
Abstract
Background:
Rates of amyloid-β (Aβ) accumulation have been characterized across the cognitively normal to typical Alzheimer’s dementia spectrum, but little is known about Aβ accumulation in atypical Alzheimer’s disease (AD) and other neurodegenerative diseases, such as frontotemporal lobar degeneration (FTLD).
Objective:
We aimed tocharacterize longitudinal Aβ accumulation anddetermine the influence of age, apolipoprotein E (APOE) genotype, disease duration, and sexin atypical AD and FTLD.
Methods:
322 patients (138 atypical AD, 184 FTLD) underwent Pittsburgh compound B PET scanning, with 73 having serialPiB-PET scans (42 atypical AD, 31 FTLD). Global Aβ standard uptake value ratios were calculated for every scan. Mixed effects models were used to assess the effect of age, APOE genotype, disease duration, and sex on baseline and change measures of Aβ.
Results:
Atypical AD showed higher baseline Aβ than FTLD. Rate of Aβ accumulation was not associated with baseline Aβ in either group. Older age was associated with greater baseline Aβ and faster rates of accumulation in FTLD. In patients under age 70, atypical AD showed faster rates of accumulation than FTLD. APOE ɛ4 genotype was associated with greater baseline Aβ in FTLD but did not influence rates of accumulation. Rates of Aβ accumulation were faster in FTLD patents with time from onset-to-PET≤4 years. Female sex was associated with faster rates of accumulation in atypical AD.
Conclusion:
Accumulation of Aβ is observed in atypical AD and FTLD, although different demographic factors influence accumulation in these diseases providing insight into potentially different biological mechanisms of Aβ deposition.
Keywords
INTRODUCTION
Deposition of the protein amyloid-β (Aβ) is one of the hallmark pathological features defining Alzheimer’s disease (AD). Aβ can be detected during life using positron emission tomography (PET) ligands which provides us with the opportunity to make a biomarker-based diagnosis of AD early in the disease course and to assess accumulation of Aβ over time in order to model disease progression. Most of what we have learned about Aβ accumulation in vivo has come from longitudinal studies assessing clinically normal individuals and patients with mild cognitive impairment and typical AD. Progressive Aβ accumulation has been observed in AD, mild cognitive impairment, and also in clinically normal individuals [1–8], with Aβ accumulation in normal cohorts greater in people with higher Aβ burden at baseline [5, 9]. Some studies have also suggested that rate of Aβ accumulation is greater in apolipoprotein E (APOE) ɛ4 carriers [2, 10].
While much has been learned about Aβ in typical AD, much less is known about Aβ in atypical clinical presentations of AD, or in other neurodegenerative diseases such as frontotemporal lobar degeneration (FTLD) that can overlap clinically with atypical AD. Two of the most common atypical clinical variants of AD are posterior cortical atrophy [11, 12] and logopenic aphasia [13]; these clinical syndromes are highly associated with underlying AD pathology [14, 15]. FTLD is the second most common cause of dementia in people under age 65 and is an umbrella term for a number of different pathologies that target the frontal and temporal lobes; these pathologies include primary tauopathies, TDP-43 proteinopathies, and fused in sarcoma proteinopathies [16]. Clinical syndromes that are highly associated with FTLD pathology include semantic dementia [17], non-fluent/agrammatic primary progressive aphasia [13], primary progressive apraxia of speech [18], and progressive supranuclear palsy [19]. Although FTLD is typically the underlying pathology in these cases, we and others have shown that Aβ deposition is still observed on PET scanning during life in FTLD cases [20–23], and Aβ deposition at autopsy is observed in 37% [24]. However, the rate that Aβ accumulates over time in atypical AD and FTLD is unclear and it is unknown whether demographic features, such as age, sex, disease duration, or APOE genotype play any role in determining rate of Aβ accumulation. Understanding Aβ accumulation in these patients will be crucial to help interpret Aβ-PET findings and in predicting which patients may show the greatest rates of Aβ accumulation, and hence, potentially, the fastest progression of underlying AD pathology.
