
Abstract
The Mexican American population is among the fastest growing aging population and has a younger onset of cognitive decline. This group is also heavily burdened with metabolic conditions such as hypertension, diabetes, and obesity. Unfortunately, limited research has been conducted in this group. Understanding methylation alterations, which are influenced by both genetic and lifestyle factors, is key to identifying and addressing the root cause for mild cognitive impairment, a clinical precursor for dementia. We conducted an epigenome-wide association study on a community-based Mexican American population using the Illumina EPIC array. Following rigorous quality control measures, we identified 10 CpG sites to be differentially methylated between normal controls and individuals with mild cognitive impairment annotated to PKIB, KLHL29, SEPT9, OR2C3, CPLX3, BCL2L2-PABPN1, and CCNY. We found four regions to be differentially methylated in TMEM232, SLC17A8, ALOX12, and SEPT8. Functional gene-set analysis identified four gene-sets, RIN3, SPEG, CTSG, and UBE2L3, as significant. The gene ontology and pathway analyses point to neuronal cell death, metabolic dysfunction, and inflammatory processes. We found 1,450 processes to be enriched using empirical Bayes gene-set enrichment. In conclusion, the functional overlap of differentially methylated genes associated with cognitive impairment in Mexican Americans implies cross-talk between metabolically-instigated systemic inflammation and disruption of synaptic vesicular transport.
INTRODUCTION
Alzheimer’s disease (AD) affects 5.8 million Americans and is currently the sixth most common cause of death [1]. AD is a neurodegenerative disease that manifests as a result of intracellular tau tangles and extracellular amyloid-β plaques within the brain, causing cellular inflammation and neuronal loss, which lead to changes in memory and personality [1]. Mild cognitive impairment (MCI), a common precursor to AD, affects over a quarter of individuals over the age of 65, with a third of these individuals developing AD later in life [2].
Within the United States, the Mexican American elderly population is experiencing rapid growth and is predicted to increase 6-fold by 2050 [3]. This population has an earlier age of AD onset [4] and are typically diagnosed at more advanced stages of disease relative to non-Hispanic whites [3, 4]. Further, Mexican Americans are disproportionately burdened with comorbid conditions such as depression [5, 6], cardiovascular disease [7], and metabolic conditions (e.g., metabolic syndrome, type 2 diabetes, obesity) [4, 7]. Other factors such as education and socio-economic status have also been suggested to play a role, where fewer years of education and lower economic status are known to increase one’s risk for AD [8].
There are several critical differences in the etiology of AD when comparing Mexican Americans and non-Hispanic whites, and these distinctions may be key to understanding the observed disparities in the population. Mexican Americans are less likely to carry the APOE ɛ4 allele which is the one of the highest predisposing genetic factors for AD among non-Hispanic whites [4, 8]. Additionally, while a vascular/inflammatory phenotype predominates risk for cognitive decline among non-Hispanic whites, risk among Mexican Americans is due primarily to metabolic factors [9]. In a study of serum biomarkers from 363 Mexican Americans (AD = 49, NC = 314), the biomarker profile of AD among Mexican Americans was weighted heavily for metabolic markers as opposed to the inflammatory-laden profile that has previously been observed among non-Hispanic whites [10].
Cognitive impairment is highly heterogeneous, with both environmental/lifestyle and genetic factors playing a role. Since AD pathology can initiate up to 20 years before the first visible symptoms [1], changes in methylation patterns may signify relevant early molecular responses to the disease. Methylation at CpG (cytosine-phosphate-guanine) sites throughout the genome is known to affect downstream expression of various genes and has been implicated in normal aging and disease processes [11, 12]. Differential methylation relating to AD and MCI has already been reported in several populations [11–14]. While metabolic stress detected in the peripheral blood is associated with cognitive impairment, the exact mechanisms are unknown [15]. Wang et al. have identified epigenetic changes in the brain of AD patients that exacerbate synaptic plasticity within functional networks overlapping multiple brain regions [16]. Moreover, changes in DNA methylation patterns have been shown to be highly correlated between peripheral blood and brain [17]. Since metabolic burden affects both the peripheral and central nervous system [18], detecting epigenetic signatures may help identify risk regions that overlap metabolic burden and cognitive dysfunction.
Here we sought to investigate genome-wide differential methylation at site- and region-specific levels to identify epigenetic factors that may confer risk for cognitive impairment in Mexican Americans.
METHODS AND MATERIALS
Samples and cohort design
This study was approved under the North Texas Regional IRB #2012–083. All participants provided written consent. Peripheral blood buffy coat of fasted-state Mexican American participants (n = 90) enrolled in the Health & Aging Brains of Latino Elders (HABLE) study was obtained.
Each participant underwent an interview (i.e., medical history, medications, health behaviors), neuropsychological testing, blood draw, and medical examination. Global cognition was assessed via the Mini-Mental State Examination (MMSE) [19] and disease severity rated according to the Clinical Dementia Rating scale [20] sum of boxes scores (CDR-SB) [21, 22]. An informant interview was conducted for each research participant to obtain information regarding his/her activities of daily living (basic and instrumental). All information was presented at a weekly consensus review conference with diagnoses of AD [23] and MCI [24] assigned according to published criteria. Cognitively normal control (NC) participants performed within normal limits on psychometric assessment [25].
Phenotypic differences between the two groups (Normal Controls & Mild Cognitive Impairment) were tested for two-tailed significance using independent t-test for continuous variables, and chi-square test for categorical variables (Table 1). Participants were matched on sex and age. Peripheral blood samples were handled in accordance to the UNTHSC Institutional Biosafety Committee approved protocol IBC-2018-0078. Forty-five individuals (both male and female) diagnosed with MCI (based on a battery of neuropsychological tests) were selected. Forty-five age and sex-matched healthy individuals were then chosen to complete the study population. Metabolic risk score (0–5) was calculated for each individual following adapted International Diabetes Federation (IDF) guidelines based on obesity, elevated triglycerides, reduced HDL cholesterol, hypertension diagnosis, and high fasting glucose [26]. Waist circumference was used to diagnose obesity, with 35.4 inches being the cut-off value for Mexican American adults [27]. Missing data was handled according to Masconi et al., 2015 [28].
