decreased the protein expression of immunoglobulin binding protein (BiP) and C/EBP-homologous protein (CHOP), but also significantly reversed these defects in axonal transport. These results suggested that the neurotoxic effects of apoE4(1–272) fragment found in AD subjects, at least in part, through triggering ER stress and inducing tau hyperphosphorylation, led to the enduring impairment of axonal transport.
Keywords
INTRODUCTION
Alzheimer’s disease (AD), as the most common cause for senile dementia [1], is a progressive and devastating age-related neurodegenerative disorder characterized clinically by memory loss, cognitive decline, pathologically by the occurrence of brain senile plaques and neurofibrillary tangles (NFTs) [2, 3]. It is well known that apolipoprotein E4 (apoE4) is one of the most important genetic risk factors for AD [4]. The apoE4 allele, which is found in 40–65% of cases of sporadic and familial AD, increases the occurrence and lowers the age of onset of the disease in a gene dose-dependent manner [5, 6]. It is currently believed that apoE4 is a key pathological chaperone molecule in the pathogenesis of AD such as NFTs formed by tau phosphorylation and senile plaques formed by amyloid β-protein (Aβ) aggregation [7]. ApoE4 is mainly expressed and secreted by astrocytes and microglia in brain tissue, and apoE4 also can be induced in neurons [8]. Tesseur et al. reported that apoE4 only induced an increase of tau phosphorylation in neurons, but this phenomenon was not observed in astrocytes, suggesting that the effect of apoE4 on tau phosphorylation was neuron-specific [9]. Early studies had shown that apoE4 could be cleaved at the Met272 site to generate apoE4(1–272) fragment, which was found in the brains of AD patients and transgenic mice expressing apoE4 [10]. Similarly, in apoE4(1–272) transgenic mice, apoE4(1–272) in neurons promotes hyperphosphorylation of tau and formation of NFTs [11]. Therefore, we speculate that apoE4(1–272) in neurons is an effector molecule of apoE4 which can lead to the pathogenesis of AD.
More and more evidence have suggested that axonal damage caused by axonal transport impairment may eventually lead to AD-related pathological changes [12]. In the brains of AD patients, axonopathy occurs in the early Braak stage, not in the late Braak stage [13]. In the apoE4 transgenic mouse model, axonopathy appears earlier than amyloid deposits and NFTs [5]. In addition, studies have confirmed that hyperphosphorylation of tau protein plays a central role in neuronal axonal transport disorders, leading to abnormal protein accumulation and degeneration in neurons [14]. However, it is still not clear whether neuron-specific apoE4(1–272) fragment can induce axonal impairment and cause neuron degeneration.
In recent years, endoplasmic reticulum (ER) stress has been found to play an important role in AD. In eukaryotic cells, the ER is the main organelle for the folding, processing, and secreting proteins. Depletion of calcium ions, accumulation of unfolded or misfolded proteins in the ER or excessive accumulation of normal proteins can trigger ER stress, which causes unfolding protein response (UPR) to maintain cells homeostasis. However, prolonged ER stress can initiate programmed cell death [15]. In vitro studies have shown that ER stress can induce Aβ aggregation during amyloid-β protein precursor (AβPP) metabolism [16]. In addition, it is also demonstrated the presence of UPR and ER stress at the site of NFT formation in the brains of AD patients [17]. All this evidence suggests that ER stress may contribute to the pathological changes of AD. However, whether aberrant neuron-specific apoE4(1–272) fragment can trigger UPR/ER stress, which leads to axonopathy and further affects pathological changes of AD, is still to be further elucidated.
In the present study, we investigated the possible mechanism of neurotoxic effects of apoE4(1–272) fragment. We found that neuron-specific apoE4(1–272) fragment could trigger ER stress both in vitro and in vivo, eventually resulted in the AD-like neuropathological changes, including tau hyperphosphorylation, axonal impairment, and spatial learning and memory impairment.
MATERIALS AND METHODS
Antibodies and chemicals
The monoclonal antibodies (mAb) tau-5 was from Epitomics Biotechnology (Epitomics, USA). Polyclonal antibody (pAb) pS262 and (pAb) pS202 reacting respectively to phosphorylated tau at Ser262 and Ser202 were from Abcam (Cambridge, UK). Immunoglobulin binding protein (BiP), glycogen synthase kinase-3β (GSK-3β), and GAPDH mAb were also from Abcam (Cambridge, UK). The p-GSK-3β mAb was from CST reacting to phosphorylated GSK-3β at Ser9. Cyclin-dependent kinase-5 (Cdk5) pAb and C/EBP-homologous protein (CHOP) were from Santa Cruz Biotechnology (Santa Cruz, CA). ER stress inhibitor 4-phenylbutyrate (4-PBA) and GSK-3β inhibitor lithium chloride (LiCl) were from Sigma Chemical Co. (St Louis, MO, USA). The bicinchoninic acid (BCA) protein detection kit, chemiluminescent substrate kit and phosphorcellulose units were from Pierce Chemical Company (Rockford, IL, USA). The Cdk5 substrate Histone H1, phosphorylase-b and immunoglobulin G were also from Santa Cruz Biotechnology. Dulbecco’s modified Eagle’s medium (DMEM), Opti-MEM, Lipofectamine 2000, and fetal bovine serum were from Invitrogen Corporation (Gaithersburg, MD, USA). Neuron-specific human apoE4(1–272) fragment expression vectors were constructed by Cyagen Ltd. (Guangdong, China), which were subcloned into a pRP.EX3d-Syn1-IRES-EGFP vector or a pEGFP-N1 vector.
Cell cultures and transfection
Mouse neuroblastoma N2a cells (N2a cells) were cultured with 1:1 mixture of DMEM and Opti-MEM containing 5% fetal bovine serum (Gibco, Grand Island, NY, USA) in a humified incubator aerated with 95% air and 5% CO2 at 37°C. The medium was replaced every other day, and cells were plated at an appropriate density according to each experimental scale. ApoE4(1–272) fragment and Enhanced Green Fluorescent Protein (EGFP) expression plasmid were transiently transfected into N2a cells using Lipofectamine 2000, according to the manufacturer’s instruction (Invitrogen, Carlsbad, CA, USA). After 24 h, the transfection efficiency of various plasmid was determined by fluorescence microscopy (Zeiss LSM710). Images were acquired using the LSM-710 confocal microscope (Carl Zeiss, Jena, Germany). The chemical chaperone 1 mM 4-PBA or 10 mM LiCl was added after 8 h transfection, and then for another 16 h before they were harvested for real-time PCR or western blot analysis.
Animals and treatments
Neuron-specific human apoE4(1–272) fragment transgenic mice and age/sex-matched control mice were purchased from Cyagen Ltd. These mice were housed in the Experimental Animal Center of Tongji Medical College, and provided an SPF environment with an alternating 12 h light/dark cycle under 22±2°C. These mice were raised five per cage, and replaced with clean bedding to avoid a damp environment. Behavioral testing was carried out prior to the week of sacrifice. Brains were removed and hippocampal tissues were harvested for further tests. In our study, all animal experiments were performed on the 12- to 14-month-old female mice and all euthanasia were performed under pentobarbital sodium (40 mg/kg, ip) anesthesia, and efforts were taken to minimize animal suffering. Our study was performed in accordance with the Animal Care and Use Committee affiliated with the Huazhong University of Science and Technology.
Identification of apoE4(1–272) fragment transgenic mice
ApoE4(1–272) fragment transgenic mice were identified by PCR, using primers as follows: forward primer, 5′-ACGTAAACGGCCACAAGTTC-3′; reverse primer, 5′-GATCTTGAAGTTCACCTTGATGC-3′, with a 440 bp length product. Mouse tail biopsies were digested overnight at 55°C with 1 mg/ml proteinase K in 50 mmol/L Tris-HCI (pH 8.5), 1 mmol/L EDTA, 0.5% sodium dodecyl sulfate, 0.1 mol/L NaCl. The enzyme was inactivated by boiling for 10 min, and diluted samples were analyzed by polymerase chain reaction with the following program: 2 min at 95°C, followed by 34 cycles of 1 min at 95°C, 30 s at 60°C, 45 s at 72°C, and a final extension of 15 min at 72°C. PCR products were analyzed on 1% agarose gels to confirm the positive mice.
