
Abstract
Background:
Greater body weight has been associated impairments in neurocognition and greater dementia risk, although the mechanisms linking weight and neurocognition have yet to be adequately delineated.
Objective:
To examine metabolic mechanisms underlying the association between obesity and neurocognition.
Methods:
We conducted a secondary analysis of weight, neurocognition, and the potentially mediating role of metabolic and inflammatory biomarkers among 160 participants from the ENLIGHTEN trial of vascular cognitive impairment, no dementia (CIND). Neurocognition was assessed using a 45-minute assessment battery assessing Executive Function, Verbal and Visual Memory. We considered three metabolic biomarkers: insulin resistance (homeostatic model assessment [HOMA-IR]), plasma leptin, and insulin-like growth factor (IGF-1). Inflammation was assessed using C-reactive protein. Multiple regression analyses were used.
Results:
Participants included 160 sedentary older adults with CIND. Participants tended to be overweight or obese (mean BMI = 32.5 [SD = 4.8]). Women exhibited higher BMI (p = 0.043), CRP (p < 0.001), and leptin (p < 0.001) compared with men. Higher BMI levels were associated with worse performance on measures of Executive Function (β= –0.16, p = 0.024) and Verbal Memory (β= –0.16, p = 0.030), but not Visual Memory (β= 0.05, p = 0.500). Worse metabolic biomarker profiles also were associated with lower Executive Function (β= –0.12, p = 0.050). Mediation analyses suggested leptin was a plausible candidate as a mediator between BMI and Executive Function.
Conclusions:
In overweight and obese adults with vascular CIND, the association between greater weight and poorer executive function may be mediated by higher leptin resistance.
Keywords
INTRODUCTION
Numerous epidemiological and mechanistic studies have demonstrated that greater body mass index (BMI) is associated with lower neurocognitive function [1, 2] and greater risk of Alzheimer’s disease and related dementias (ADRD) [3–5]. Evidence increasingly indicates that obesity and related cardiometabolic risk factors increase the risk of ADRD [6–8], with obesity increasing the risk of AD by 80%, independent of traditional risk factors [9]. The impact of obesity on ADRD outcomes is particularly important given its increasing prevalence, affecting nearly half of adults in the United States [6, 10].
Impaired metabolic function has been implicated as an important mechanism linking greater BMI levels to worse neurocognitive outcomes [11–14], with studies in both animal and human samples demonstrating that impairments in metabolic biomarker profiles, including insulin sensitivity [15], leptin [13, 16], and insulin-like growth factor (IGF) [17–19], are associated with worse neurocognition. Leptin is an adipocyte-secreted hormone thought to be involved in both regulation of appetite and cognitive function [20, 21]. Chronically elevated levels of leptin are believed to impair the brain’s ability to effectively modulate leptin levels in response to changes in homeostatic mechanisms, leading to leptin resistance [22]. Similarly, peripheral levels of IGF-1 have previously been associated with neurocognition [23–25], with lower IGF-1 levels associating with worse neurocognition and a greater likelihood of cognitive decline. Although impaired metabolic function, including insulin and leptin resistance, has been postulated to mediate the association between greater BMI and impaired neurocognition, results have been conflicting [26–28], and few studies have examined these associations in clinical samples at risk for ADRD [29], We therefore examined the associations between greater BMI, metabolic biomarkers, and neurocognition from baseline analyses of a cohort of older adults with vascular cognitive impairment participating in the Exercise and NutritionaL Interventions for neurocoGnitive HealTh EnhaNcement (ENLIGHTEN) study, which examined the independent and combined effects of aerobic exercise and the Dietary Approaches to Stop Hypertension (DASH) diet on neurocognitive functioning in older adults with CIND [30]. Results of the 6-month intervention showed that exercise was associated with improved executive function, and that combining exercise with the DASH diet produced greater improvements compared to education controls [30]. The present report describes an analysis of the metabolic mechanisms associated with BMI and neurocognition in the ENLIGHTEN sample.
