
Abstract
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder characterized by progressive cognitive impairments. Vitegnoside is a flavonoid present in the medicinal plant Vitex negundo, widely used as a folk medicine in several Asian countries including China. It possesses several biological activities, including axon outgrowth, but no evidence is available on its effect on AD. Since no effective treatment is available to cure AD, the effect of vitegnoside on this disease was investigated. The human neuroblastoma SH-SY5Y cell line carrying the Swedish mutation that induces AβPP overexpression was used as an in vitro AD cell model. AβPP overexpression does not induce toxicity per se unless triggered by copper. Vitegnoside promoted neuroprotection through the improvement of cell viability, maintenance of cytomembrane integrity and nuclear homogeneity in these cells, but these effects were not observed in the copper-treated SH-SY5Y cells without AβPP overexpression used as the wild-type control, indicating that vitegnoside exerted neuroprotection under copper-triggered Aβ toxic conditions. Vitegnoside failed to decrease AβPP expression, Aβ40/42 levels, and oxidative stress due to copper-induced Aβ toxicity. However, its administration protected the mitochondrial function and restored the imbalance between pro-apoptotic and anti-apoptotic proteins. Additionally, vitegnoside inactivated p38 MAPK/MK2, JNK/c-Jun, and downstream NF-κB inflammatory transductions. Furthermore, the inactivation of p38 MAPK/JNK signaling contributed to vitegnoside-mediated neuroprotection resulting from pharmacological inhibition of p38 MAPK/JNK and in silico interaction prediction. Our study revealed the neuroprotective effect of vitegnoside and its potential mechanisms against copper-induced Aβ neurotoxicity. These findings highlighted the potential therapeutic effect of vitegnoside against AD progression.
INTRODUCTION
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder characterized by progressive cognitive impairments and memory loss [1]. A large number of studies proved that amyloid-β (Aβ) overgeneration and subsequent aggregation in the brain are the critical causative factor for inducing AD [2], characterized by mitochondrial dysfunction, increased oxidative stress, inflammatory response, and neuronal death [3]. Besides, abnormally high levels of metal ions, such as copper and aluminum, are present in amyloid plaques of AD patients, indicating a potential role in Aβ-associated toxicity in vivo [4, 5]. Indeed, these metal ions not only induce the generation of off-pathway Aβ aggregates synchronously with cytotoxicity, but also lead to harmful effects inducing neuronal cell damage. Therefore, inhibition of Aβ-mediated neurotoxicity induced by metal ions might be considered as a potential and safe strategy in the prevention and treatment of AD.
With regards to mitochondrial dysfunction, mitochondrion-induced apoptosis is characterized by translocation of cytochrome c from the mitochondrion to the cytosol and subsequent activation of caspase-9, followed by a series of apoptotic events such as nuclear fragmentation and chromatin condensation [6]. Additionally, mitochondrial dysfunction exacerbates oxidative stress by increasing reactive oxygen species (ROS) production [7]. In turn, excessive ROS primarily damages mitochondrion, since they induce structural and functional modification of its DNA, proteins, and lipids [8, 9]. Activation of MAPK signaling pathways in response to Aβ-induced oxidative stress also induces neuronal apoptosis through the mitochondrion-dependent pathway [10]. The representative mammalian MAPK pathways, represented by p38 MAPK, c-Jun N-terminal kinase (JNK), and extracellular signal-regulated kinase 1/2 (ERK1/2), induce the transcription factor nuclear factor-kappaB (NF-κB), which in turn regulates the expression of proinflammatory cytokines and inflammatory mediator genes, thus triggering neuroinflammation [11]. Thus, therapeutic strategies aiming at blocking Aβ-induced pathological cascades involving mitochondrial dysfunction, apoptotic response, and inflammatory reaction, might be effective in the treatment of this disease.
Vitegnoside (Fig. 1) is one of the polyphenolic compounds present in the medicinal plant Vitex negundo (VN), which is widely used as a folk medicine in several Asian countries including China, Japan, and Indonesia [12]. Several studies reported that VN possesses extensive biological activities, such as anti-inflammatory, anti-oxidant, anti-osteoporotic, anti-hyperglycemic, and hepatoprotective effects [13–15]. Recently, the effect of VN methanolic extracts on neurons was demonstrated, resulting in keeping the long neurites in length of the primary hippocampal neurons [16]. However, to the best of our knowledge, no studies are available investigating whether VN or its bioactive compounds have the ability to prevent AD pathogenesis. Therefore, in this study, the neuroprotective effect of vitegnoside, one of the bioactive flavonoids isolated from VN, was investigated on copper-induced Aβ toxicity in human neuroblastoma SH-SY5Y cells carrying the Swedish mutation that induces amyloid-β protein precursor (AβPP) overexpression, and the underlying mechanisms were explored.
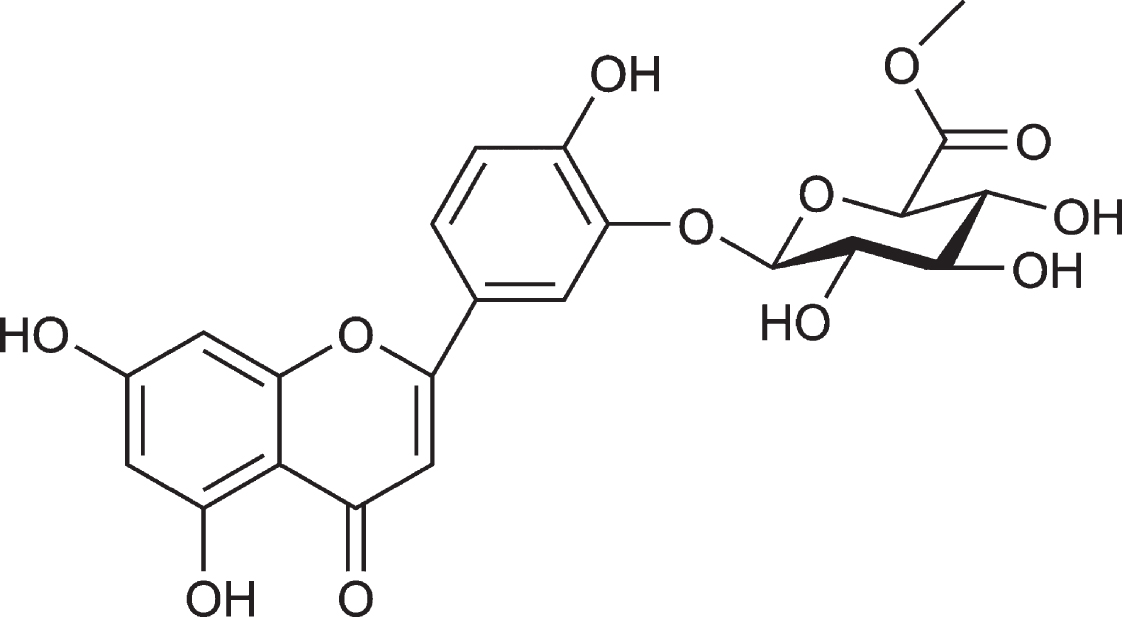
Chemical structure of vitegnoside. The molecular formula of vitegnoside is C22H20O12.