PCR Primer sequences for experimental and reference genes
Western blotting
The N2a cells or hippocampal tissue homogenates were lysed by RIPA (Tris-HCL, PH 7.5, 50 mM; Nacl, 150 mM; EDTA, 1 mM; 1% NP–40; 0.1% SDS; 1% Triton X-100) with protease inhibitor PMSF and phosphatase inhibitor cocktail added. The total lysate samples were centrifuged at 4°C, 12,000 rpm for 12 min, and the supernatants were mixed immediately by adding 5×loading buffer and then heated to 100°C for 10 min. The samples were aliquoted and stored at –80°C. Equal amounts of protein (30μg) were separated on SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (PVDF, Millipore, CA). The membrane was blocked with 5% bovine serum albumin in TBS/Tween-20 (TBST) for 1 h at room temperature, and then the membrane was incubated overnight with primary antibodies tau-5 (1:500), p-Tau-262 (1:1000), p-Tau-202 (1:1000), p-GSK-3β (1:1000), GSK-3β (1:1000), Cdk5 (1:1000), BiP (1:1000), CHOP (1:1000), and GAPDH (1:1000) at 4°C. On the 2nd day, after washing with TBST, the membrane was incubated with horseradish peroxidase-conjugated secondary antibody (1:2000; Cell Signaling Technology) for 2 h at room temperature and visualized by using the enhanced chemiluminescence substrate system (Santa Cruz, CA, USA). Quantification of protein bands was performed via scanning with the Bio-Rad GelDocTM XR and Chemi DocTM XRS systems and the intensities of the bands were measured using Image J analysis software.
RNA isolation and real-time RT-PCR
The total RNA was extracted from the hippocampus of the experimental mice or N2a cells with the Trizol reagent (Invitrogen) according to the manufacturer’s protocol. The concentration and purification of RNA were measured with NanoDrop (Thermo). Total RNA (1μg) was reverse-transcribed with the reverse transcription kit (Toyobo Co. Ltd., Osaka, Japan). The relative quantity of mRNA was determined by real-time RT-PCR using an ABI Prism 7300 sequence detector (Applied Biosystems, Foster City, CA), which was performed in triplicate. Primers were synthesized by Tsingke Biological Technology (Beijing, China). The cDNAs (2μL) were amplified with the specific primers (Table 1). All cDNA samples were amplified using the SYBR Green Real-time PCR kit (Toyobo Co. Ltd.) according to the manufacturer’s protocol. All data were expressed as the relative ratio of target gene to the housekeeping gene GAPDH.
Morris water maze test
The cognitive function including learning and memory was assessed with a Morris water maze (MWM) test. Briefly, mice were trained in a circular water pool filled with opaque water, and temperature was maintained at 23±2°C. The pool was divided into four equal quadrants. A hidden escape square platform, submerged in the water level approximately 1 cm, was placed in the third quadrant of the pool. The mice were randomly placed in the pool from any of the three quadrants except the one with the escape platform (targeting quadrant), and allowed to swim for 60 s to find the platform. The trial was terminated when the mice found the platform within 60 s. If the mice did not find the platform within 60 s, it was guided toward target platform and allowed to stay for 2-3 s. In order to allow the mice to adapt to the pool environment, each mouse was conditioned 3 times per day for 2 days (visible platform training) and then made three times training trials per day for 6 days to find the hidden platform (hidden platform training). An automated video tracking system and software (NoldusEtho Vision 2.3.19, Netherlands) were used to record the swimming path, the platform crossing frequency, and escape latencies (the time it took the mice to reach the hidden platform). The probe test was performed on the next day (day 7) after the last training trial on day 6 with the platform removed, and the time the mice spent in the quadrant where the platform was previously located was recorded as a measure of memory retention. Behaviors of mice were tracked using EthoVision 3.0.
Analysis of Splicing in X-box binding protein(Xbp-1) mRNA
Splicing in Xbp-1 mRNA was investigated by RT-PCR and Pst I restriction enzyme digestion as the method established by Calfon [18]. After treatment, extraction of RNA and reverse transcription were carried out. cDNA of Xbp-1 was amplified using PCR with Xbp-1 specific primers (Table 1). The PCR cycles consisted of denaturation at 94°C for 1 min, annealing at 60°C for 1 min and extension at 72°C for 1 min, for 33 cycles. Pst I (New England Biolabs) restriction digestion of the 600 bp PCR products was done at 37°C for 16 h. The restriction fragments were separated on a 1% (w/v) agarose gel with ethidium bromide and visualized under ultraviolet ray illumination.
The enzymatic activity assay
The GSK-3 activity was measured using γ-32P-ATP assay. Briefly, for cell lysates, 7.5μg of protein sample was incubated for 30 min at 30°C with 200μM ATP, 2000 cpm/pmol γ-32P-ATP, 30 mM Tris-HCL (pH7.4), 10 mM MgCl2, 10 mM NaF, 1 mM Na3VO4, 2 mM EGTA, and 10 mM β-mercaptoethanol. The reaction was terminated by adding 300 mM O-phosphoric acid. The reaction mixture was applied in triplicates on a phosphocellulose paper (Pierce). The filters were washed three times with 75 mM O-phosphoric acid, dried, and counted by liquid scintillation counter. Incorporation of γ-32P was quantified in counts per minute (cpm) using a scintillation counter.
The Cdk5 activity was measured using immunoprecipitation of Cdk5. Briefly, 200μg of total cellular protein was mixed with 4μg of the anti-Cdk5 antibody and Protein G Plus-Agarose (Santa Cruz, CA, USA). The immunoprecipitates were rinsed once with kinase buffer (50 mM HEPES, pH7.0, 1 mM DTT, 10 mM MgCl2, and 1 mM cold ATP) and three times with lysis buffer. The rinsed agarose beads were incubated with kinase buffer containing 2.5μg histone H1 plus 200μM γ-32P-ATP in a final volume of 50μL at 30°C for 30 min, and the reaction products were determined using a scintillation counter.
In vivo neuronal tracing assay and semi-quantitative analysis of axonal changes
The in vivo neuronal tracing was used by the previously established method [19]. Briefly, for in vivo tracing, animals were anesthetized and fixed their heads in a standard stereotaxic apparatus (Stoelting, USA). The mice were received tracer injection [10% biotinylated dextran amine (BDA) in 0.05 M TBS (0.05 M Tris, 0.9% NaCl, pH7.6), molecular weight 10,000, Molecular Probes, Invitrogen] under pressure (Microsyringe pumpcontroller, WPI, USA), using a glass pipette with a tip of maximally 40–50 mm in diameter. The glass pipette was filled with an injection volume of 10μl. All coordinates for the injected regions were adapted from the mouse Brain in Stereotaxic Coordinates. For the apoE4(1–272) transgenic mice or non-transgenic wild-type mice, the tracer was injected into the hippocampus. After tracer injection, the mice were allowed to survive for 3–5 days in the same housing conditions. Then animals were deeply anesthetized and perfused with saline, followed by a solution of 4% paraformaldehyde in 0.1 M PBS (pH7.4) at 4°C. The brains were removed and kept in the same fixative at 4°C for overnight postfixation, equilibrated 48 h with 30% sucrose in 0.1 M PBS.
For tracer detection, brains were coronally cut in a cryostat into 40μm sections, and then sections were collected in sequential order and rinsed in 0.05 M TBS. Then these sections were incubated with the Avidin-Biotin Complex (Vector Laboratories, Burlingame, CA, USA, 1:800) in a mixture of 0.05 M Tris, 0.9% NaCl, 0.25% gelatin, and 0.5% Triton-X100, pH7.4 for 2 h at room temperature. After several rinses with TBS, the sections were incubated with 0.05% 3,3’-diaminobenzidine tetrahydrochloride (DAB), 0.2% nickel ammoniumsulphate, and 0.003% H2O2 in 0.05 M TBS (pH7.4), mounted on gelatin-coated slides, and then dehydrated, cleared, and coverslipped.