Demographic characteristics for the sample cohort
DNA extraction and quantification
DNA was extracted from peripheral blood buffy coat using the MagBind® Blood and Tissue DNA HDQ Kit (Omega Bio-tek, Norcross, GA) on a Microlab STAR liquid handling system. DNA concentrations were quantified using Qubit®4 dsDNA BR Assay Kits with the Qubit® Fluorometer (Thermo Fisher Scientific Inc.) and the NanoDrop® spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA). For downstream applications, DNA extracts with concentrations ≥10 ng/μL were considered sufficient due to the manufacturer’s requirement of 200 ng of input DNA for both methylation and genotyping arrays. Low yield DNA extracts were concentrated using Microcon® DNA Fast Flow Filters (Sigma Aldrich, St. Louis, MO). Any extracts that remained <10 ng/mL following concentration, were excluded from downstream processing.
Single nucleotide polymorphism (SNP) genotyping
All 90 subjects were genotyped on the Infinium HTS Global Screening Array v.2 (Illumina, San Diego, CA) following manufacturer’s instructions. Intensity data files were imported into Genome Studio®. All SNPs with call rates <0.99 were excluded from analysis. Remaining SNPs were QC’d in PLINK; SNPs with > 5% missingness were removed (–geno) [29]. The QC’d SNP dataset was used to identify presence of population substructure using ADMIXTURE [30]. The cross-validation error rate was lowest for k of 1, concluding that the population did not have any sub-clusters (Supplementary Figure 1).
DNA methylation array assay
DNA methylation levels were analyzed for all 90 participants. DNA was bisulfite converted using the EZ DNA MethylationTM kit (Zymo Research, Irvine, CA). Bisulfite converted DNA was then processed on the Infinium HD MethylationEPIC Array (Illumina) following manufacturer’s guidelines. The array has an exhaustive coverage of more than 850,000 CpG sites and is highly correlated with Infinium HumanMethylation450 BeadChip and whole genome bisulfite sequencing.
Intensity data (IDAT) files were imported into Genome Studio® and technical replicates were compared to determine consistency in array processing. Technical replicates all displayed R2 values greater than the 0.98 suggested threshold (Illumina).
Data analysis and visualization
Raw IDAT files were processed using the ‘ChAMP’ Bioconductor package [31] in R, which converts fluorescent intensities into beta values (ratio of methylated:unmethylated) ranging from 0 to 1 for each individual probe. Data was normalized using the BMIQ method, followed by inspection for batch effects using singular value decomposition. Both the lowest (i.e., sample plate) and highest (i.e., array) stages in the experimental workflow were corrected for using ComBat. Due to the consistent placement of technical replicates on the beadchips, one of each replicate pair was removed from analysis allowing for further correction of variance at the array level. The final dataset for analysis after QC included 738,919 CpG sites from 90 individuals. An association test to identify significantly differentially methylated probes was performed using M-values (transformed beta-values) and adjusted for variation in proportions of the cell types lymphocytes, monocytes, and neutrophils, using limma [32]. Differentially methylated probes were considered significant based on multiple testing corrected p-value <0.05. Differentially methylated regions were analyzed using DMRcate [33]. Functionally differentially methylated modules were also analyzed using normalized, batch-corrected beta-values using FEM package [34] built-in ChAMP.
The significant genes identified were analyzed for gene ontology enrichment using empirical Bayes gene set enrichment analysis (GSEA; built-in ChAMP) [35] and the ShinyGO [36] tool set for biological processes, cellular components and molecular functions at an FDR <0.05. Visualizations were created using Gviz [37] and ggplot2 [38] in R. Candidate genes were analyzed for pathway enrichment, and subsequent figure was generated through the use of IPA (QIAGEN Inc., https://www.qiagenbio-informatics.com/products/ingenuity-pathway-analysis software).
RESULTS
Study characteristics
The cohort participants did not differ on demographic characteristics: age, sex, metabolic risk score, and APOE status (Table 1). The metabolic risk score was derived as per IDF guidelines [26] wherein the presence of hypertension, waist circumference, fasting glucose, triglycerides, and HDL levels is evaluated. Both groups had two or more metabolic risk factors.
Our study cohort is representative of the Mexican American population, which often 1) is metabolically burdened (as reflected by high metabolic risk scores), 2) has fewer years of education (on average) [4, 39], and 3) has a lower frequency of the APOE ɛ4 allele. The most prevalent APOE status within our cohort was ɛ3/ɛ3, with a single individual being ɛ4/ɛ4; this was optimal for our study as the frequency and risk effect of the APOE ɛ4 allele is often less in Hispanic populations [40]. This is consistent with other studies reporting differences in APOE ɛ4 frequency (approximately 20–25%) in Mexican American population [41, 42]. MMSE scores were lower for both normal and MCI participants than usually observed in individuals of European ancestry [4]. The lower MMSE scores and number of years of education are correlated and these differences have been reported by other research groups and confirms the disparity in ethnic populations [8, 39].
Differentially methylated sites and regions
Genome-wide methylation profiles were generated using the Illumina MethylationEPIC BeadChip, a microarray-based method which assesses over 850,000 cytosine methylation sites (CpG sites) throughout the genome. These sites are enriched for enhancer regions but also include gene bodies, promoters and exons boundaries. After QC and normalization, the transformed beta values (identifying percentage of methylation) were significant (FDR p-value <0.05) for 10 CpG sites when comparing normal and MCI individuals. The differentially methylated probes (DMPs) identified were annotated to genes using UCSC’s reference gene name (Supplementary File 1). Four of these sites were hypomethylated among participants with MCI compared to normal controls: cg25016219 (KLHL29), cg26479998 (SEPT9), cg02586267 (not mapped to a gene), and cg18978297 (CPLX3). Six CpG sites were found to be hypermethylated in individuals with MCI: cg22360048 (PKIB), cg20904111 (intergenic), cg05917713 (BCL2L2-PABPN1), cg20201669 (OR2C3), cg14179796 (CCNY), and cg22327037 (intergenic).