The aim of this study was to assess rates of Aβ accumulation and determine how demographic features influence both cross-sectional and longitudinal Aβ PET measures in atypical AD and FTLD
METHODS
Patients
A total of 322 patients with either an atypical AD or FTLD clinical diagnosis were recruited by the Neurodegenerative Research Group and underwent at least one Pittsburgh Compound B (PiB) PET scan between July 7, 2010 and April 11, 2019 as part of a number of grants led by JLW and KAJ (NIH grants R01-AG50603, R01-DC12519, R01-DC010367, R01-NS89757, R21-NS94684, and R01-DC14942, and an Alzheimer’s Association grant). All patients were recruited from the Department of Neurology, Mayo Clinic Rochester, MN. The 322 patients consisted of 138 with atypical AD and 184 with FTLD, and of these 322 patients, 73 had undergone at least two serial PiB-PET scans (42 atypical AD and 31 FTLD) (Fig. 1). The atypical AD cohort consisted of patients that showed evidence for Aβ deposition on PET and met clinical criteria for posterior cortical atrophy [11] or logopenic progressive aphasia [13] (Fig. 1). The FTLD cohort consisted of patients with primary progressive apraxia of speech [18], non-fluent/agrammatic primary progressive aphasia [13], semantic dementia [25], progressive supranuclear palsy [19], and patients with primary progressive aphasia [26] that could not be classified into one of the three variants [13] but were strongly suspected to have FTLD pathology (Fig. 1). We also included three patients in the FTLD group that had a clinical diagnosis of logopenic aphasia but were Aβ- and were found to carry progranulin gene mutations, as we have previously described [27].
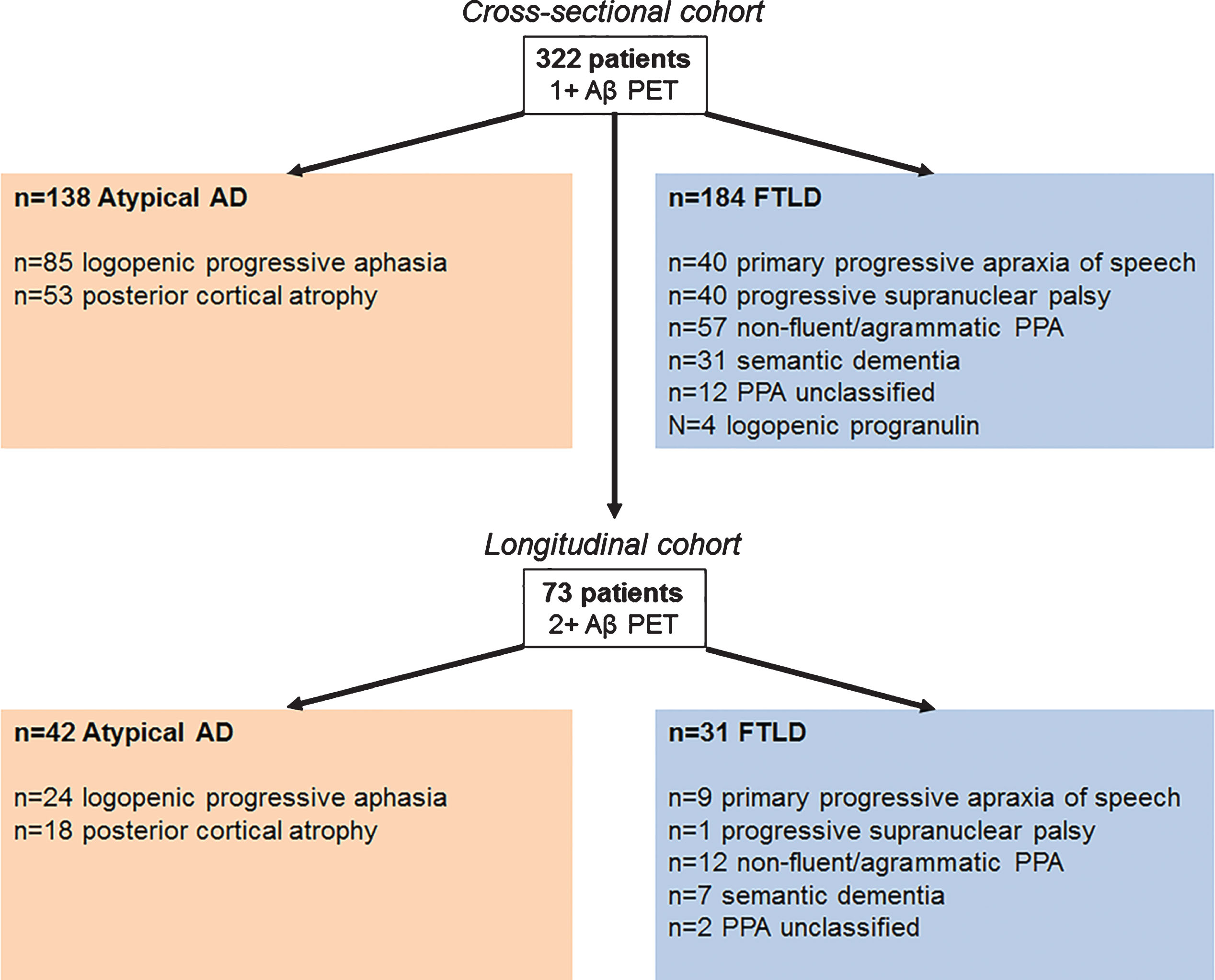
Flow chart illustrating the cross-sectional and longitudinal patient cohorts. AD, Alzheimer’s disease; FTLD, frontotemporal lobar degeneration; PPA, primary progressive aphasia.