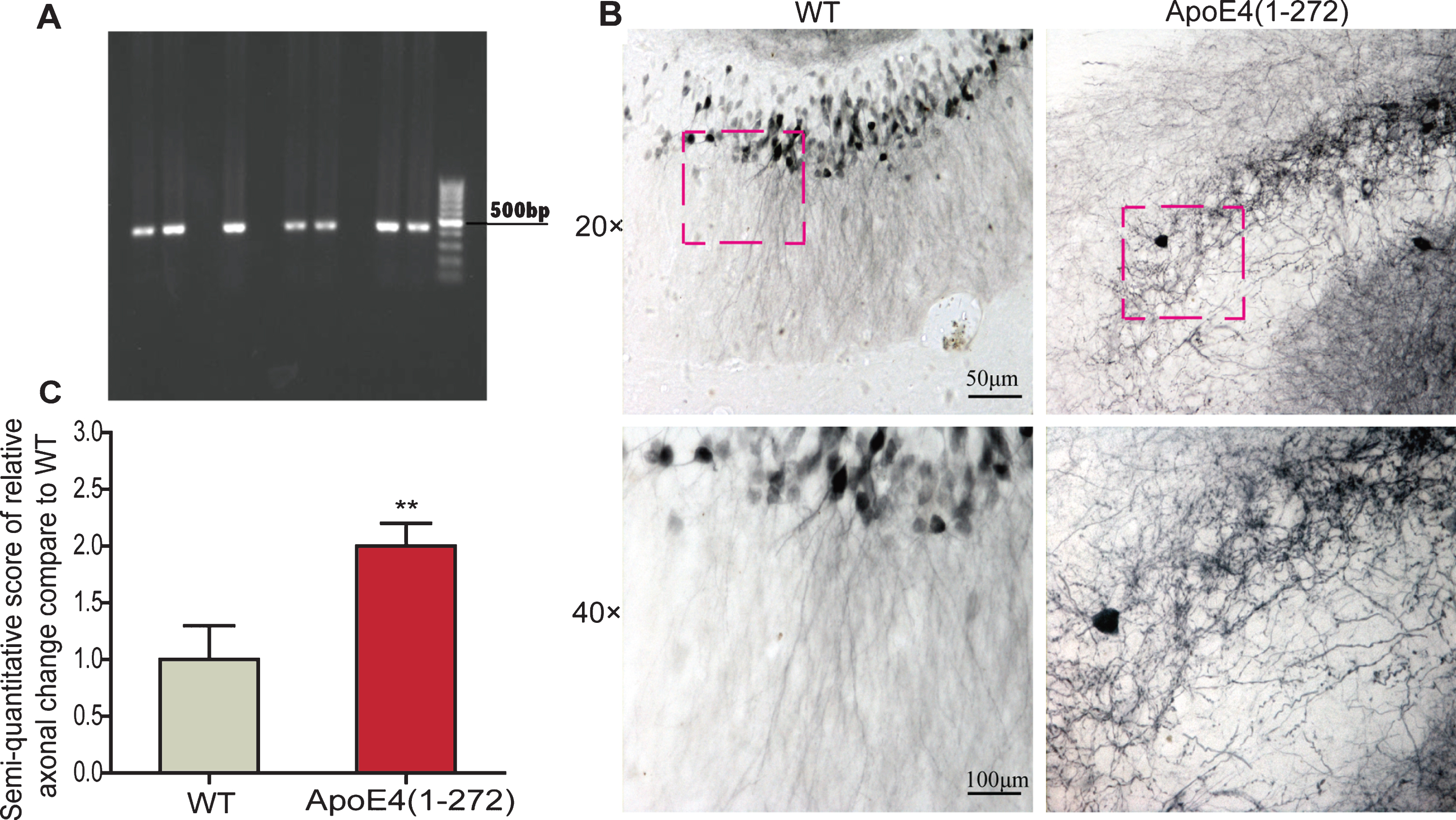
Identification of apoE4(1–272) transgenic mice and over expressing human apoE4(1–272) fragment induced axonal changes in the apoE4(1–272) transgenic mice. A) PCR analysis of genomic tail DNA using specific primers and the PCR products were separated on a 1% agarose. B) The axonal changes in the hippocampus regions in age- and sex- matched non-transgenic wild-type mice and apoE4(1–272) transgenic mice. C) The semi-quantitative score of relative axonal change of the hippocampus. (**p<0.01 versus non-transgenic wild-type mice; n = 8 per group; the significant differences from the respective values were determined by ANOVA).
A semi-quantitative analysis of the axonal changes in the hippocampal regions was performed in 16 mice (2 mice were failed to observe and assess) with the grading system. The changes were scored separately in each brain region according to our team’s previous scoring criteria [20]. Two investigators were blinded to the cases during the semi-quantitative analysis.
Live-cell imaging and image analysis
According to the manufacturer’s instruction (Invitrogen, USA), pEGFP-neurofilament middle chain (pEGFP-NFM) was transiently transfected into N2a cells using Lipofectamine 2000. The cells used for axonal transport study were cultured for 24 h before addition of apoE4(1–272) fragment constructs with or without PBA. The cells were observed by evanescent-field fluorescence after transfection of apoE4(1–272) fragment with or without 1 mM 4-PBA. Exposure time (100–300 ms) and interval time (4–7 s) were adjusted to appropriate location according to the fluorescence intensity of images. Single and time-lapsed images were observed, processed and analyzed. For these analyses, we only selected filaments that were undergoing bouts of sustained unidirectional movement. For axonal transport velocity studies, approximately 70–100 vesicles were counted in about 20 cells per sample.
Statistical analysis
Data were expressed as means±SEM and analyzed using SPSS 18.0 statistical software (SPSS Inc., Chicago, IL, USA). The statistical significance of differences was determined by one-way analysis of variance (ANOVA) following Tukey’s post hoc Test. Differences were considered significant when p < 0.05.
RESULTS
Overexpression of apoE4(1–272) induced axonopathy and cognitive deficit in vivo
In order to determine whether the overexpressing apoE4(1–272) transgenic mice existed axonal morphological changes, we used the in vivo tracing detection to observe the structure and morphology changes in the brain of 12- to 14-month-old apoE4(1–272) transgenic mice. Firstly, to verify the expression of ApoE4(1–272) in transgenic mice, the apoE4(1–272) transgenic mice were identified by PCR analysis of genomic tail DNA (Fig. 1A). Then, we found that there were particularly obvious axonal changes, especially swollen axons (Fig. 1B), or even swollen varicosities in the hippocampus of apoE4(1–272) transgenic mice. Meanwhile, there were more than one swollen varicosity at an axon. However, there were no obviously the structure and morphology changes in the hippocampus of the non-transgenic wild-type controls, and there were abundant and smoothly projected fibers (Fig. 1B). The semi-quantitative scoring for axonal changes in the hippocampus was also conducted and analyzed (Fig. 1 C). The axonal changes in the apoE4(1–272) transgenic group were significantly larger than that of the wild-type controls. Thus, our results demonstrated that the transgenic mice expressing human apoE4(1–272) fragment in neurons presented obviously axonopathy.
Axonopathy was closely associated with memory deficit. To assess whether the overexpressing apoE4(1–272) transgenic mice could cause memory impairment, we performed MWM to evaluate the learning ability and memory of the mice at 10 months of age via training with a hidden platform (Fig. 2A), and used the age- and sex-matched non-transgenic wild-type mice as controls. The results show that, compared with the non-transgenic wild-type mice, the mean escape latency time in apoE4(1–272) transgenic mice increased significantly (Fig. 2B). On the 7th day, we removed the platform and conducted the probe trial. The platform crossing frequency of the apoE4(1–272) transgenic mice was decreased significantly (Fig. 2 C). In addition, the apoE4(1–272) transgenic mice spent less time in the target quadrant than the control wild-type mice (Fig. 2D). Also, the percent distance of apoE4(1–272) transgenic mice in the target quadrant decreased significantly (Fig. 2E). Taken together, our observations demonstrated that the apoE4(1–272) transgenic mice showed significant deficits in spatial learning and memory.