We further tested the CpG sites using the DMRcate package [33] which maps to hg19 coordinates and clusters significant sites into regions. The genome-wide region analysis yielded four differentially methylated regions (DMRs): two on chromosome 5 located within TMEM232 and SEPT8, one on chromosome 12 within SLC17A8, and one on chromosome 17 within ALOX12 (Supplementary File 2). These four regions comprised of 41 significant CpG sites that were located primarily within islands, with some being located in open sea and shore regions (Fig. 2). CpG islands are 0.5–2 kb sequences with higher GC content located in the promoter region and methylation of these sites is believed to affect gene expression. Surrounding regions within the 2 kb of islands are the CpG shores, and regions which are further away from the island are considered open sea [43, 44].

(A) Manhattan plot highlighting differentially methylated probes. The annotated genes using UCSC RefSeq are shown above the CpG ids. (B) Scatter plots. The distribution of beta intensity for each of the FDR significant CpG probe is shown between the NC (normal control) and MCI (mild cognitive impairment).
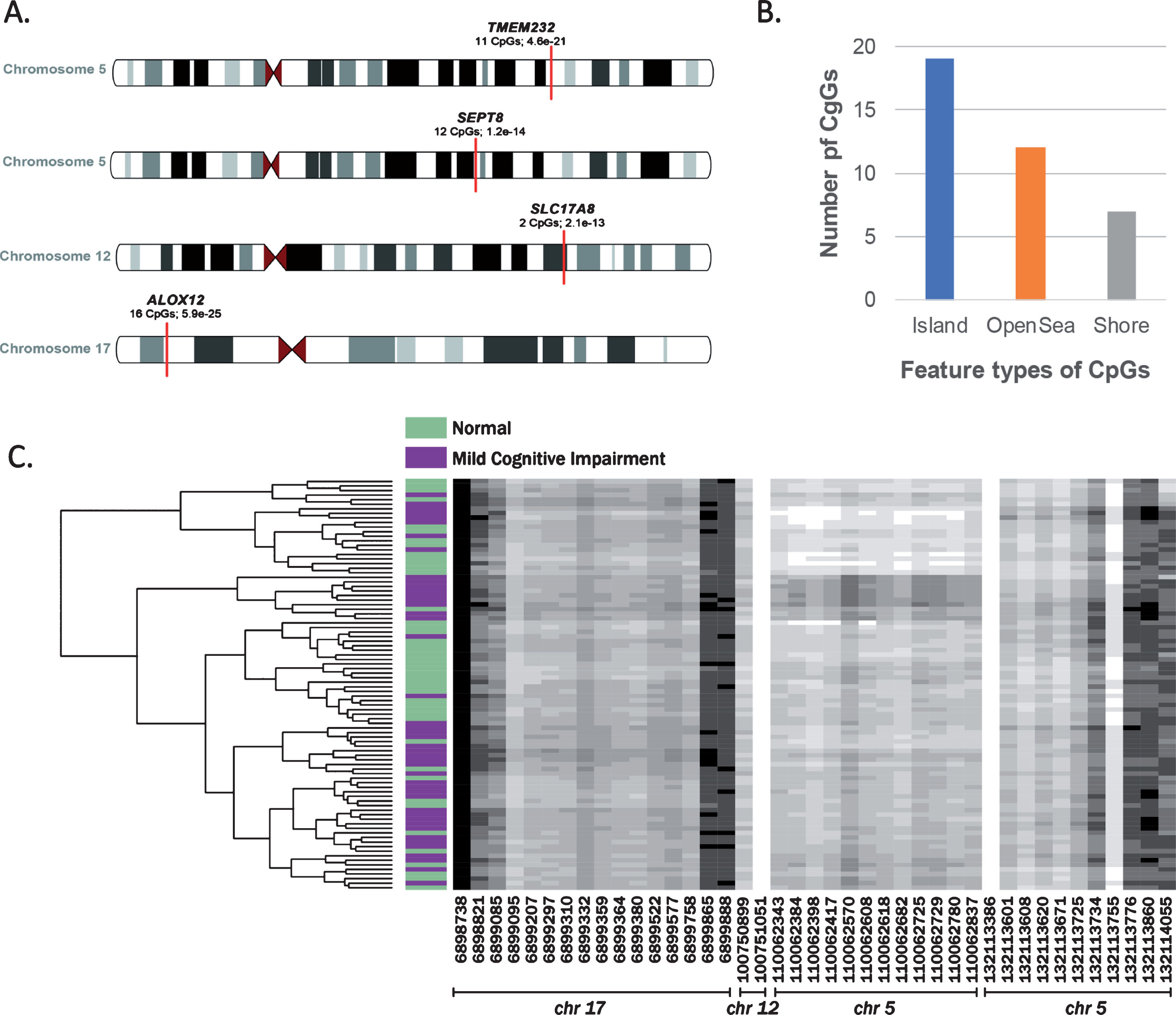
A) Differentially methylated regions. An ideogram representation of the location of differentially methylated regions and corresponding genes, number of CpGs in the regions and FDR p-value of the region. B) Classification of CpGs features. An overview of all the CpGs sites in the identified regions based on their feature type-CpG island are sites located in 0.5 kb stretches with high GC content; CpG shores are located with ±2 kb of the island and CpG opensea are intergenic sites in the genome. C) Dendrogram plot. Beta values for the CpG sites within the differentially methylated regions are represented as shades of grey. The x axis is the chromosome and cpg site location (bp) within each region. The y axis is study participants clustered based on similarity of beta values of the cpg sites (See Supplementary File 2 for details).