MATERIALS AND METHODS
Cell cultures and treatments
The human neuroblastoma SH-SY5Y cell line was purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA) and grown in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) at 37°C in humidified 5% CO2 atmosphere. The SH-SY5Y cells express a low level of AβPP and produce a small quantity of Aβ in physiological status and they are generally regarded as reliable in vitro models to imitate AD pathological changes combined with transfection of exogenous humanized genes following exposure to a certain of stimuli. In our study, the SH-SY5Y cells were stably transfected with the human Swedish mutation that induces AβPP overexpression using Lipofectamine® 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA). The stably transfected cells were selected by G418 resistance that were called APPswe cells, and were used as the in vitro AD cell model. AβPP overexpression does not induce toxicity in an in vitro culture unless triggered by metal ions such as copper. APPswe cells were grown in DMEM/F12 containing 10% FBS (Invitrogen) at 37°C in humidified 5% CO2 atmosphere.
Vitegnoside was kindly provided by the Xinjiang Institute of Materia Medica (Urumqi, China), with a 97% purity obtained by high performance liquid chromatography. APPswe cells were randomly divided into two groups with or without 200 μM copper for 24 h and the regular SH-SY5Y cells were used as a wild-type (WT) control treated with or without 200 μM copper for 24 h as well. These two cell types were further divided into several subgroups receiving vitegnoside at different concentrations: 0 μM, 0.3 μM, 1.0 μM, 3.0 μM, 10.0 μM, and 30.0 μM. With regards to the evaluation of the mechanism of action, cells were pre-incubated for 6 h with 20 μM SB203580 (Sigma-Aldrich, St. Louis, MO, USA), a selective inhibitor of p38 MAPK, or 20 μM SP600125 (Sigma-Aldrich, St. Louis, MO, USA), a selective inhibitor of JNK, prior to the treatment with 10.0 μM vitegnoside, which was added after the replacement of the culture medium with fresh one.
Cell viability assay
Cells were seeded in a 96-well plate at a density of 8000 cells/well in 200 μL medium/well and subjected to all treatments described in the previous paragraph. Cell viability was assessed by MTS assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay – MTS; Promega, Madison, WI, USA). Cells were incubated for 2 h at 37°C with an appropriate amount of MTS according to the manufacturer’s instructions. The soluble formazan product was detected at 490 nm by a Spark 20 M multimode microplate reader (Tecan Group Ltd., Mannedorf, Switzerland).
YO-PRO-1, rhodamine 123, and Hoechst 33342 staining assay
YO-PRO-1, a nucleic acid binding fluorescent dye, is designed to distinguish apoptotic cells from live cells, since it selectively passes through the plasma membrane of apoptotic cells and labels them with a moderate green fluorescence [17]. YO-PRO-1 was used to detect the membrane permeability of APPswe cells after vitegnoside plus copper treatment according to the protocol described in the Cell cultures and treatments section. Mitochondrial membrane potential (MMP) was measured using the potential-sensitive dye rhodamine 123 (Rh123). Nuclear DNA was stained by Hoechst 33342 (Dojindo Laboratory, Kumamoto, Japan). Fluorescent images were acquired by a Cellomics ArrayScan VTI High-content Analysis (HCA) Reader (Thermo Fisher Scientific Cellomics, Pittsburgh, PA, USA) combined with the Cell Health Profiling BioApplication module. The values were finally quantified as the mean fluorescent intensity that was compared between control group and treated groups.
Aβ expression determination
The expression of Aβ40 and Aβ42 was examined using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) in APPswe cell homogenates after vitegnoside and copper treatment described in the Cell cultures and treatments section. Briefly, APPswe cells cultured in 6-well plates with a density of 1×106 cells/well were washed twice with phosphate buffer before protein collection using ice-cold lysis buffer (pH 7.4) containing 50 mM Tris-HCl, 20 mM ethylene diamine tetraacetic acid, 0.1% sodium dodecyl sulfate, 100 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 2 mM sodium pyrophosphate, 1 μg/mL pepstatin A, 100 μg/mL leupeptin, and 1×protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN, USA). The lysates were centrifuged and the supernatant was collected for detection of Aβ40 and Aβ42 expression in accordance with the manufacturer’s instructions.
Oxidative stress measurement
The level of intracellular ROS was measured using 2’,7’-dihydrodichlorofluorescein diacetate (DCFH2-DA) staining, since DCFH2-DA is cell-permeable and intracellularly hydrolyzed to DCFH, where it is oxidized to the fluorescent product DCF by the ROS. Thus, the amount of fluorescence is positively correlated to the ROS amount into the cell. Fluorescent images were acquired using 485 nm excitation and 535 nm emission by a Cellomics ArrayScan VTI HCA Reader with the Morphology Explorer BioApplication and quantified as the mean fluorescent intensity that was compared between control group and treated groups. The activity of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) was measured using commercially available assay kits (Nanjing Jiancheng Bioengineering, Nanjing, China) according to the manufacturer’s instructions.
Phosphorylation assay
APPswe cells were plated with a density of 1×106 cells/mL and treated with copper and vitegnoside described in Cell cultures and treatments. Then cells were centrifuged and resuspended in an appropriate volume of Hank’s balanced salt solution (HBSS; Thermo Fisher Scientific, Pittsburgh, PA, USA) containing 5% FBS (Invitrogen). Next, cells were lysed with Cell Lysis Buffer 5× from the p38 MAPK [Thr180/Tyr182], JNK [Thr183/Tyr185], and ERK1/2 [Thr202/Tyr204 and Thr185/Tyr187] (Total/phospho) Multispecies InstantOne™ ELISA Kits (Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s instructions. The absorbance of each sample was measured at 490 nm by a Spark 20 M multimode microplate reader (Tecan Group Ltd., Mannedorf, Switzerland).