Overexpression of apoE4(1–272) led to the hyperphosphorylation of tau in vivo and in vitro
To explore whether the pathological changes of neurons and memory impairment, induced by apoE4(1–272) in our models, were associated with the tau hyperphosphorylation, we investigated tau phosphorylation levels at Ser202 and Ser262 sites in apoE4(1–272) transgenic mice via western blotting of brain homogenates or N2a cells expressing apoE4(1–272) constructs. The results showed that tau became hyperphosphorylated at all of the two sites in hippocampus (Fig. 3A, B) of 12- to 14-month-old apoE4(1–272) transgenic mice, compared to the age- and sex- matched non-transgenic wild-type mice. Similarly, the activation of tau sites was found in N2a cells (Fig. 3 C, D) transfected with apoE4(1–272) constructs.
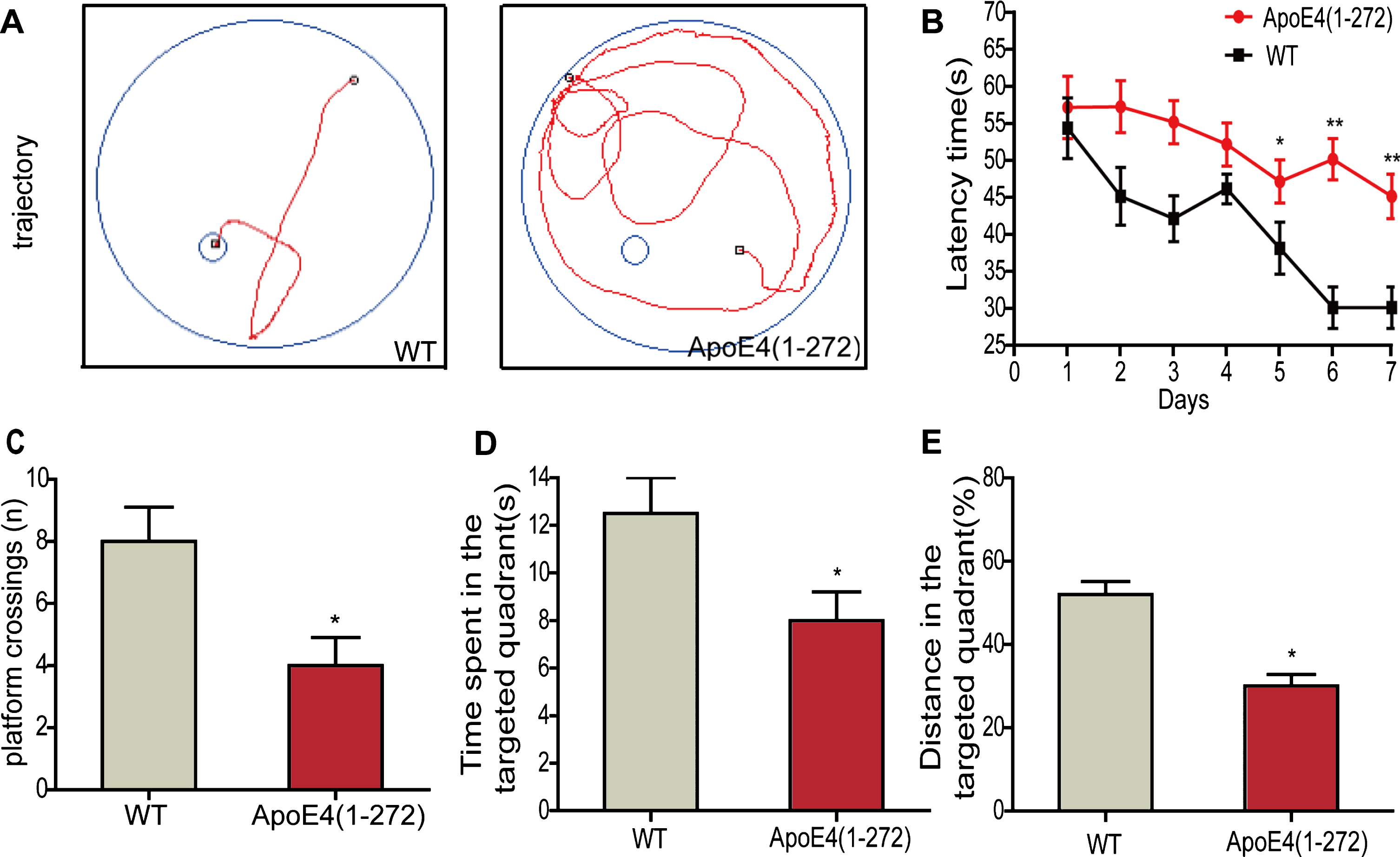
Overexpressing human apoE4(1–272) fragment induced the cognitive deficit in the apoE4(1–272) transgenic mice via MWM test. A) The trajectory of mice in MWM. B) Escape latency analysis for escaping in the 6-days consecutive training trials. C) The frequency of crossing over the former station during the probe trial. D) The time spent in target quadrant during the probe trial. E) The percentage of distance in the target quadrant during the probe trial. (**p<0.01, *p<0.05 versus non-transgenic wild-type mice; n = 8 per group; the significant differences from the respective values were determined by ANOVA).

Overexpressing human apoE4(1–272) fragment led to tau hyperphosphorylation in apoE4(1–272) transgenic mice and N2a cells. A, B) Phosphorylation levels of tau (Ser202 and Ser262) were detected by western blot in the hippocampus of 12–14-month-old apoE4(1–272) transgenic mice and the age- and sex-matched non-transgenic wild-type mice. C, D) Phosphorylation levels of tau (Ser202 and Ser262) in N2a cells transfected with apoE4(1–272) fragment construct or EGFP vector. (**p<0.01, *p<0.05 versus non-transgenic wild-type mice or N2a cells transfected with EGFP vector; n = 3 for western blotting; the significant differences from the respective values were determined by ANOVA).
Overexpression of apoE4(1–272) triggered ER stress in vivo and in vitro
To investigate whether the apoE4(1–272) could induce ER stress, we firstly detected whether the ER stress existed in the brain of apoE4(1–272) transgenic mice. We then examined the expression changes of ER stress markers by western blotting, real-time PCR, and Xbp-1 splicing in the brain of 12-to 14-month-old apoE4(1–272) transgenic mice and control mice. As shown in Figure 4, the BiP and CHOP protein levels in the hippocampus were significantly higher than the non-transgenic control group (Fig. 4A, B). Besides, the mRNA expressions of BiP and CHOP (Fig. 4D) were also agreement with the protein levels, which significantly increased in the hippocampus of apoE4(1–272) transgenic mice (p < 0.01). In addition, as we all known that unconventional splicing of mRNA of Xbp-1 by the ER stress sensor IRE1 was one of typical markers of ER stress [21]. During the splicing of Xbp-1 mRNA, an endoribonuclease could cut off a 26-nucleotide intron, which contained the Pst1 restriction enzyme cutting site. Once the mRNA of xbp-1 was spliced, it would prevent the digestion of mRNA for Xbp-1 by Pst1. In our results, we found that before Pst1 digestion there was only about 600 bp mRNA for Xbp-1 in the hippocampus of transgenic mice and non-transgenic mice. After Pst1 digestion, there were mainly about 300 bp fragments in non-transgenic mice group, but only a little in the apoE4(1–272) transgenic mice (Fig. 4 C). These data indicated that there was a significant splicing of mRNA for Xbp-1 in the hippocampus of the apoE4(1–272) transgenic mice. Taken together, our data suggested that neuron-specific expression human apoE4(1–272) fragment also triggered ER stress in vivo.