Differentially methylated modules
We next analyzed differential methylation by clustering CpG sites into known gene modules of protein-protein interactions (PPI) from pathway commons database. The methylated CpG sites are mapped to genes and weighted based on the distance to transcription start sites. These genes, driven by weighted averages of methylation levels of their CpG sites, are evaluated for association with the phenotype. The protein-protein interaction network with significant genes are considered functionally methylated modules [34]. The functional methylation analysis identified four gene-sets (modules) to be significantly hypomethylated in MCI compared to NC: RIN3 (also known as SLC24A4), SPEG, CTSG, and UBE2L3 (Supplementary File 3). The RIN3 and SPEG cluster identified overlapping significant genes: RIN3, SPEG, XDH, and KNDC1; while in the CTSG cluster, we found CTSG, F2RL1, and MMRN1 to be significant. Lastly, in the UBE2L3 gene-set, we found RNF144A, UBE2L3, and NEDD4L to be significant (Fig. 3).
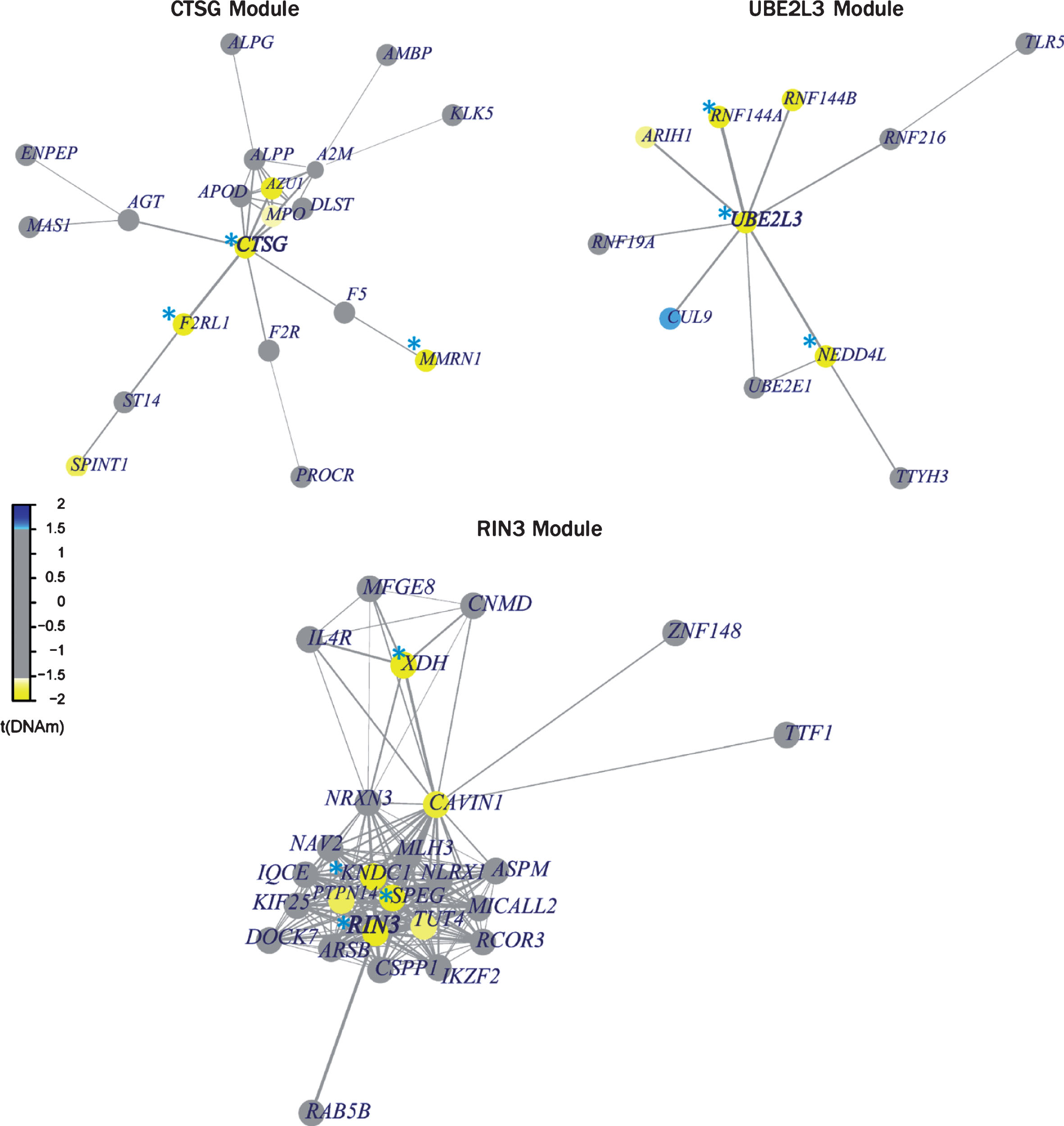
Differential methylated gene modules. The differentially methylated genes within the set are shown in yellow/blue on the legend; yellow signifies hypomethylated and blue signifies hypermethylated. Each of the module was found to be significant based on its overall p < 0.05. Individual significance of each gene (p < 0.05) in the module is highlighted with blue asterisk.
Gene ontology and pathway enrichment test
Genes annotated from significant differentially methylated CpG sites, regions, and modules were analyzed for their involvement in pathways and processes. Several biological processes were found to be significantly enriched, which clustered primarily on immunological processes and systemic arterial blood pressure (Fig. 4). Four KEGG pathways—synaptic vesicle cycle, bacterial invasion of epithelial cells, inflammatory mediator regulation of TRP, and ubiquitin mediated proteolysis—were found to be enriched. Using IPA to assess enriched disease pathways, we found neuronal cell death and metabolic disease (insulin-resistance) among the most significant pathways (Fig. 5), in addition to the aforementioned processes.

GO enrichment results for biological processes. Significant processes are shown in the bar plot. The y-axis shows names of the processes, and x-axis shows number of genes, and each observation is labelled with the number of genes present in the respective gene category. The color of bars corresponds to the scale of FDR corrected p-values.