AβPP expression, apoptotic, MAPK, and NF-κB pathway detection by cellular immunofluorescence
APPswe cells were seeded in a 96-well plate at a density of 8000 cells/well in 200 μL medium/well and subjected to all treatments described in the Cell cultures and treatments section. Cellular immunofluorescence was performed using Cellomics ArrayScan VTI HCA Reader (Thermo Fisher Scientific Cellomics) as previously described [18]. Briefly, APPswe cells were fixed with 4% paraformaldehyde (Beijing Chemical Works, Beijing, China) for 30 min, permeabilized with 0.3% Triton X-100 for 10 min, blocked with 3% bovine serum albumin for 30 min at room temperature, and incubated with polyclonal rabbit primary antibodies overnight at 4°C as follows: anti-human AβPP primary antibody (1:200, Cell Signaling Technology, Danvers, MA, USA), anti-Bcl-2 (1:200, Abcam Cambridge, MA, USA), anti-Bax (1:100, Abcam), anti-cytochrome c (1:100, Abcam), anti-p-JNK1/2 (T183/Y185) (1:100, Abcam), anti-p-ERK1/2 (T202/Y204) (1:200, Cell Signaling Technology), anti-p38 MAPK (T180/Y182) (1:200, Cell Signaling Technology), anti-p-MK2 (Thr334) (1:100, Cell Signaling Technology), anti-p-c-Jun (Ser63) (1:100, Abcam), and anti-p-NF-κB p65 (Ser536) (1:100, Abcam). The secondary antibodies used for 2 h incubation at room temperature were goat anti-rabbit conjugated with Alexa Fluor 488 or Alexa Fluor 546 (1:1000, Invitrogen). Fluorescent images were acquired by a Cellomics ArrayScan VTI HCA Reader using the Morphology Explorer BioApplication and the Cytoplasm to Nucleus Translocation BioApplication for protein expression and translocation, respectively. The nuclear and cytosolic fluorescence intensities of p-c-Jun, AβPP, cytochrome c, Bcl-2, and Bax were quantified as the mean fluorescent intensity that was compared between control group and treated groups. Translocation of p-JNK1/2, p-ERK1/2, p-p38 MAPK, p-MK2, and p-p65 into the nucleus was evaluated by the Mean_CircRingAvgIntenDiff value (difference in average fluorescence intensity between nucleus and cytoplasm) that was compared between control group and treated groups.
Detection of caspase-3 and caspase-9 activity
Caspase-3 and caspase-9 activity was measured in APPswe cell homogenates using the Caspase-Glo 3 and Caspase-Glo 9 assay kits (Promega) according to the manufacturer’s instructions. APPswe cell homogenates were obtained as described in the Aβ expression determination section.
TNF-α and IL-6 by ELISA
APPswe cells were seeded in 96-well plates at a density of 8000 cells/well in 200 μL medium/well and subjected to different treatments according to the Cell cultures and treatments section. TNF-α and IL-6 concentration in the culture medium was measured using ELISA kits (Invitrogen). Optical density was determined at 450 nm and the results were expressed as pg/mL.
Statistical analysis
Statistical analysis was performed using the SPSS software (Version 18.0; SPSS, Inc., Chicago, IL, USA). Comparisons among groups were performed using one-way ANOVA followed by appropriate post-hoc tests. Each experiment was repeated at least three times. Results are expressed as mean±S.E.M. p < 0.05 was considered statistically significant.
RESULTS
Vitegnoside protects APPswe cells against copper-induced Aβ toxicity
In order to better resemble Aβ-induced neurotoxicity in the AD pathogenesis, copper-treated APPswe cells were used as an in vitro AD cell model because Aβ overexpression does not induce neurotoxicity in vitro without copper [18, 19]. Cell viability was measured to evaluate the protective effect of vitegnoside. As shown in Fig. 2A, no significant difference in cell viability was observed among the normally cultured wide-type SH-SY5Y cells and APPswe cells treated with vitegnoside ranging from 0.3 μM to 30.0 μM. As depicted in Fig. 2B, cell viability was only significantly decreased in APPswe cells which were exposed to culture medium containing 200 μM copper for 24 h compared to the control group (p < 0.01), and the viability was higher in copper-injured APPswe cells treated with vitegnoside ranging from 1.0 μM to 30.0 μM (p < 0.01–0.001) compared to the APPswe cells treated with copper alone. No significant difference was observed among SH-SY5Y cells treated with copper and vitegnoside or not. As indicated above, the protective effect of vitegnoside was concentration-dependent in APPswe cells treated with copper-inducing Aβ toxicity, and the safe, non-toxic concentrations at 1.0 μM, 3.0 μM, and 10.0 μM, were chosen for further investigation regarding the beneficial effects of vitegnoside in the AD cell model.

Vitegnoside protection against copper-induced Aβ toxicity on APPswe cells. A) Effect of vitegnoside on the viability of WT SH-SY5Y cells and APPswe cells (n = 8). B) Vitegnoside increased cell viability of APPswe cells treated with copper, as evaluated by MTS assay (n = 8). C) Representative images of YO-PRO-1 and Hoechst 33342 staining indicating the membrane and nuclear protective effect of vitegnoside, respectively (20×magnification). D, E) Vitegnoside decreased the mean fluorescence intensity of YO-PRO-1 (D) and Hoechst 33342 (E) in APPswe cells after copper injury (n = 4). Results are expressed as mean±S.E.M. **p < 0.01, ***p < 0.001 versus control, #p < 0.05, ##p < 0.01, ###p < 0.001 versus copper.
The protective effects of vitegnoside on membrane and nucleus were evaluated by YO-PRO-1 and Hoechst dye, respectively. Lower levels of mean fluorescence intensity representing membrane integrity and nucleus uniformity were observed in control cells, while copper-treated APPswe cells showed both higher cellular and nuclear mean fluorescence intensity compared to the control cells (both p < 0.001, Fig. 2C–E), suggesting a significant increase of membrane permeability and nucleus damage in APPswe cells when treated with copper. Vitegnoside treatment at concentrations of 1.0 μM, 3.0 μM, and 10.0 μM reduced the cellular and nuclear fluorescence intensity due to copper treatment (p < 0.05–0.001) because of reduced morphological changes in the nucleus such as condensation and shrinkage, indicating that vitegnoside was effective in preserving a relative intact structure of the cell membrane and nucleus, which was in agreement with the protective effect on cell viability.
Vitegnoside inhibits mitochondrion-mediated apoptosis of APPswe cells due to copper-induced Aβ toxicity
Mitochondrion takes part in multiple events involved in Aβ toxicity during AD pathogenesis, such as redox imbalance and apoptotic responses [20]. The mitochondrial function of APPswe cells was evaluated using the fluorescent dye Rh123 in an HCA reader. The mean fluorescent intensity of Rh123 significantly increased in copper-treated APPswe cells compared to the control group (p < 0.001, Fig. 3A, B), indicating that copper caused the loss of MMP. As expected, vitegnoside at 1.0 μM, 3.0 μM, and 10.0 μM reduced the sharp increase of Rh123 fluorescence observed in copper-treated APPswe cells in a concentration-dependent manner (all p < 0.001), demonstrating the protective effect of vitegnoside on mitochondrial function.

Vitegnoside protection against copper-induced Aβ toxicity in APPswe cells by alleviating mitochondrial dysfunction and abolishing mitochondrion-mediated apoptosis. A) Representative images of rhodamine 123 (Rh123) staining indicating mitochondrial membrane potential (MMP) and expression of apoptotic markers, cytochrome c and Bax and Bcl-2 in the same field (20×magnification). B–E) Mean fluorescence intensity of Rh123 (B), cytochrome c (C), Ratio of Bax and Bcl-2 (D). E) Caspase-9/3 activity. Results are expressed as mean±S.E.M. n = 4. **p < 0.01, ***p < 0.001 versus control, #p < 0.05, ##p < 0.01, ###p < 0.001 versus copper.