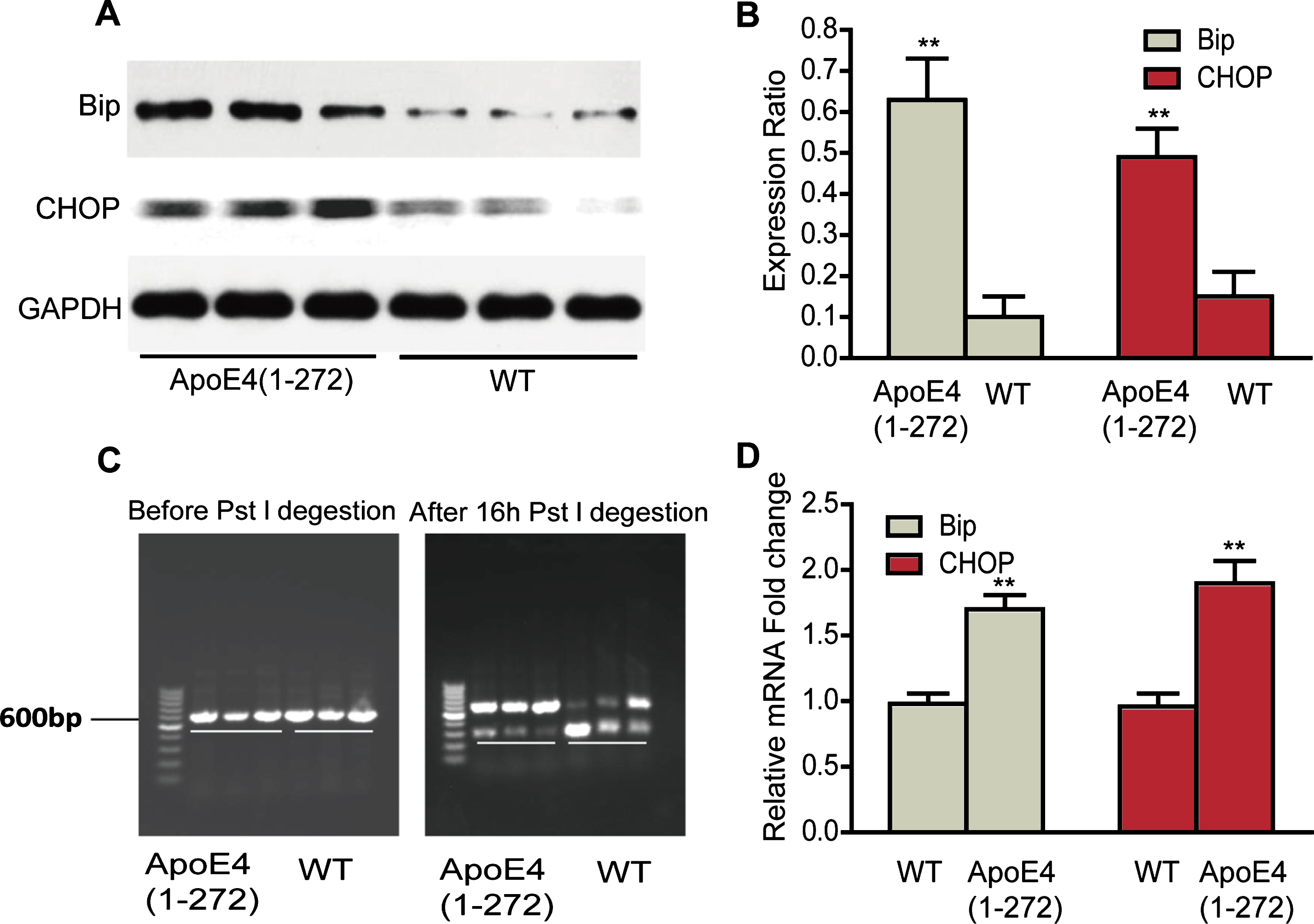
Overexpressing human apoE4(1–272) fragment triggered ER stress in hippocampus of apoE4(1–272) transgenic mice. A, B) ER stress markers, BiP and CHOP, were analyzed by western blotting. C) The xbp-1 mRNA alternative splicing before and after Pst 1 restriction enzyme digestion. D) The relative mRNA expressions of BiP and CHOP were quantified by real-time PCR. (**p<0.01 versus non-transgenic wild-type mice; n = 3 for western blotting; n = 8 for real-time PCR; the significant differences from the respective values were determined by ANOVA).
Next, to further demonstrate whether the human apoE4(1–272) could induce ER stress in vitro, we expressed the human apoE4(1–272) constructs or EGFP vector in N2a cells, and examined the expression of ER stress markers. We found that the protein expression of BiP and CHOP in the human apoE4(1–272) significantly increased than that of vector group (Fig. 5A, B). Besides, the mRNA expression of BiP and CHOP was also significantly increased in apoE4(1–272) transfected group (Fig. 5 C), comparing with the vector group. These results also confirmed that human apoE4(1–272) could trigger ER stress in vitro.
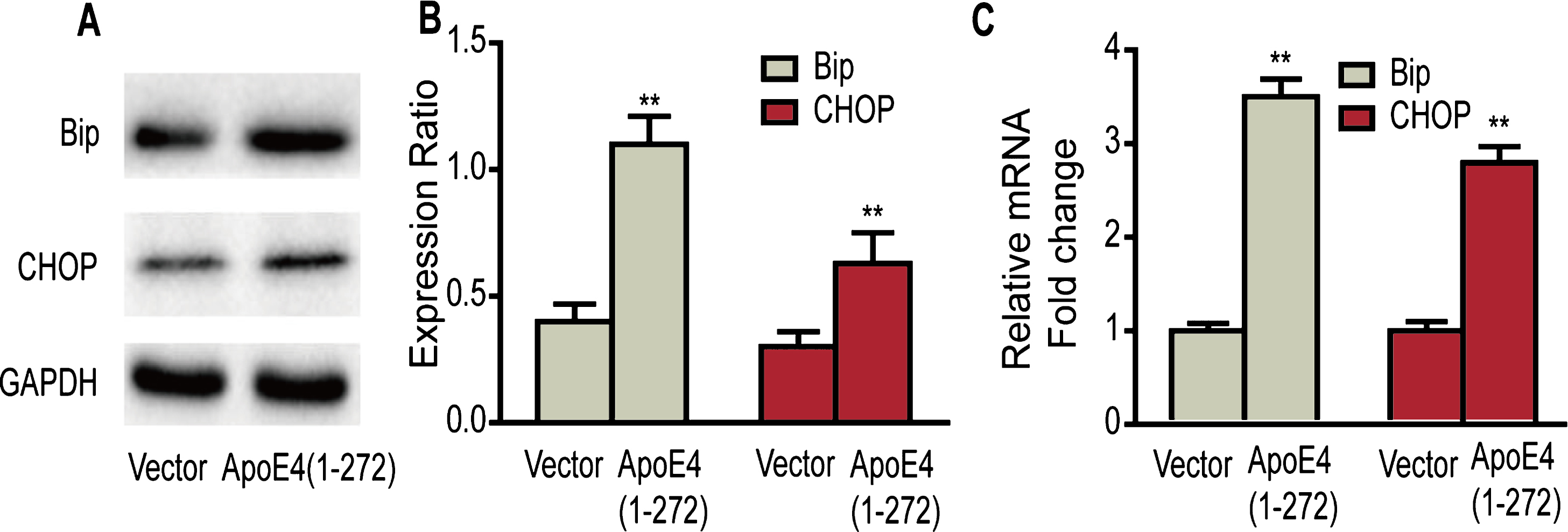
Overexpressing human apoE4(1–272) fragment triggered ER stress in N2a cells. A, B) ER stress markers, BiP and CHOP, were analyzed by western blotting. C) The relative mRNA expressions of BiP and CHOP were quantified by real-time PCR. (**p<0.01 versus Vector; n = 3 for western blotting; the significant differences from the respective values were determined by ANOVA).