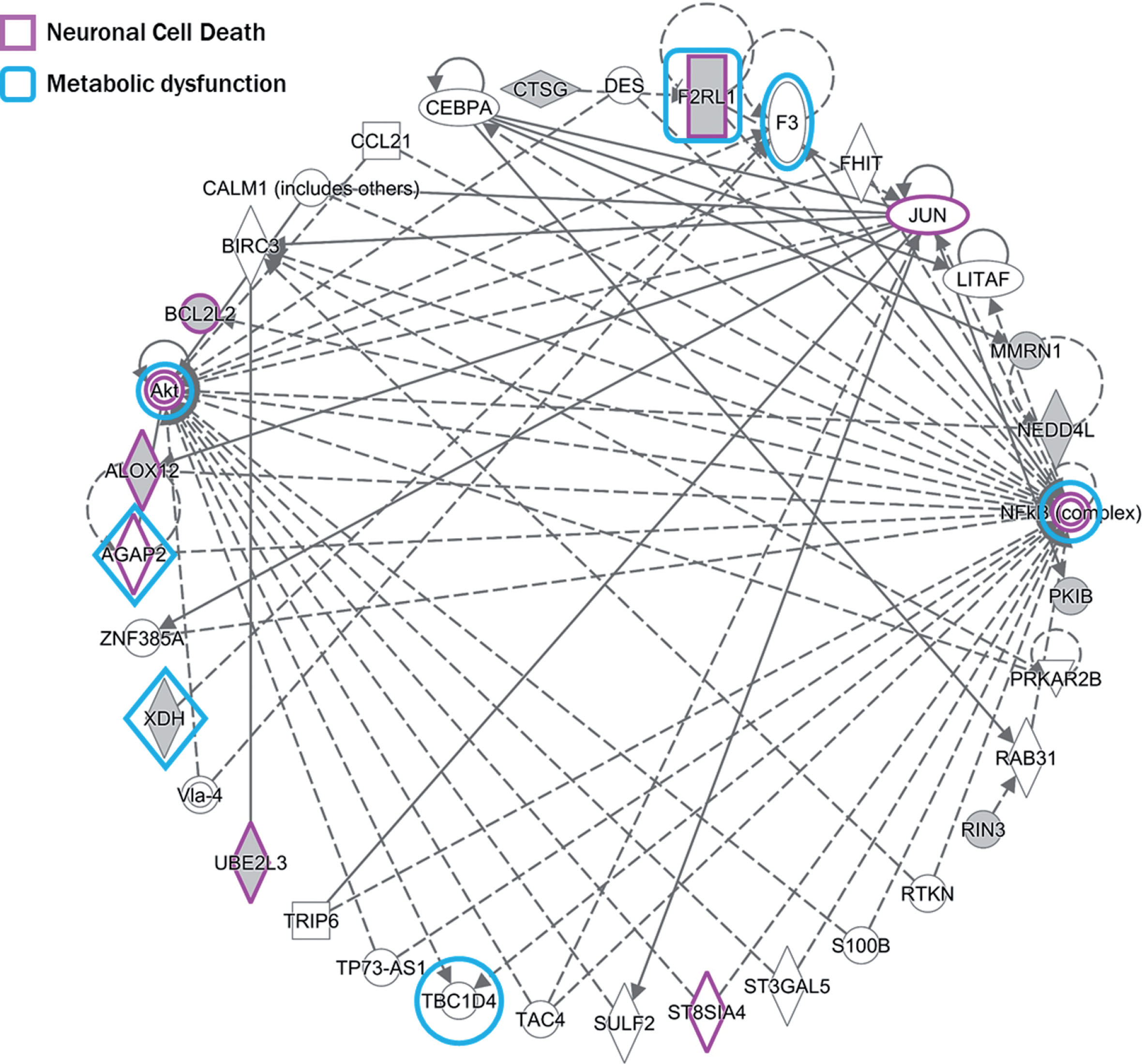
Pathway enrichment. Genes that were identified for neuronal cell death pathway are highlighted in purple and genes identified for metabolic dysfunction/insulin resistance are highlighted in blue.
Bayes gene-set enrichment analysis
Traditional GSEA methods, as reported above, use singular CpG sites/regions which represent genes for enrichment in the network. The empirical Bayes method [35] for gene set enrichment of differential methylation overcomes this singularity by analyzing average methylation of all CpG sites (i.e., untransformed beta-values) mapped to a gene. These genes are ranked based on degree of differential methylation. The ranked list is then tested for biological enrichment using the annotated gene sets in the Molecular Signatures Database (MsigDB, containing 17,810 gene sets).
We found a total of 1,450 gene-sets (8.14% of MsigDB sets) to be significantly enriched (adjusted FDR p-value < 0.05). The gene sets were sorted by area under curve (AUC) and significance.
The gene sets with the highest classification accuracy (AUC ∼72–78%) were attributed to the following gene sets, among others: Regulation of protein, Rho GTPase activity, and Aging (Table 2 in Supplementary File 5).
The gene-sets with the highest statistical significance were from experimentally-derived datasets, previously associated with H3K27 methylation in various experimental models (Table 1 in Supplementary File 5). The top gene sets were: ‘BENPORATH_ES_WITH_H3K27ME3’ (AUC – 60%; p-value: 5.1e-27), ‘MEISSNER_BRAIN_HCP_WITH_H3K4ME3_AND_H3K2 7ME3’ (AUC – 59.3%; p-value: 7.5e-21), and ‘BENPORATH_PRC2_TARGETS’ (AUC – 61%; p-value: 2.76e-19). The experimentally-derived gene sets contained ∼1,100 genes, and the top three gene set members were analyzed using the Panther database [45] for representative pathways in order to understand their biological relevance (Supplementary File 5). We found four pathways to be common in all three gene-sets: 1) Alzheimer disease-presenilin pathway, 2) Ionotropic glutamate receptor pathway, 3) Heterotrimeric G-protein signaling pathway, and 4) Cadherin signaling pathway (Fig. 6).