Subsequently, the mitochondrion-dependent apoptotic markers were examined in APPswe cells, such as the pro-apoptotic proteins cytochrome c, Bax, caspase-9/3, and anti-apoptotic protein Bcl-2. Copper increased the expression of cytochrome c, ratio of Bax/Bcl-2, and activity of caspase-9/3 in APPswe cells compared to the control group (p < 0.01–0.001, Fig. 3A, C–E). Vitegnoside treatment at 1.0 μM, 3.0 μM, and 10.0 μM significantly decreased the copper-induced cytochrome c expression, Bax/Bcl-2 ratio, and caspase-9/-3 activity in the APPswe cells compared with the copper-treated groups (p < 0.05–0.001). These results suggested that a mitochondrion-associated signaling pathway was involved in vitegnoside-mediated neuroprotection in APPswe cells.
Vitegnoside is not effective on copper-induced AβPP and Aβ40/42 expression and redox imbalance in APPswe cells
Aβ deposition, resulting in senile plaques, is the major biochemical feature of AD. As a 40- to 42-amino-acid peptide, Aβ is formed via the AβPP by β-secretases and γ-secretases [21]. To evaluate the effect of vitegnoside on amyloid formation, AβPP expression and Aβ40/42 levels in APPswe cells injured by copper were evaluated. AβPP immunostaining showed that AβPP expression was significantly upregulated in APPswe cells treated with copper compared to the control group (p < 0.001, Fig. 4A, B), along with the increased level of cellular soluble Aβ40 and Aβ42 peptides (both p < 0.001, Fig. 3C). Vitegnoside treatment did not alter AβPP expression and Aβ40/42 levels at any of the tested concentrations, suggesting that the protective effects of vitegnoside on APPswe cells was probably not due to the inhibition of Aβ generation.
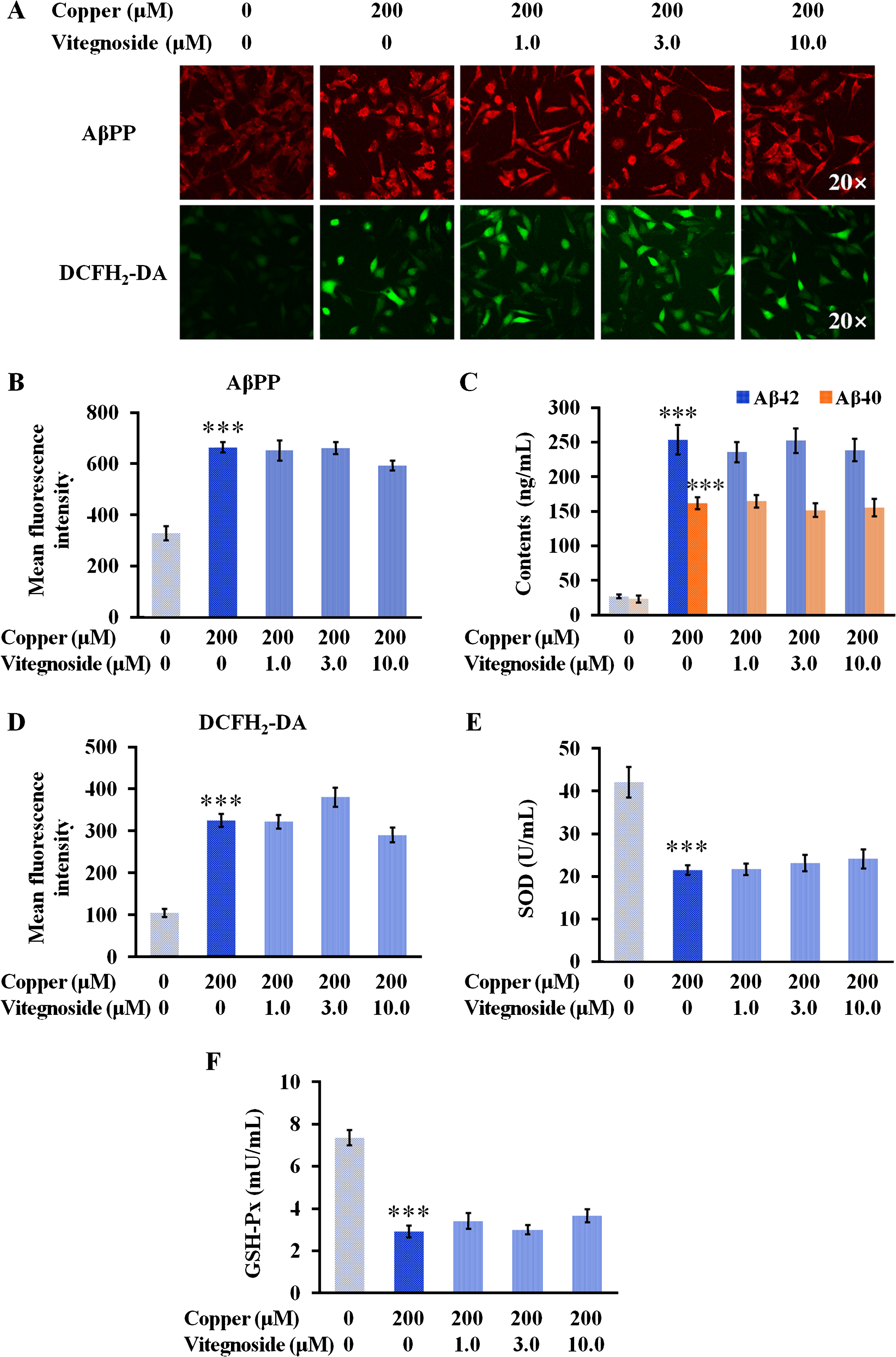
Vitegnoside is not effective on copper-induced AβPP and Aβ40/42 expression, and redox imbalance in APPswe cells. A) Representative images of AβPP expression and DCFH2-DA staining (20× magnification). B) Mean fluorescence intensity of AβPP. C) Cellular soluble Aβ40/42 level in APPswe cells. D) Mean fluorescence intensity of DCFH2-DA. E) SOD and F) GSH-Px activity in APPswe cells. Results are expressed as mean±S.E.M. n = 4. ***p < 0.001 versus control.
It is known that Aβ enhances ROS generation, which is considered as one of the earliest biochemical effects in the progress of AD [22] and promotes AβPP processing and Aβ overproduction in a positive feedback [23, 24]. Copper exposure markedly increased the generation of ROS compared to the control (p < 0.001, Fig. 4A, D), which was not reduced by any of the concentrations of vitegnoside used. Since the oxidative balance plays a role in Aβ toxicity [23], the activity of two important antioxidant enzymes, GSH-Px and SOD, was further evaluated by biochemical assays. The results revealed that vitegnoside did not improve the activity of GSH-Px and SOD at any of the tested concentrations (Fig. 4E, F). These results demonstrated that vitegnoside, ranging from 1.0 μM to 10.0 μM, did not possess the ability of ameliorating redox imbalance due to Aβ toxicity triggered by copper through the enhanced activity of GSH-Px and SOD that could scavenge ROS overproduction.