METHODS
Participant recruitment and eligibility
ENLIGHTEN was a randomized clinical trial of inactive older adults with subjective memory complaints, objective evidence of cognitive impairment, and at least one additional cardiovascular disease (CVD) risk factor (besides being sedentary) [30]. Participants were recruited from newspaper and television advertisements, physician referrals, and patient mailings through the Duke University Alzheimer’s Disease Prevention Registry (ADPR) located in the Joseph & Kathleen Bryan Alzheimer’s Center and through the recruitment database of the Duke Center for Aging and Human Development. Participants were older adults (age ≥55 years), had subjective cognitive complaints, and evidence of objective cognitive weakness from a screening battery that included tests of verbal fluency and the Montreal Cognitive Assessment (MoCA).
Procedures
Neurocognition
Neurocognitive functioning was assessed using a 45–60-min test battery recommended by the Neuropsychological Working Group for vascular cognitive disorders [31, 32]. A more complete description of the instruments along with normative data are available for the interested reader [33–35].
Metabolic function
Insulin resistance: Insulin resistance was defined using the Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) [43–45]. Optimal ranges for healthy adults tend to range from 0.5–1.4, with higher levels indicating greater insulin resistance [46]. All participants completed assessments of HOMA-IR.
Leptin: Plasma leptin levels were analyzed from EDTA plasma samples, assayed using solid phase quantitative ELISA according to a standard protocol [29, 48].
Insulin like growth factor (IGF-1): IGF-1 is an essential neurotrophic factor that is produced both peripherally and in the brain. Data on both leptin and insulin like growth factor were obtained in an ancillary biomarker study, for which 132 individuals (83%) provided data.
Inflammation
High-sensitivity C-reactive protein (hsCRP). hsCRP was quantified by Lab Corp using commercial enzyme-linked immunosorbent assay kits (R&D Systems). Values exceeding 10 mg/dL were truncated at 10 mg/dL, consistent with current guidelines for the analysis of inflammatory markers [49].
Data analysis
Analyses were conducted using general linear models within SAS 9.4. Analyses of baseline weight and metabolic biomarkers were conducted using hierarchical linear regression modeling in which HOMA-IR, leptin, and IGF were all combined into a mean rank composite [50]. In order to test for possible mediation, these individual biomarkers were examined independently in a final step due to their plausibly distinct contributions to neurocognitive functioning using the PROCESS MACRO [51]. Within each neurocognitive domain, separate rank-based composites were created combining results from all neurocognitive subtests within the domain. This method had been advocated for increasing power, while simultaneously lowering type-I error rates [52, 53]. All analyses controlled for age, education, gender, race, CVD medication burden, and CVD history, with BMI as the predictor of interest. Missing data were accounted for using Markov chain Monte Carlo multiple imputation methods available in SAS (PROC MI) and 100 imputations. Assumptions regarding linearity, independence and distribution of residuals were assessed in all analyses and both leptin and IGF-1 were log-transformed prior to analysis.
RESULTS
The cohort was comprised of 160 participants (Table 1). All patients provided HOMA-IR data, while 132 individuals (83%) had biomarker data on leptin and IGF-1. Participants who did not provide biomarker data had greater baseline Memory performance (p = 0.002), had a lower CVD medication burden (p = 0.014), were more likely to be male (p = 0.050), and tended to have greater Executive Function compared to those who participated (p = 0.087). Examination of baseline BMI and metabolic profiles revealed that approximately two-thirds of the sample was obese (mean BMI = 32.5 [SD = 4.8]) and nearly half of participants demonstrated HOMA-IR levels consistent with impaired insulin resistance (mean HOMA-IR = 4.2 [SD = 2.7]). Women demonstrated higher BMI (women: 33.0 [SD = 4.8] versus men: 31.4 [SD = 4.5], p = 0.043), leptin (women: 61.1 [SD = 50.2] versus men: 16.0 [SD = 14.1], p < 0.001), and CRP levels (women: 3.5 [SD = 4.1] versus men: 1.7 [SD = 1.7], p < 0.001) compared to men, but did not differ in HOMA-IR (p = 0.461) or IGF-1 (p = 0.869).