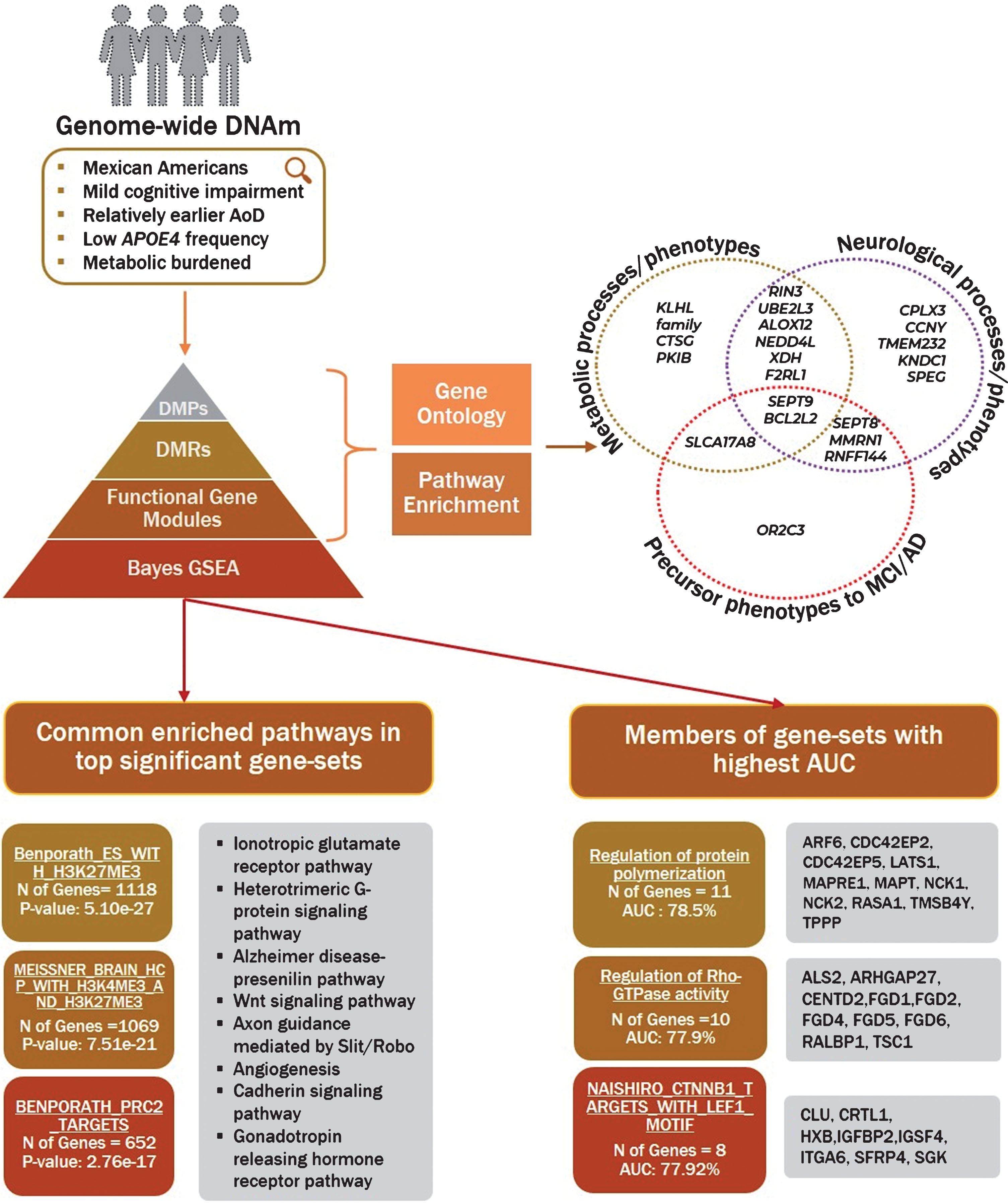
Visual summary of identified genes and pathways. Visual representation of methodology and an overview of findings converging on phenotypes and pathways. The figure shows known characteristics of the population, followed by levels of analysis performed for identification of differential methylation with cognitive impairment. The Venn diagram highlights the genes involved in key processes based on research studies reported in the discussion section. The gene enrichment was performed on differentially methylated probes, regions and modules. The top three gene sets identified using Bayes gene enrichment are sorted by statistical significance (left) and AUC (right) with corresponding details for each is highlighted in the box.
DISCUSSION
The etiology of MCI/AD in Mexicans-Americans is distinctive from non-Hispanic whites. Much of this disparity has been attributed to the elevated prevalence of metabolic dysfunction within this population, with 43% of individuals with dementia also suffering from diabetes, stroke, or both [8]. While several studies have reported an association between metabolic burden and cognitive impairment, the exact mechanisms linking the two are poorly understood [46]. In the last 20 years, based on PubMed query, we found only 71 articles that were published in the genetics of AD in Mexican-American population. There are no publications listed when we replace the term genomics and its associated terms with ‘methylation’ or ‘epigenetics’ (Supplementary Figure 2). Studying alterations in genome-wide methylation profiles associated with cognitive decline in a metabolically burdened admixed population may help identify changes that occur as a result of both genetic and lifestyle factors. Understanding these factors will be vital for development of effective diagnostic and therapeutic strategies.
Genes implicated in differentially methylated sites and regions
Here we found 10 differentially methylated CpG sites significantly associated with MCI in our Mexican American cohort. The hypomethylated sites were in genes KLHL29, SEPT9, and CPLX3. KLHL29 has been reported to be associated with diabetes [47], while other members of the Kelch-like gene family have been implicated in hypertension [48]. Septin 9 (SEPT9), known to be involved in cell cycle regulation, has primarily been associated with neuralgic amyotrophy, a peripheral nervous system disorder characterized by weakness and atrophy of the upper extremities [49]. SEPT9 variants have also been associated with systolic blood pressure [50]. The hypomethylation of SEPT9 observed in our analysis was also reported by Dayeh and colleagues, who found hypomethylation of SEPT9 on multiple CpG sites and an upregulation of SEPT9 in human pancreatic islets from type 2 diabetes donors [51]. Complexin 3 (CPLX3) is involved in regulation of SNARE-associated synaptic vesicle fusion, thereby impacting neurotransmitter transporter activity and localization [52]. The CpG site within CPLX3 was found to be hypomethylated in our study based on peripheral blood, and Annese et. al have reported CPLX3 to be upregulated in the hippocampal region of individuals with late-onset AD [53].