All patients underwent a standardized neurological evaluation by a behavioral neurologist (KAJ, JGR, HB) which included testing for cognitive impairment using the Montreal Cognitive Assessment Battery (MoCA) [28], functional severity using the Clinical Dementia Rating-FTLD Scale [29], parkinsonism severity using the Movement Disorder Sponsored revision of the Unified Parkinson’s Disease Rating Scale part III (MDS-UPDRS III) [30], and ideomotor apraxia severity using the Western Aphasia Battery-Revised limb apraxia scale [31]. The FTLD patients also all underwent a detailed speech and language assessment by a speech-language pathologist (JRD, HMC, RLU, EAS), as previously described in detail [18], and diagnosis was rendered by consensus of at least two of the speech/language pathologists. Apolipoprotein E genotyping was performed for 301 of the 322 patients (69 of the 73 patients with longitudinal Aβ PET).
This study was approved by the Mayo IRB and all patients consented to participate in the study.
Neuroimaging analysis
All PET scans were acquired with a PET/CT scanner (GE Healthcare) while operating in 3D mode. Patients were injected with PiB of approximately 628 MBq (range, 385–723 MBq) and after a 40-to-60-min uptake period a 20-min PiB scan was obtained. Emission data was reconstructed into a 256×256 matrix with a 30-cm FOV. All patients also underwent a 3T volumetric head MRI within two days of the PiB-PET scan, which included a 3D magnetization prepared rapid acquisition gradient echo sequence (MPRAGE). All MPRAGE scans underwent corrections for intensity inhomogeneity [32] and gradient unwarping [33] before analysis.
The PiB-PET scans were registered to the corresponding MPRAGE scan using a 6 degrees-of-freedom registration in SPM12. The Mayo Clinic Adult Lifespan Template (MCALT) (https://www.nitrc.org/projects/mcalt/) was then transformed into the native space of each MPRAGE using ANTs software [34]. Median Aβ uptake was calculated for the following regions-of-interest defined using MCALT: inferior parietal, superior parietal, supramarginal gyrus, angular gyrus, cingulate [anterior, mid, posterior, and retrosplenial], precuneus, superior frontal, middle frontal, orbitofrontal, inferior frontal [operculum + triangularis], medial frontal, fusiform, lateral temporal [inferior, middle, and superior temporal gyri + Heschl], and temporal pole. Uptake was calculated from the grey matter in each region and divided by uptake in the cerebellar crus grey matter to calculate standard uptake value ratios (SUVRs). A global Aβ SUVR was calculated as the weighted average from the regions-of-interest. Aβ positivity was defined as a global Aβ SUVR >1.48. For patients with serial PiB-PET, all global PiB SUVRs were calculated for each time-point separately without reference to the other time-points.
Statistical analysis
Participant characteristics and clinical variables were compared between atypical AD and FTLD groups using Wilcoxon rank sum tests or Fisher’s exact tests as appropriate. p < 0.05 was considered statistically significant. Within-subject regression models were used to calculate rates of change in global Aβ SUVR (expressed as SUVR points change per year). We also used log SUVR values to estimate change on an annual percentage scale. We used cut-offs to determine whether a patient is increasing (>2% per year) or declining (< –2% per year) over time. The 2% cut-off is based on a previous publication that reported within-subject measurement error of 2–4% [35].