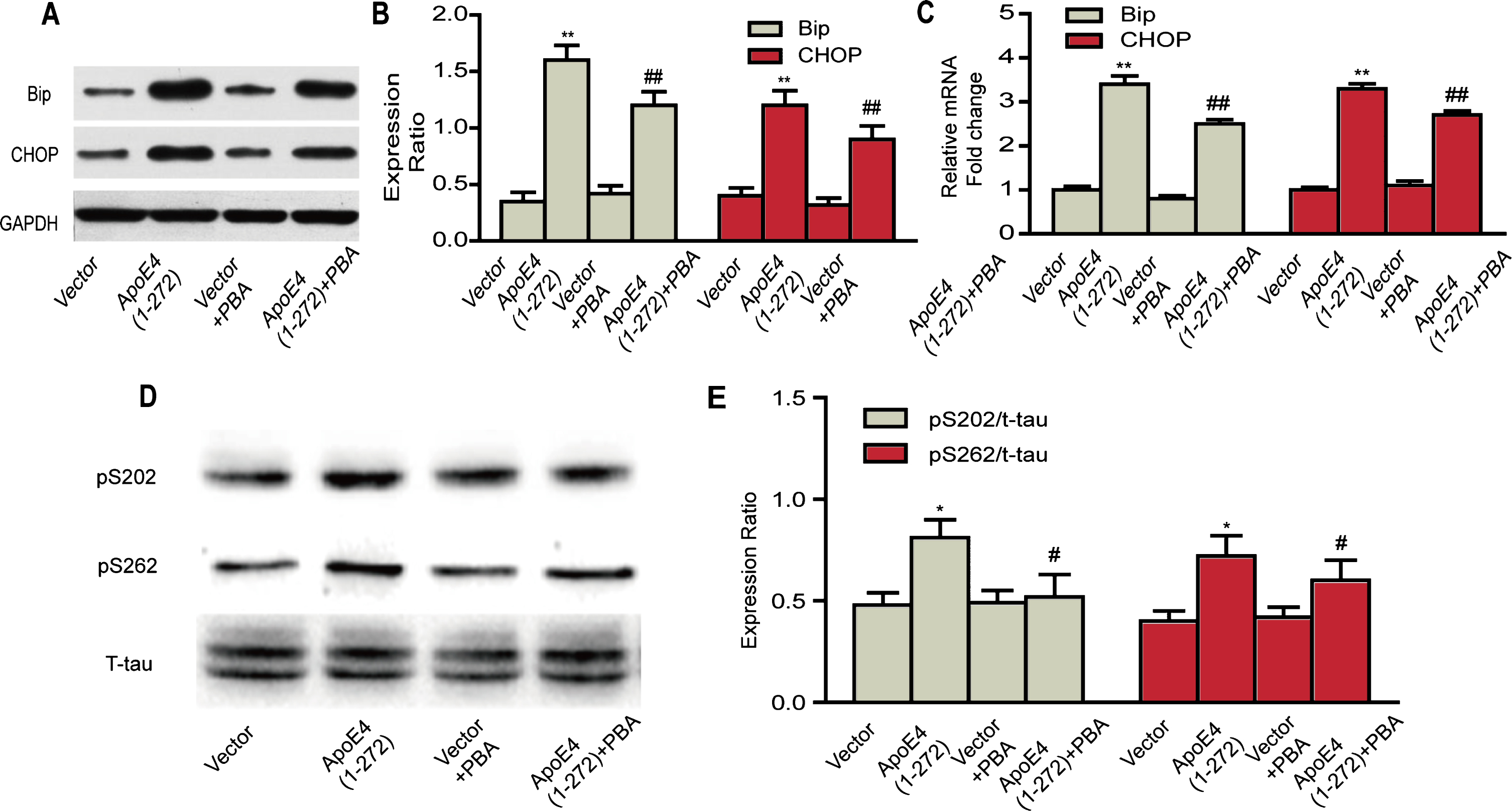
Inhibitory effects of PBA on human apoE4(1–272) fragment induced-ER stress and tau hyperphosphorylation in N2a cells. A, B) Protein levels of BiP and CHOP were analyzed by western blotting. C) The relative mRNA expressions of BiP and CHOP were quantified by real-time PCR. D, E) Phosphorylation levels of tau (Ser202 and Ser262) were detected by western blotting. (**p<0.01, *p<0.05 versus Vector; # #p<0.01, #p<0.05 versus ApoE(1–272); n = 3 for western blotting; n = 8 for real-time PCR; the significant differences from the respective values were determined by ANOVA).
Inhibitory effects of PBA on apoE4(1–272)-induced ER stress and tau hyperphosphorylation in vitro
Based on our above results, we demonstrated that over expressing apoE4(1–272) led to the ER stress and tau hyperphosphorylation. Then, we explored whether the ER stress protector 4-PBA could relieve these situations. Firstly, the N2a cells were transiently transfected with human apoE4(1–272) constructs or EGFP vector, and treated with 1 mM 4-PBA for another 16 h, then cells were harvested to examine the expressions of ER stress markers by western blot analysis or real-time PCR. We found that treatment with 1 mM 4-PBA partly rescued the upregulation of BiP and CHOP both at the protein levels (Fig. 6A, B) and mRNA levels (Fig. 6C), and there was a significant difference between apoE4(1–272) group and apoE4(1–272)+PBA group. No significant change was seen between the Vector group and the Vector+PBA group both at the mRNA and protein levels. These results suggested that PBA could effectively reduce the ER stress activity in N2a cells.
In addition, we examined the phosphorylation levels of tau at Ser202 and Ser262 sites by western blotting. The results showed that expressed apoE4(1–272) constructs in N2a cells significantly increased phosphorylation levels of tau at two sites, and treated with 1 mM 4-PBA partly rescued the tau hyperphosphorylation. Likewise, no significant change was seen between the Vector group and the Vector+PBA group (Fig. 6D). Our observations demonstrated that human apoE4(1–272) induced hyperphosphorylation of tau in N2a cells, which would be closely associated with the ER stress triggered by over expression of apoE4(1–272) fragment.
PBA rescued apoE4(1–272) induced-GSK3 activity in vitro
To unravel the molecular mechanism by which apoE4(1–272) caused tau hyperphosphorylation, we further examined the activity of two key enzyme, GSK-3 and Cdk5, which were involved in the phosphorylation of tau [22, 23]. Meanwhile, we also observed the effects of 4-PBA on their activity. N2a cells were transiently transfected with human apoE4(1–272) constructs or EGFP vector, with or without 1 mM 4-PBA, then we detected the protein expression levels of p-GSK-3β ser9, GSK-3β, and Cdk5. We also used the 32P-labeling assay to detect the activity of GSK-3 and Cdk5, respectively. Our results showed that expressed human apoE4(1–272) in N2a cells significantly increased the protein expression level of p-GSK-3β ser9 (Fig. 7A, B) and activity of GSK-3 (Fig. 7C), while 1 mM 4-PBA also significantly rescued its activity (Fig. 7A, C). However, no significant changes were seen in the protein level and activity of Cdk5 after overexpressing human apoE4(1–272) in N2a cells (Fig. 7A, D). In addition, to further demonstrate whether apoE4(1–272)-induced tau phosphorylation was regulated by GSK-3 activity, we used GSK-3 inhibitor. N2a cells were transiently transfected with human apoE4(1–272) constructs or EGFP vector, with or without 10 mM LiCl, then we detected the phosphorylation levels of tau at Ser202 and Ser262 sites by western blotting. Our results showed that LiCl rescued apoE4(1–272) induced-tau phosphorylation (Fig. 7E, F). Thus, our results indicated that hyperphosphorylation of tau, caused by apoE4(1–272), might be associated with upregulation of GSK-3β activity by ER stress induced by apoE4(1–272) in neurons.