Background and metabolic characteristics of the sample
Body weight, metabolic function, and neurocognition
Examination of body weight and neurocognition revealed that greater BMI was associated with lower scores of Executive Function (β= –0.16, p = 0.024) (Fig. 1A) and Verbal Memory (β= –0.16, p = 0.030), but were not consistently associated with either Visual Memory (β= –0.08, p = 0.241). Greater BMI levels also were strongly associated with worse metabolic biomarker profiles (β= 0.49, p < 0.001) and greater CRP (β= 0.30, p < 0.001). Follow-up examination of individual metabolic biomarkers revealed that greater BMI was associated with greater HOMA-IR levels (β= 0.31, p < 0.001), leptin (β= 0.41, p < 0.001), and IGF-1 (β= –0.18, p = 0.041). Worse metabolic biomarker profiles also were associated with lower Executive Function (β= –0.12, p = 0.054), but were not consistently associated with either Verbal (p = 0.176) or Visual Memory (p = 0.292). Follow-up analyses revealed that both HOMA-IR (β= –0.15, p = 0.032) (Fig. 1B) and leptin (β= –0.18, p = 0.039) (Fig. 1 C) were associated with lower Executive Function, whereas IGF-1 was not (β= 0.01, p = 0.978). Elevated CRP also was associated with lower Executive Function (β= –0.14, p = 0.047). None of the observed associations appeared to vary by either gender (ps > 0.482) or APOE status (ps > 0.406). For example, in exploratory analyses of potential gender differences, we found that the association between BMI and Executive Function was comparable between men and women (βs = –0.16 and –0.13, p = 0.717 for interaction) (Supplementary Figure 1). Similarly, although the associations between CRP, HOMA-IR, and Executive Function appeared slightly stronger for men than women, neither of these associations varied consistently by gender (CRP: βs = –0.07 and –0.16, p = 0.977 for interaction; HOMA-IR: βs = –0.06 and –0.16, p = 0.424 for interaction) (Supplementary Figure 2). Finally, the association between leptin and Executive Function also did not vary consistently by gender, although the association appeared slightly stronger in men compared to women (βs = –0.22 and –0.14, p = 0.467 for interaction) (Supplementary Figure 3).
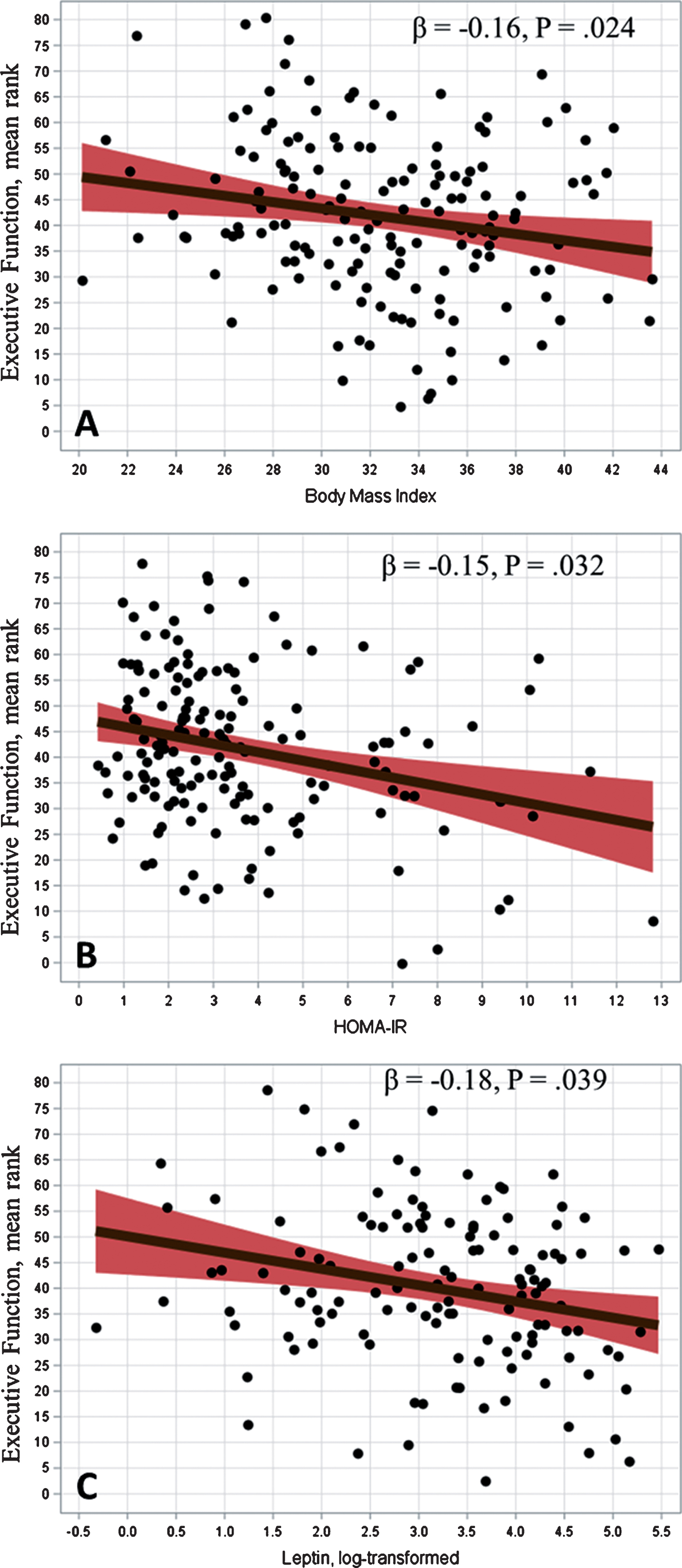