Vitegnoside inhibits MAPK signaling pathways and NF-κB-associated inflammatory reaction of APPswe cells due to copper-induced Aβ toxicity
Activation of MAPKs signaling is involved in an enhanced cellular sensitivity to Aβ-induced neuronal apoptosis, found both in AD patients’ brains at an early-stage and in multiple AD-related in vivo and in vitro models [25, 26]. To assess whether p38 MAPK, JNK, and ERK were potential signaling pathways associated with vitegnoside neuroprotection, cellular MAPK phosphorylated levels and the phosphorylation of their signaling transduction were evaluated using the specific InstantOne™ ELISA and fluorescence-based cellular immunostaining technology on the HCA platform.
Among the p38 MAPK signaling pathways, the p38 MAPK/MK2 axis has a role in apoptosis and inflammation following oxidative stress [27]. In copper-treated APPswe cells, the phosphorylated p38 MAPK was increased at a cellular level and a rapid nuclear translocation from the cytoplasm at 2 h was observed, as illustrated by an increased absorbance of p-p38 MAPK (but the total p38 MAPK absorbance was not altered) detected by ELISA and a significant increase of p-p38 MAPK in Mean_CircRingAvgIntenDiff values (p < 0.01 and p < 0.001, Fig. 5A–D). The subsequent substrate, phosphorylated MK2, was also activated and translocated from the nucleus to the cytoplasm, demonstrated by a remarkable decrease of p-MK2 in Mean_CircRingAvgIntenDiff values (p < 0.001, Fig. 5C, E). Vitegnoside was able to lower the cellular level of p-p38 MAPK and suppress the translocation of the cytosolic p-p38 MAPK to the nucleus at the concentrations of 1.0 μM, 3.0 μM, and 10.0 μM in a dose-dependent manner (p < 0.05–0.001, Fig. 5B–D), in accordance with the inhibitory effect on the nuclear translocation of p-MK2 to the cytoplasm at the same concentrations (all p < 0.001, Fig. 5C, E). These results indicated that vitegnoside treatment inhibited the activation of p38 MAPK/MK2 signaling pathway.
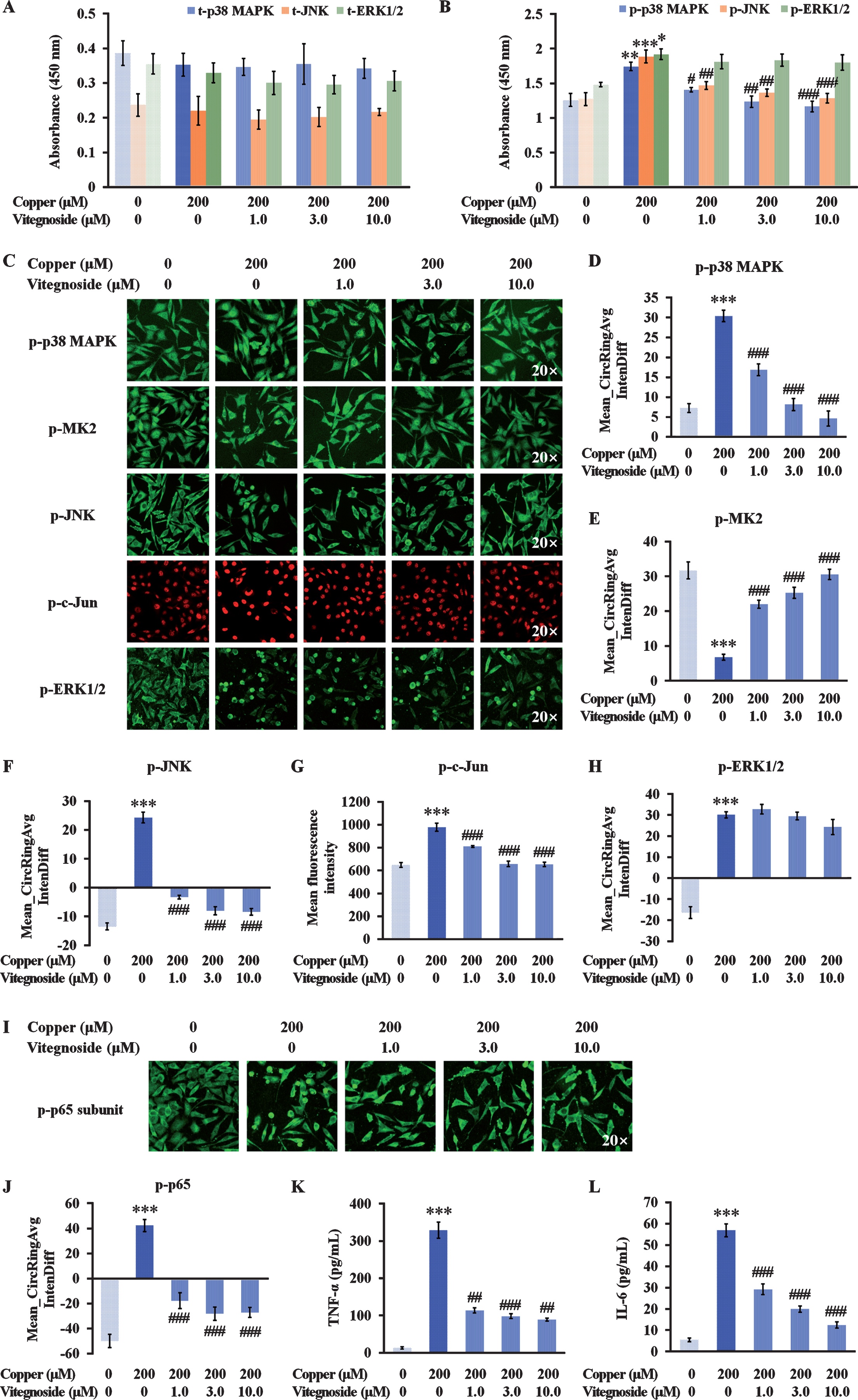
Vitegnoside protection against copper-induced Aβ toxicity in APPswe cells by inhibiting p38 MAPK and JNK signaling pathway and subsequent NF-κB-associated inflammatory reaction. A) Absorbance of total p38 MAPK, JNK, and ERK1/2 in APPswe cell lysates. B) Absorbance of phosphorylated p38 MAPK, JNK, and ERK1/2 in APPswe cell lysates. C) Representative images of p-p38 MAPK, p-MK2, p-JNK, p-c-Jun, and p-ERK1/2 immunofluorescence in APPswe cells (20× magnification). D) Mean_CircRingAvgIntenDiff values describing the translocation of cytosolic p-p38 MAPK, E) p-MK2, and F) p-JNK to the nucleus. G) Mean fluorescence intensity of p-c-Jun expression in nuclei. H) Mean_CircRingAvgIntenDiff values describing the translocation of cytosolic p-ERK1/2 to the nucleus. I) Representative images of p-p65 expression (20× magnification) and J) Mean_CircRingAvgIntenDiff values describing the translocation of cytosolic p-p65 to the nucleus. K) Release of TNF-α and L) IL-6 as detected by ELISA. Results are expressed as mean±S.E.M. n = 4. *p < 0.05, **p < 0.01, ***p < 0.001 versus control, #p < 0.05, ##p < 0.01, ### p < 0.001 versus copper.