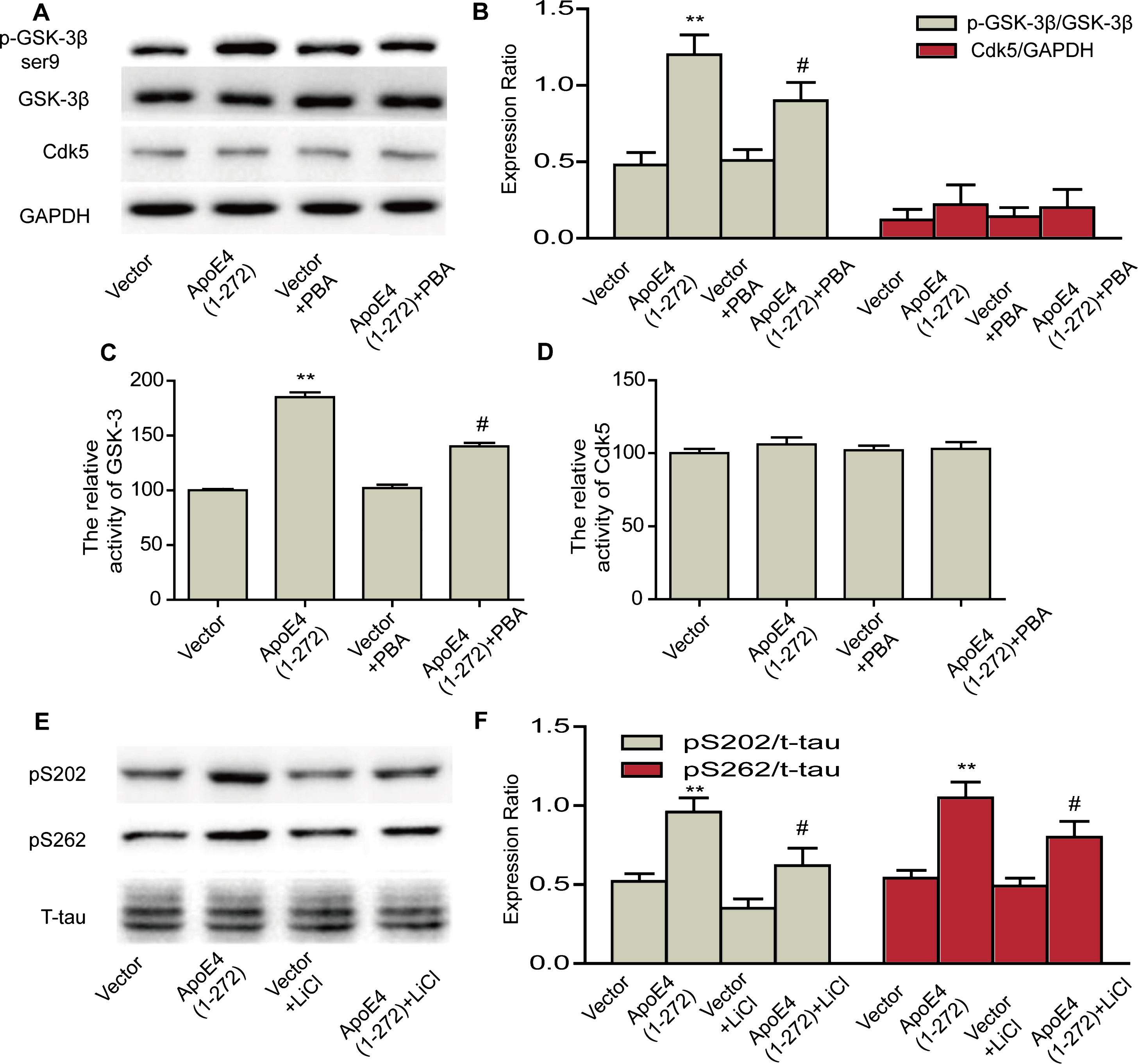
PBA rescued human apoE4(1–272) fragment induced-GSK3 activity in N2a cells. A, B) Protein levels of p-GSK-3β ser9, GSK-3β, and Cdk5 were analyzed by western blotting. C) Relative activity of GSK-3 in N2a cells. D) Relative activity of Cdk5 in N2a cells. E, F) Protein levels of pS202 and pS262 were analyzed by western blotting. (**p<0.01 versus Vector; #p<0.05 versus ApoE(1–272); n = 3 for western blotting; n = 5 for assay of activity; the significant differences from the respective values were determined by ANOVA).
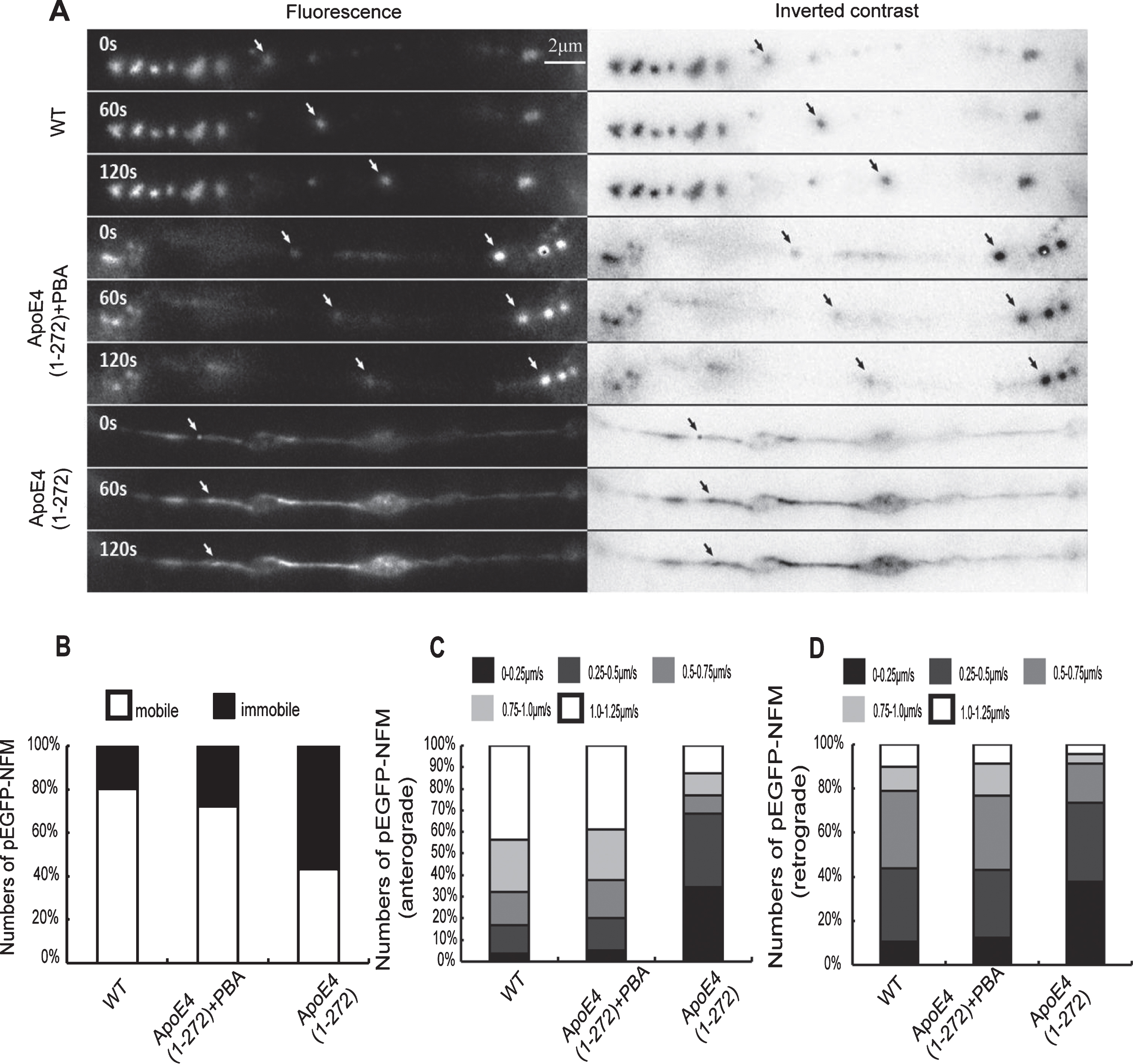
PBA effectively improved human apoE4(1–272) fragment induced-axonal transport impairment in N2a cells. A) A forward-moving eGFP-NFM particle in wild-type N2a cells and a relatively stationary particle in apoE4(1–272) fragment-transfected N2a cells. PBA reversed apoE4(1–272) fragment-induced slowing of neurofilament axonal transport significantly. B, C, D) Quantitative analyses showed that the proportion of the neurons with immobile and slow-moving eGFP-NFM increased significantly after apoE4(1–272) fragment-transfected and this was rescued by treatment with PBA (1 mM). About 90–100 vesicles in ~20 cells for each treatment were counted. (Bar = 2μm).