Body mass index and executive function (A). Insulin sensitivity and executive function (B). Leptin and executive function (C). Values are adjusted for age, education, gender, ethnicity, MoCA score, CVD medication burden, and CVD history.
Mediation analyses
Mediation analyses were conducted to examine the extent to which the metabolic biomarkers might explain the BMI and Executive Function association. In an initial step, greater BMI was associated with lower Executive Function (p = 0.039), greater HOMA-IR levels (p < 0.001), greater leptin (p < 0.001), lower IGF-1 (p = 0.041), and greater CRP levels (p < 0.001). In addition, greater levels of HOMA-IR and leptin were associated with greater CRP (β= 0.18, p = 0.055; β= 0.32, p < 0.001). In bootstrapped mediation models, elevated HOMA-IR partially mediated the association between obesity and executive dysfunction (indirect effect = –0.03 [–0.08, 0.01]), as did greater levels of CRP (indirect effect = –0.03 [–0.09, 0.01]), whereas IGF-1 did not show a consistent, indirect association that explained the observed relationship between greater BMI and Executive Function (indirect effect = –0.00 [–0.03, 0.04]). In contrast, examination of leptin demonstrated that higher leptin levels mediated the association between obesity and Executive Function (indirect effect = –0.07 [–0.16, –0.01]) (Fig. 2).

Mediation models examining the role of HOMA-IR and leptin to the BMI and executive function association.
DISCUSSION
Our findings add to existing data linking excess body weight and impaired metabolic function to diminished neurocognitive performance among older adults at risk for ADRD. We found that greater BMI levels, HOMA-IR, and leptin were all associated with lower executive function. We also found that our data were consistent with metabolic dysfunction—particularly leptin resistance—mediating the association between BMI and executive function. Our findings underscore the potential importance of metabolic dysfunction and particularly leptin to the association between greater BMI and impaired executive function, suggesting that elevated leptin may mediate this previously demonstrated association [54]. The present findings extend previous evidence by demonstrating that elevated levels of leptin, which may suggest leptin resistance [4], appear to associate with worse executive function even after accounting for other relevant metabolic and inflammatory markers [55–57].