Activation of JNK and ERK1/2 pathways was also observed in copper-treated APPswe cells. As depicted in Fig. 5A–C, F, and G, phosphorylated JNK level was increased and translocated into the nucleus, phosphorylation that peaked at 2 h after copper exposure, as shown by the high absorbance of p-JNK by ELISA and an increase in Mean_CircRingAvgIntenDiff values (both p < 0.001), coincidently with the increased phosphorylation of c-Jun as indicated by the mean fluorescent intensity in the nucleus (p < 0.001). Vitegnoside treatment significantly suppressed JNK activation as revealed by the suppressed expression and nuclear translocation of cytosolic p-JNK and the decreased expression of the downstream p-c-Jun (p < 0.01–0.001). Although the expression and translocation of the phosphorylated ERK1/2 to the nucleus were both promoted by copper (p < 0.05 and p < 0.001, Fig. 5B, C, and H), vitegnoside treatment did not result in a remarkable effect neither on the inhibition of p-ERK1/2 expression nor on the nuclear translocation. Combined together, these results suggested that p38 MAPK and JNK signaling pathway, but not ERK1/2, might be involved in the neuroprotective effect of vitegnoside on APPswe cells against copper-induced Aβ toxicity.
Regulated partly by the MAPK family, NF-κB has a dominant role in the neuroinflammation involved in the progression of AD [28]. After the translocation of the p65 subunit into the nucleus, NF-κB enhances the production and secretion of cytokines and chemokines [28, 29]. p65 translocation from the cytoplasm to the nucleus in the copper-treated cells resulted in a drastic increased in the Mean_CircRingAvgIntenDiff value (p < 0.001, Fig. 5I, J). This translocation was significantly decreased by vitegnoside treatment, as shown by the decreased Mean_CircRingAvgIntenDiff value at 1.0 μM, 3.0 μM, and 10.0 μM in a dose-dependent manner (all p < 0.001). These results were consequently associated to an increased TNF-α and IL-6 level in the culture supernatant of the APPswe cells treated with copper (both p < 0.001, Fig. 5K, L). Vitegnoside reduced the increase of these two cytokines in APPswe cell supernatant at each tested concentration (p < 0.01–0.001).
p38 MAPK/JNK pathways are involved in vitegnoside-mediated neuroprotection in copper-induced Aβ toxicity of APPswe cells
To further examine whether p38 MAPK and JNK activation contributed to vitegnoside-mediated neuroprotection against copper-induced Aβ toxicity, APPswe cells exposed to copper and treated with vitegnoside were also pre-treated with SB203580, a specific p38 MAPK inhibitor, and SP600125, a specific JNK inhibitor.
As shown in Fig. 6A and B, the pharmacological inhibition of p38 MAPK with SB203580 and JNK with SP600125 reduced the ability of vitegnoside, at any concentration used, to improve cell viability reduced by copper-induced Aβ toxicity (p < 0.05–0.01), indicating the involvement of p38 MAPK and JNK in the beneficial effect of vitegnoside in the AD cell model.
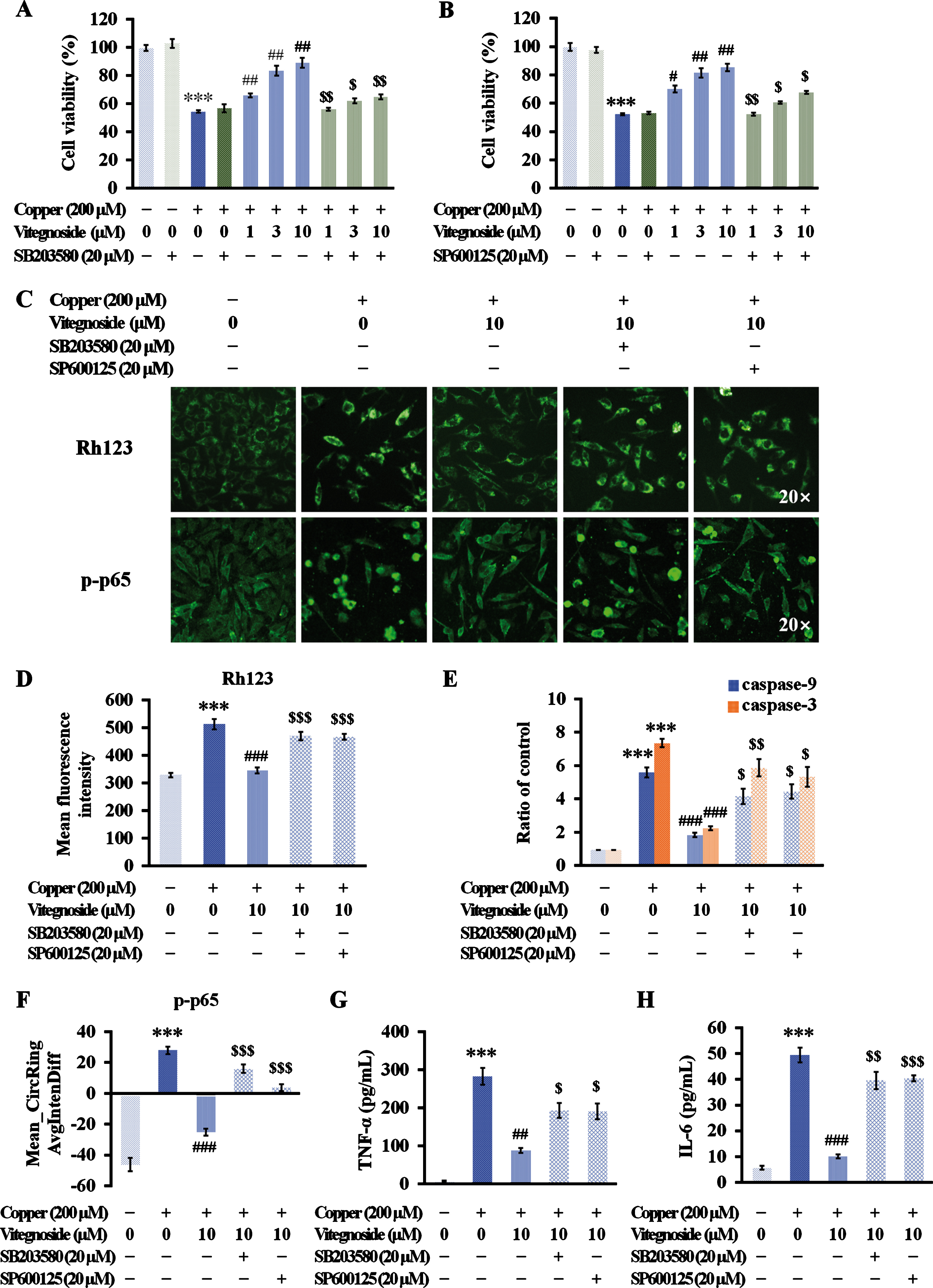
p38 MAPK/JNK pathways involved in vitegnoside-mediated neuroprotection in APPswe cells against copper-induced Aβ toxicity. A, B) Vitegnoside treatment did not exert any protective effect on inhibiting the decrease of cell viability due to copper-induced Aβ toxicity when p38 MAPK and JNK were pharmacologically inhibited by SB203580 (A) and SP600125 (B), respectively. C) Representative images of Rh123 and p-p65 staining (20×magnification). D) Mean fluorescence intensity of Rh123 demonstrating the abolished protective effect of vitegnoside on MMP when copper-injured APPswe cells were pre-treated with SB203580 and SP600125. E) Suppressive effect of vitegnoside on caspase-9 and -3 activity reduced by SB203580 and SP600125 treatment. F–H) The inhibitory effect of vitegnoside on p-p65 translocation and TNF-α and IL-6 release reduced by SB203580 and SP600125 treatment, as shown by (F) Mean_CircRingAvgIntenDiff values and (G) TNF-α content and (H) IL-6 content in the culture medium. n = 6. ***p < 0.001 versus control; #p < 0.05, ##p < 0.01, ###p < 0.001 versus copper; $p < 0.05, $$p < 0.01, $$$p < 0.001 versus copper + vitegnoside.