The hypermethylated CpG sites were in genes PKIB, BCL2L2-PABPN1, OR2C3, and CCNY. PKIB is a kinase inhibitor in PKA signaling known to suppress insulin secretion under chronic hyperglycemia [54]. Interestingly, PKA signaling also regulates mitochondrial function/morphology as well as neuronal survival, and has been suggested as a therapeutic target for treatment of neurodegenerative diseases [55]. BCL2L2 (BCL-w or BCL2-w), encoding an anti-apoptotic protein, was hypermethylated in our study, and has been reported to be downregulated in AD brains afflicted with increased phosphorylation of tau proteins [56]. Furthermore, variants in BCL2L2-PABPN1 have been reported to be associated with basal metabolic rate and body mass index in obese Korean women [57]. Mutations in PABPN1 encoding a poly A binding protein, have been associated with increased protein aggregation, a pathological hallmark of many neurodegenerative diseases [58]. Olfactory deficits have been reported to precede clinical symptoms of AD and MCI [59], and our study identified hypermethylation of OR2C3, encoding a GPCR that binds to the extracellular domain that generates signal to the olfactory sensory neurons in the brain [60]. OR2C3 binding results in additional activation of Akt, MAPK, and Rho-signaling pathways [60]. Therefore, it is plausible that hypermethylation of sites within OR2C3 may cause downregulation of its expression resulting in olfactory deficits and neuronal dysregulation [61]. The CpG site within Cyclin Y (CCNY) was observed to be hypermethylated in our study; downregulation of CCNY has been reported to inhibit the formation of new synapses during synaptic remodeling [62]. CCNY also regulates Wnt-signaling pathways, which have been found to be altered in the prefrontal cortex of individuals with AD [63].
We also identified four regions within TMEM232, SEPT8, SLC17A8, and ALOX12 that were significantly differentially methylated. ALOX12, arachidonate 12-oxidoreductase, is an enzyme involved in the generation of lipid metabolites and loss of function mutants of ALOX12 (mouse model) have demonstrated sensitization of pancreatic beta cells to oxidative stress [64] leading to declines in insulin secretion, an event that precedes onset of type 2 diabetes [65]. Interestingly, ALOX12 has also been reported to modulate glutamate-induced degeneration of neurons [66], and SNPs within this gene have been implicated in loss of prefrontal cortical thickness in individuals with posttraumatic stress disorder [67]. Vesicular glutamate transporter 3 (otherwise known as VGLUT3 or SLC17A8) has been shown to be co-expressed with glutamate receptor GABRG3 in peripheral glutamatergic neurons; together they are suspected to play a compensatory role in response to loss of glycinergic activity which occurs during hearing loss [68], a precursor symptom to MCI and AD [69]. Another member of the Septin gene family, SEPT8, was found here to be associated with MCI. SEPT8 is believed to regulate the transportation of synaptic vesicles through interaction with different SNARE-complex components [70]. Dysfunction of synaptic vesicle transmission in hippocampal and cortical regions has been associated with cognitive decline [71]. Additionally, multiple variants in TMEM232, encoding a transmembrane protein, have been implicated in various neuropathies [72]. SNPs in this gene have also been associated with schizophrenia, depression, and bipolar disorder in a Chinese population [73].
Genes implicated in differential methylated modules
We found four gene-sets in our study, RIN3, SPEG, CTSG, and UBE2L3, to be significantly hypomethylated in individuals with MCI. Four genes, RIN3, SPEG, XDH, and KNDC1, were hypomethylated in both RIN3 and SPEG gene-sets. Hypomethylation of CpG sites in the 3’UTR of RIN3 has also been observed in peripheral blood of individuals with sporadic early onset AD [74]. Interestingly, in our study, the functional module analysis which weights location of CpG sites from transcription start sites in an overall gene-based analysis, also identified hypomethylation of RIN3 derived from peripheral blood of individuals with MCI, suggesting that hypomethylation at this site may serve as an early indicator of cognitive decline. Due to its involvement in regulation of endocytic trafficking and processing of amyloid-β protein precursor [75], hypomethylation of RIN3 may also serve as a compensatory response to amyloid toxicity. This postulation is supported by Boden et al. who observed increased hypomethylation of RIN3 in AD brains in comparison to blood, which they suggest to be due to prolonged exposure of the brain to an amyloid-rich environment [74]. In a study by Stage et al., a single SNP within RIN3 (rs10498633) was found to be associated with gray matter density within the medial temporal lobe, a phenotypic change detectable even in early stages of cognitive impairment [76]. Other variants within RIN3 have also been associated with metabolic syndrome in Japanese individuals [77]. KNDC1, encodes a Ras-guanine nucleotide exchange factor, and Ji et al. have shown KNDC1 upregulation to stimulate p53-oxidative stress responses [78]. Other studies focusing on 5-hydroxymethylation in AD have found KNDC1 to be enriched in GO terms related to neurogenesis and signal transduction [79]. Remarkably, a genome-wide association study of 1 million individuals implicated variants in KNDC1 to be associated with ‘self-reported educational attainment’, a highly correlated risk factor for cognitive impairment [80]. Xanthine dehydrogenase (XDH), a hydroxylase involved in oxidative purine metabolism, is capable of converting to xanthine oxidase, a major producer of reactive oxygen species (ROS) [81]. Elevated ROS is a hallmark of both AD and type 2 diabetes [82]. Upregulation of XDH activity has also been demonstrated in the brain of rats with late-stage diabetes [83]. Hypomethylation of CpG sites within XDH may allow for increased expression permitting potential downstream elevation in ROS.