PBA effectively improved apoE4(1–272) induced-axonal transport impairment in vitro
Our above results showed that overexpressing apoE4(1–272) led to the swollen axons, varicosities, and enlarged fibers in the hippocampus of apoE4(1–272) transgenic mice. Additional experiments were conducted to analyze the impact of PBA on the apoE4(1–272)-induced axonal transport impairment in vitro. We transiently transfected the cultured N2a cells with pEGFP-NFM, and this could facilitate us direct observation of neurofilament axonal transport by live fluorescence imaging. When N2a cells were transfected with pEGFP-NFM for 24 h, many fluorescence particles were seen to move along axons. Most of the particles were moving at an average velocity of about 1.10μm/s in an anterograde direction, while only a few particles moved at a slower speed of 0.25μm/s in a retrograde direction (average velocity). Very few immobile fluorescence particles were observed (Fig. 8). To determine whether apoE4(1–272) could induce axonal transport impairment and whether PBA influence this situation, we transiently transfected the cultured N2a cells with apoE4(1–272) constructs in the presence or absence of 1 mM 4-PBA and monitored neurofilament movement along the axons. We found that the N2a cells transfected with apoE4(1–272) constructs induced a decrease by 58% in the velocity of EGFP-NFM. However, treatment of the cells with 1 mM 4-PBA antagonized this effect by 28%. These data indicated that the treatment of the N2a cells with apoE4(1–272) fragment slowed the velocity of EGFP-NFM transport and PBA reversed apoE4(1–272) fragment-induced axonal transport impairment significantly (Fig. 8).
DISCUSSION
Growing evidence have demonstrated that apoE4 plays an important role in the pathogenesis of AD. ApoE is a key protein involved in lipid metabolism through its high affinity interaction with cell surface receptors [24, 25]. ApoE has two seemingly independent domains separated by a protease sensitive hinge region, which contains a N-terminal domain (residues 1–191) and a C-terminal domain (residues 216–299) [26, 27]. The amino-terminal region forms a four-helix bundle spanning residues 24–164. The carboxyl-terminal region is also highly α-helical, but its tertiary structure is very polymorphic and participates in inter-domain interactions with the amino-terminal domain as well as with lipids [28–30]. Early researches had been reported that ApoE4 could be cleaved by a protease to generate bioactive C-terminal-truncated forms of apoE4, such as apoE4(1–272) fragment [31]. The truncated apoE4(1–272) fragment was generated inside neurons and in AD brains. Many researches had demonstrated that the accumulation of apoE4(1–272) fragments were neurotoxic. Therefore, we speculated that apoE4(1–272) in neurons was an effector molecule for apoE4 and caused AD lesions.
Early studies had confirmed that axonopathy triggered by axonal impairments was thought to contribute significantly to the early stages in the pathogenesis of AD [13]. In AD patients, axonal pathology in the brain was visible in the form of dystrophic neurites. These defects were usually manifested by axonal swelling or spheroids, which corresponded to axonal enlargements and abnormal accumulation of axonal proteins and cytoskeletal proteins, like neurofilaments or hyperphosphorylated tau. These characteristic changes including axonal and bulging swelling, axonal transport disorders, and axonal leakage also had been described in different APP-based transgenic AD mouse models [32]. Accumulating evidence suggested that early axonal lesions were closely related to cognitive decline [33]. Here in our studies, we showed the apoE4(1–272) transgenic mice were with good breeding performance at early stages. However, at about 9 months, apoE4(1–272) mice showed abnormal performance such as weight loss, skin infections, poor activity, and even death. By MWM, we found that the 12- to 14-month-old apoE4(1–272) mice exhibited significant deficits in spatial learning and memory. Moreover, there were prominent axonal morphological changes in the hippocampus of apoE4(1–272) transgenic mice (Fig. 1B). These results suggested that the cognitive dysfunction in apoE4(1–272) transgenic mice might be related to neuronal axon lesions.
Intracellular NFTs consisting of hyperphosphorylated tau is an important pathological feature of AD. Increasing evidence has confirmed that the abnormal hyperphosphorylation of tau and NFTs are critical for the impairment of axonal transport during the pathological process of AD, which can mediate axonopathy and contribute to the cognitive deficit [34]. Microtubules serve as a track for the axonal transport. Tau protein binds to microtubules, promotes tubulin assembly into microtubules, and stabilizes neuronal microtubules. Hyperphosphorylated tau disrupts the stability of microtubules and influences microtubules organization [35]. In the current study, we demonstrated that theapoE4(1–272) fragment could induce axonal transport impairment in vitro and cause tau hyperphosphorylation in vivo and in vitro (Figs. 8 and 3). Tau phosphorylation process can be regulated by tau kinases such as GSK-3β [36]. In addition, we further found that the hyperphosphorylation of tau induced by apoE4(1–272) fragment could be regulated by GSK-3 activity. Our results showed that apoE4(1–272) fragment-induced neuronal axonal lesions might be closely associated with aberrant hyperphosphorylation of tau, which was due to activation of the GSK-3β triggered by apoE4(1–272) fragment.
ER stress has been shown to contribute to various disorders in the central nervous system [37]. It was also demonstrated the presence of UPR and ER stress at the site of NFT formation in the brains of AD patients [17]. Accordingly, our results demonstrated that apoE4(1–272) fragment triggered significant ER stress in vivo and in vitro (Figs. 4 and 5). Furthermore, we found that the inhibition of ER stress via PBA efficiently downregulated tau hyperphosphorylation and the activation of GSK-3β. In addition, PBA reversed apoE4(1–272) fragment-induced axonal transport impairment significantly. Our results indicated that the ER stress due to the gradual accumulation of toxic effects by apoE4(1–272) was an early cause of subsequent events.
Our findings have implications for understanding how the apoE4(1–272) fragment contributes to the pathogenesis of AD in some extent. Combining these previous findings with our present results, we speculate a possible pathway in which the human apoE4(1–272) fragment participates in the pathogenesis of AD. First, under the pathophysiological conditions, the neurons express apoE4, then the later will be cleaved into the apoE4(1–272) fragment. The apoE4(1–272) fragment loses the secretion function and stays in the cytosol, which may interact with the ER or directly trigger ER stress. At young stages, the stress may be at a low level. The effects of ER stress (induced by apoE4(1–272) fragment), which causes tau phosphorylation, are also not serious. However, with aging, oxidative stress, head trauma, or stroke, the effects of stress accumulate over time. ER stress will be excessively active and lead to hyperphosphorylation of tau. Later, the hyperphosphorylation of tau affects the stability of microtubules, which eventually results in the dysfunction of axonal transport in neurons. Because of the impairment of axonal transport, lots of substrates, such as the organelles and biomolecules, do not effectively transport from cell bodies to terminal (synapse or plasma membrane) or from terminal toward the cell body and are accumulated in the axons, which will result in axonopathy (swollen axons or even swollen varicosities). The latter will further lead to the impairment of learning and memory and participate in the pathogenesis of AD. Our findings also provide a new clue to connect apoE4 with ER stress and it is also a potential therapeutic target or biomarker for AD.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (81873725, 31670778, 81371416), the Fundamental Research Funds for the Central Universities (HUST: 2016YXMS191) to J.C; the Shanxi Provincial Key research and development Program (2018SF-090) to F.X.