In addition to its role in neuronal viability improvement, mitochondrion-associated apoptosis was also observed after the use of the above inhibitors. However, after pre-treatment with SB203580 and SP600125, the effects of vitegnoside on the loss of MMPs and increased caspase-9 and -3 activity both induced by copper, were significantly reduced (p < 0.05–0.001, Fig. 6C–E).
Considering that NF-κB signaling plays a role in the transduction with MAPK, the effect of vitegnoside on NF-κB-associated inflammatory responses was determined after the use of the above inhibitors. After SB203580 and SP600125 treatment, vitegnoside did not inhibit the nuclear translocation of p-p65 and consequent reduction of TNF-α and IL-6 that were increased in the presence of copper (p < 0.05–0.001, Fig. 6C, F–H). Thus, these results suggested a specific role of p38 MAPK and JNK in the protective effect of vitegnoside against mitochondrial apoptosis and inflammation induced by copper.
DISCUSSION
In the present study, we aimed at investigating the neuroprotective effect of vitegnoside in an AD cell model and its mechanisms of action. Two major contributions are provided by this study associated to vitegnoside-mediated neuroprotection. First, to our knowledge, this is the first study exploring the beneficial effect of vitegnoside against Aβ toxicity in AD. Second, this study revealed that after copper-induced Aβ toxicity, vitegnoside exerted a protection against mitochondrion-mediated apoptosis and NF-κB-induced inflammation via p38 MAPK and JNK inhibition. These findings provide novel evidence and insights for a potential therapeutic approach.
Transition metals including copper are associated with the aggregation of Aβ plaques and involved in Aβ-mediated neurotoxicity by increasing AβPP expression and Aβ secretion in the AD brain [30]. To better mimic AD pathological state, a copper-treated APPswe cell system was used as an in vitro AD cell model, since it is widely used and known to induce Aβ toxicity, instead of directly subject cells to an Aβ treatment [18, 32]. As expected, copper-induced Aβ toxicity in APPswe cells was exerted by the noticeable induction of oxidative stress, mitochondrial dysfunction, caspase-related apoptosis, and inflammatory responses, while these pathological changes slightly appeared in the SH-SY5Y cells in the presence of copper (Supplementary Figure 1), indicating that copper-induced Aβ toxicity played a role in neurotoxicity in this model.
In this study, a protective effect of vitegnoside was revealed against copper-induced Aβ toxicity in APPswe cells exerted by an increase of cell viability and the preservation of membrane integrity and nucleus uniformity observed by both the morphology and fluorescence intensity of the cells. By contrast, when copper-treated SH-SY5Y cells were used as the WT control, the parameters such as the recovery of cell viability, nuclear condensation, mitochondrial damage, and inflammatory cytokine release that we previously analyzed were not distinctly observed after vitegnoside treatment (Supplementary Figures 1 and 2), suggesting that vitegnoside could exert its neuroprotective effect under copper-induced Aβ toxicity, and not under copper treatment in WT cells in which copper was not toxic. Therefore, the underlying mechanism of vitegnoside effect was discussed using our AD cell model as follows.
The mechanism of preventing the coordination of copper and Aβ toxicity was explored by the evaluation of the following three aspects: 1) copper-induced AβPP overexpression and Aβ overgeneration; 2) Aβ-induced neurotoxicity correlated with copper associated to oxidative stress indicated by ROS production and oxidative markers; and 3) mitochondrion-dependent apoptosis, MAPK signaling, and NF-κB-induced inflammatory cytokines.
AβPP and Aβ are both copper-binding molecules [33], and the interaction between copper ions and copper-binding domain can modulate Aβ production [34, 35]. As showed by our results, the AβPP and Aβ expressions were both increased by copper treatment at high concentrations, which is in agreement with the evidence that increase in cellular copper level upregulates AβPP gene expression [36, 37]. However, vitegnoside was not effective on altering AβPP and Aβ, suggesting that its protective effect was not involving this mechanism. AβPP is redistributed from the intracellular compartment to the plasma membrane in response to high copper levels [38], and acquires a dimeric configuration [35]. For this reason, this failure might be due to a limited or no affinity of vitegnoside to certain residues resulting by the action of cleavage enzymes.
The effect of copper in potentiating Aβ effect on AD is not only due by its involvement in Aβ aggregation, but also in the oxidative stress. The redox imbalance was due to ROS overproduction and the decreased activity of the antioxidant enzymes GSH-Px and SOD following copper treatment in APPswe cells. VN extracts possess considerable potential anti-oxidant effects through their free radical scavenging activities thanks to their diverse phenolic constituents [39]. However, vitegnoside, as a natural flavonoid present in VN, could not significantly scavenge the excess of ROS in our model at each concentration used, suggesting that vitegnoside did not have a sufficient effect on reducing ROS overproduction or improving redox defense in response to Aβ-associated neurotoxicity. In view of these results underlining that the amyloidogenic processing or redox balance was not the mechanisms contributing to vitegnoside-mediated neuroprotection, vitegnoside might act on other mechanisms involved in AD development.