Cathepsin G (CTSG) is an anti-inflammatory protease found in azurophil granules of blood neutrophils that functions to breakdown pathogens and inflammatory tissues. Specifically, CTSG has been shown to play a role in abating neuroinflammation in AD through cleavage of amyloid-β [84]. The hypomethylation of CTSG observed in our study was reported to be upregulated in the hippocampus of hypertensive aged rats suggesting perhaps that neuroinflammation may bridge hypertension and cognitive dysfunction [85]. The G-protein coupled receptor F2RL1 (otherwise known as PAR2) also accelerates neurodegeneration via inflammatory mediators [86]. Interestingly, upregulation of PAR2 has been reported to be induced by alpha-synuclein [87]. PAR2 has also been shown to be upregulated in adipose tissues and is a contributor to insulin resistance and metabolic dysfunction [88], while the upregulation of MMRN1 has been associated with cognitive deficits in Parkinson’s disease [89].
The UBE2L3 gene-set revealed several ubiquitin-related proteins to be hypomethylated. UBE2L3 (E2F1) encodes a ubiquitin-conjugating enzyme, while NEDD4L and RNF144A encode E3 ubiquitin ligases. The expression of UBE2L3 is upregulated in neurons of AD brains [90] and is also increased in obesity for maintenance of metabolic homeostasis [91]. SNPs within NEDD4L have been associated with essential hypertension, a known comorbidity for AD [92]. Hypomethylation and upregulation of mRNA expression of RNF144 was reported in liver samples of individuals with type 2 diabetes [93]. Further, the interactions between these enzymes are important for maintaining synaptic plasticity via selective degradation of abnormal or misfolded proteins [94].
Shared processes and pathways of differentially methylated genes based on gene-set enrichment
Three genes, F2RL1, ALOX12, and UBE2L3, were identified in both KEGG and IPA’s knowledge base highlighting their role in inflammation, neuronal cell death, and metabolic processes. We also conducted GSEA analysis using empirical Bayes approach which clusters differentially methylated probes into genes followed by overall ranking of genes based on differential methylation and testing for enrichment of gene-sets. Several experimentally derived gene-sets stored in MSigDB were found to be enriched in our genome-wide methylation analysis. The gene members of each of the top three gene sets coincided for four pathways: Alzheimer disease-presenilin, Ionotropic glutamate receptor, Heterotrimeric G-protein signaling, and Cadherin signaling pathway.
The Alzheimer disease-presenilin pathway consists of the presenilin gamma-secretase complex which is involved in the cleavage of the amyloid-β protein which is known to form plaques in brains of AD individuals [45]. The glutamatergic receptors are involved in neurotransmission, and overexcitation of the glutamatergic synapses results in neuronal cell death [95] in AD. The Heterotrimeric G-protein pathway consists of G-protein coupled receptors [45], which are known to interact with BACE1 secretase molecule and are involved in phenotypes of vision and olfaction [96]. Lastly, the molecules in cadherin signaling such as N-cadherin[45] are required for maintaining synaptic plasticity, which is otherwise deregulated by amyloid-β and increases tau and p38 phosphorylation in AD [97].
Conclusion: Reported genes and pathways converge between cognitive dysfunction, metabolic burden, and inflammation
Our results point to modification of genes involved in metabolic, neuroinflammatory, and AD-precursor phenotypes (Fig. 6). The data presented here links peripheral metabolic dysregulation with cognitive decline. Metabolic syndrome which includes obesity, diabetes, and hypertension has been known to provoke poor cerebral blood flow, resulting in vasoconstriction and endothelial dysregulation; this leads to production of ROS and inflammation [98]. Insulin receptors are expressed in multiple brain regions, including the olfactory bulb, hypothalamus, and hippocampus, and are sensitive to changes in the peripheral system due to metabolic conditions [99]. Insulin resistance alters cAMP/PKA signaling pathways which not only induces inflammation from oxidative stress, but also affects synaptic plasticity of hippocampal neurons [100]. In the presence of hypertension, homeostatic dysfunction results in microglia-induced inflammation in the brain [101]. Metabolic syndrome–obesity induces low-grade systemic inflammation which is detected by brain [102] causing disruption of the synaptic transporter activity [103, 104]. While our findings do not incriminate any single pathway for pathogenicity of cognitive dysfunction, they highlight genes and pathways involved in cross-talk between peripheral metabolic burden and neuroinflammation. We postulate that insults sustained from metabolic dysregulation results in low grade inflammation (reported earlier by our group) [10] affecting synaptic vesicle activity which ensues cognitive deficits in Mexican Americans. Therefore, it is possible that cognitive decline in Mexican Americans is a manifestation of genetic/lifestyle factors of metabolic stress as seen here in their epigenetic profile.
There are several limitations to our study. First, our study is limited in sample size; therefore, the genes reported here should be considered as preliminary findings and should be replicated in larger cohorts. We have also tested cognitive-associated methylation changes in peripheral blood, which may not be parallel to brain-specific methylation alterations. Epigenetic variations in conjunction with RNA expression in larger cohorts of Hispanic individuals will provide much needed characterization of the pathogenesis of cognitive impairment.
Despite the limitations, we have strengthened our study design by implementing a very rigorous and conservative QC approach, correcting for multi-level batch effects, adjusting for cell composition and incorporating balanced demographic characteristics, representative of the Mexican American population. Our study has found several novel epigenetic markers associated with metabolic burden and cognitive dysfunction which contribute toward addressing the critical health disparity faced by Mexican Americans.
Footnotes
ACKNOWLEDGMENTS
The research team thanks the local Fort Worth community and participants of the Health & Aging Brain Study. Research reported in this publication was supported by the National Institute on Aging of the National Institutes of Health under Award Numbers R01AG054073 and R56AG054073. We would also like to acknowledge the NIH – Neurobiology of Aging T32 grant AG020494 for supporting this research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.