Previous studies have demonstrated that greater BMI is associated with subclinical impairments in executive function [58, 59], as well as amplifying age-associated changes in brain morphology, including cortical volume loss [8, 61], greater white matter hyperintensities [62], and a 2-3 fold increase in subsequent ADRD risk [6–8]. The adverse impact of elevated BMI on the brain is thought to be mediated by underlying metabolic abnormalities, which are closely linked to BMI and have also been associated with increased risk of ADRD [27, 63–70]. For example, the presence of obesity has been reported to increase ADRD risk independent of comorbid vascular risk factors [71] or genetic risk [10], with some investigators demonstrating a 3-4 fold increased risk of dementia among obese adults compared to their normal weight counterparts [8, 71–75].
Despite the widely documented association between greater BMI and impaired neurocognition, relatively few studies have examined metabolic biomarkers as putative explanatory mechanisms underlying these associations [76] and even fewer have attempted to elucidate these associations in clinical populations [54, 55]. A large body of evidence has demonstrated that greater BMI levels may associate with ADRD through their association with impaired insulin signaling [3, 77], with some investigators going as far as introducing additional nomenclature to cast AD as ‘type-III diabetes’ [78, 79]. In contrast, the potential role of leptin has only recently gained attention, with the majority of existing data coming from animal studies [4, 80] and few in clinical samples among humans [54, 55]. Previous data among individuals with both MCI [81] and AD [82] have demonstrated that elevated leptin levels are present among individuals with clinically impaired neurocognition, as well as associating with peripheral markers of metabolic risk. Not all studies have produced similar findings, however, and a recent examination of older women in the Study of Osteoporotic Fractures demonstrated that the associations between peripheral leptin and prospective risk of MCI and dementia may vary by BMI level [54].
Dysregulation of leptin has also been widely implicated in impaired CNS signaling within brain areas critical for energy homeostasis, memory, and learning [22, 83]. Several central nervous system (CNS) mechanisms have been suggested to mediate the association between impaired metabolism, leptin, and neurocognition. Chronically elevated leptin levels, as observed in the presence of obesity, may result in downregulation of CNS leptin signaling, suppressing the ability of CNS leptin to disrupt Aβ accumulation and toxicity [4]. Finally, elevated leptin has also been suggested to impair neurocognition by increasing systemic inflammation [22, 56], although our findings demonstrated that the observed association between leptin and executive function was not explained by elevated levels of CRP. Although our findings may be interpreted as suggesting that leptin resistance associated with worse executive function because higher levels of peripheral leptin may suggest leptin resistance, previous studies in non-obese samples have also found that leptin deficiency associated with neurocognitive decline [4, 84]. Similar to previous reported findings with insulin resistance [85], where peripheral levels of metabolic function may vary in their association with CNS metabolism depending on the individual differences in metabolic status, such as obesity or diabetes.
Limitations
First, the sample size in the present study is smaller than previously published cohort studies and the present findings will therefore require replication [86]. Second, we did not obtain cerebrospinal fluid or other CNS markers of leptin, which limit any inferences we can make between the observed association of peripheral, plasma levels of leptin and alterations in CNS functioning. As noted above, because our sample was comprised primarily of obese older adults, the present pattern of findings may not generalize to other samples without significant metabolic comorbidities. Third, individuals in the present study were selected based on their diminished neurocognitive function, the presence of CVD risk factors, and their motivation to participate in a diet and exercise study, potentially limiting the generalizability of our study findings. Finally, as with all observational studies, the associations observed in the present study are potentially subject to confounding with unmeasured variables.
In summary, our findings suggest that metabolic dysfunction mediates the association between greater BMI and executive function. Future studies should examine whether modifying metabolic function through behavioral or pharmacologic treatment improves neurocognitive function among individuals with elevated BMI at risk for neurocognitive impairment. In particular, examination of behavioral interventions among individuals with leptin resistance would inform whether targeted, preventive interventions hold promise in lowering the incidence of neurocognitive impairment among at-risk adults.