Mitochondria, both target and source of ROS, are implicated in AD as critical organelle mediating its pathogenesis. Persistent mitochondrial dysfunction contributes to apoptosis via the mitochondria-dependent caspase cascade induced by the release of cytochrome c into the cytosol with subsequent activation of caspases that initiate and execute apoptosis [40]. The antiapoptotic protein Bcl-2 maintains mitochondrial membrane integrity and prevents the release of cytochrome c from mitochondria. Conversely, the pro-apoptotic protein Bax induces mitochondrial injury that leads to cell death [41]. The present study revealed improvements on MMP preservation due to vitegnoside treatment in APPswe cells subjected to copper. Furthermore, vitegnoside treatment inhibited the release of proapoptotic cytochrome c, maintained the equilibrium between Bcl-2 and Bax, decreased the subsequent activation of caspase-9 and -3 and ultimately reduced nuclear condensation caused by the disruption of MMPs due to copper-induced Aβ toxicity in APPswe cells. These results demonstrate that vitegnoside effectively inhibited mitochondrion-mediated apoptosis and provided new evidence that this active compound from VN exerts an anti-apoptotic function.
The neurotoxic response to Aβ-associated stimuli, such as oxidative stress, is correlated with the activation of three subfamilies of MAPKs, p38 MAPK, JNK, and ERK1/2 [25, 27]. In response to the activation of p38 MAPK/MK2, JNK/c-Jun, ERK1/2 signaling pathways in APPswe cells due to copper treatment, our results demonstrated a remarkably inhibitory effect on their activation exerted by vitegnoside, although resulting ineffective in inhibiting ERK1/2 activation. p38 MAPK and JNK are activated by Aβ-associated toxicity, redox imbalance, and/or inflammation responsible in AD, thus, blocking them may help in the development of new therapeutic approaches that could improve AD patients’ health status [18, 43]. Therefore, our results on the neuroprotective mechanism of vitegnoside are highlighting the involvement of p38 MAPK and JNK in AD.
The pathological role of p38 MAPK/MK2 and JNK/c-Jun cascades in apoptosis and inflammation has been described in detail [18]. p38 MAPK participate directly by activating cytochrome c release [44] and causing the mitochondrial translocation of Bax [45], while activated JNK influences Bcl-2 phosphorylation [46]. The unbalanced Bax/Bcl-2 ratio affected by p38 MAPK and JNK also contributes to mitochondrial outer membrane permeabilization, and sequentially activates caspase-9 and caspase-3, thus causing apoptotic cell death through the mitochondrion-dependent pathway [47]. Consistent with the above knowledge, the inhibitory effect of vitegnoside on p38 MAPK and JNK signaling activation in this study involved a mitochondrial function preservation.
Furthermore, an inhibitory effect of vitegnoside on the activation of NF-κB signaling was observed, along with a reduced expression of TNF-α and IL-6 in the APPswe cell model. Indeed, the activation of MAPKs can further activate NF-κB, inducing an overproduction of proinflammatory cytokines which subsequently triggers an inflammatory response and initiates an apoptotic cascade [48]. JNK and p38 MAPK both act in NF-κB activation, the former through the phosphorylation of the β-subunit of IκB kinase complex (IKKβ) in response to inflammatory stimuli [49, 50], the latter to allow IκB degradation or promote p65 translocation into the nucleus [51]. Therefore, suppression of NF-κB-mediated inflammatory cytokine release by vitegnoside treatment might be due to the inactivation of p38 MAPK and JNK signaling pathway.
To explore this possibility, two inhibitors were used: SB203580, a selective inhibitor of p38 MAPK, with the ability of inactivating MK2 signaling [52], and SP600125 that is a selective inhibitor of JNK suppressing the phosphorylation of JNK by competitive binding to the JNK ATP-binding site [53]. Our results revealed that when the two pathways were inactivated by these inhibitors, vitegnoside could not exert its action of improving the cell viability of APPswe cells reduced by copper treatment. Importantly, the mitochondrial function preservation and the caspase signaling inhibition exerted by vitegnoside were also blocked by the inhibition of p38 MAPK and JNK, indicating that vitegnoside protective mechanism was exerted through targeting p38 MAPK and JNK. More importantly, inhibition by SB203580 and SP600125 markedly abolished vitegnoside-induced suppression of NF-κB-mediated inflammatory response triggered by copper, as shown by the inability to block p65 translocation and the inability to decrease TNF-α and IL-6 release. Additionally, an in silico molecular docking for interaction analysis between vitegnoside and p38 MAPK/JNK was utilized, and it was found that vitegnoside may interact with p38 MAPK/JNK pathways by the efficient binding performances with the favorable interaction energy scores (Supplementary Figure 3). Altogether, it is reasonable to deduce that vitegnoside neuroprotection on copper-induced Aβ toxicity might be due to the inactivation of p38 MAPK/JNK-dependent mitochondrial apoptosis and inflammatory response. However, the potential use and proposed regulatory mechanisms of vitegnoside for the prevention and/or treatment of AD need to be further defined in an appropriate AD-related animal model.
In conclusion, our study identified the neuroprotective effects of vitegnoside and its potential mechanisms (Fig. 7) on one of the AD deficits, copper-triggered Aβ toxicity, in APPswe cells. These findings highlighted the potential therapeutic beneficial effect of vitegnoside through attenuating p38 MAPK/JNK signaling during the progression of AD.

Proposed mechanisms of vitegnoside-mediated neuroprotection against copper-triggered Aβ toxicity via p38 MAPK/JNK-dependent mitochondrion-mediated apoptosis and NF-κB inflammatory pathway. Vitegnoside exerted the action of improving cell viability, preserving mitochondrial function, and inhibiting caspase signaling of APPswe cells through inactivating p38 MAPK and JNK. Moreover, vitegnoside suppressed NF-κB-mediated inflammatory response, as shown by the inability to block p65 translocation and to decrease TNF-α and IL-6 release, via the inhibition of p38 MAPK and JNK. Thus, it is suggested that vitegnoside neuroprotection on copper-induced Aβ toxicity may be due to the inactivation of p38 MAPK/JNK-dependent mitochondrial apoptosis and inflammatory response. AβPP, amyloid-β protein precursor; Aβ, amyloid-β; ROS, reactive oxygen species; MAPK, mitogen-activated protein kinase; JNK, c-Jun N-terminal protein kinase; MK2, MAPKAP kinase-2; Bax, Bcl-2 associated X protein; Bcl-2, B cell lymphoma-2; cyto c, cytochrome c; NF-κB, nuclear factor kappa-B; IκB, inhibitor of NF-κB; TNF-α, tumor necrosis factor alpha.
Footnotes
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (No. U1803281 and 81673411), China; the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2018RC350013), China; and Chinese Academy Medical Sciences (CAMS) Innovation Fund for Medical Science (2017-I2M-1-016), China.