We used longitudinal linear mixed effects models to separately evaluate the effects of age, APOE ɛ4, disease duration, and sex on rate of Aβ change in both diagnosis groups. Age at first Aβ PET and disease duration were classified into age ≤70, or >70 years old, disease duration ≤4, or >4 years. The time variable for the models was defined as years from first Aβ PET. We fit longitudinal models including data from all individuals with one or more Aβ PET, including all serial Aβ PET available in each patient (249 with only one PiB and 73 with serial PiB [59 with two Aβ PET, 12 with three Aβ PET, and 2 with four Aβ PET], total n = 322 patients). A longitudinal linear mixed effects model can be considered as simultaneously estimating both cross-sectional parameters (i.e., mean Aβ within a group) and longitudinal, or within-subject change, parameters (i.e., annual change in Aβ within a group) [32]. Individuals with one Aβ PET contributed information to the cross-sectional parameters whereas individuals with multiple Aβ PETs contributed to both cross-sectional and longitudinal parameters.
Estimated Aβ rates of accumulation for different groups were obtained via interactions in the linear mixed effects models. Each model included all subjects and a three-way interaction between the predictor of interest, diagnosis group (atypical AD versus FTLD), and time. This resulted in a separate Aβ rate estimate for both levels of a covariate within each diagnosis group and allowed us to compare rates for different levels of a covariate within a diagnosis or to compare diagnosis group rates within a level of a covariate. For purposes of comparison, we also fit linear mixed effects models but with the log of Aβ SUVRs as the response, so that estimated annual change can be interpreted on an annual percentage scale. All of our linear mixed effects models included subject-specific random intercepts. We included continuous age at Aβ PET as an adjustment variable in all models except the ones in which age group was analyzed.
We also performed correlation analysis between baseline Aβ SUVRs and subject specific slopes of Aβ SUVRs stratified by diagnosis within subjects with serial Aβ PET scans. Correlation coefficients were compared using Fisher’s Z transformation which transforms correlation coefficients (r) into z-scores. The significance of the difference between two correlation coefficients was assessed through the observed Z test statistic.
Analyses were performed with R statistical software (version 3.4.4; R Foundation for Statistical Computing, Vienna, Austria) and models were fit with the lmerTest package (version 3.0.1), which extends R’s generalized linear mixed model package “lme4” with p-values for fixed effects.
RESULTS
The atypical AD and FTLD groups did not differ for sex, education, and disease duration (Table 1). Atypical AD was younger at onset and scan than FTLD in the cross-sectional cohort, but age did not differ across groups in the longitudinal cohort. Atypical AD had a higher proportion of APOE ɛ4 carriers, and performed worse on the MoCA and CDR-FTLD than the FTLD group. Conversely, FTLD performed worse on the MDS-UPDRS III than atypical AD.
Patient characteristics
Data shown are N (%) or median [IQR]. p-values for continuous variables are from Wilcoxon Rank Sum test. p-values for categorical variables are from Fisher Exact test. MoCA, Montreal Cognitive Assessment battery; CDR-FTLD, FTLD modified version of the Clinical Dementia Rating Scale; MDS-UPDRS III, Movement Disorder Society sponsored revision of the Unified Parkinson’s Disease Rating Scale part III; WAB, Western Aphasia Battery-Revised.
Global Aβ SUVR was higher in atypical AD compared to FTLD (Table 1). Approximately 50% of both cohorts showed stable Aβ over time, with 43% of atypical AD and 35% of FTLD showing Aβ accumulation over time of greater than 2% per year. Very few patients showed declining Aβ over time. There was no significant difference in rate of Aβ accumulation between groups, either when expressed as annualized SUVR change or annualized percentage change, although rates were slightly higher in atypical AD. Rate of Aβ accumulation was not associated with baseline Aβ SUVR in atypical AD (rank correlation [rho] = –0.05, p = 0.77), FTLD (rho = –0.27, p = 0.15), or in the cohort as a whole (rho = 0.21, p = 0.0.08) (Fig. 2).
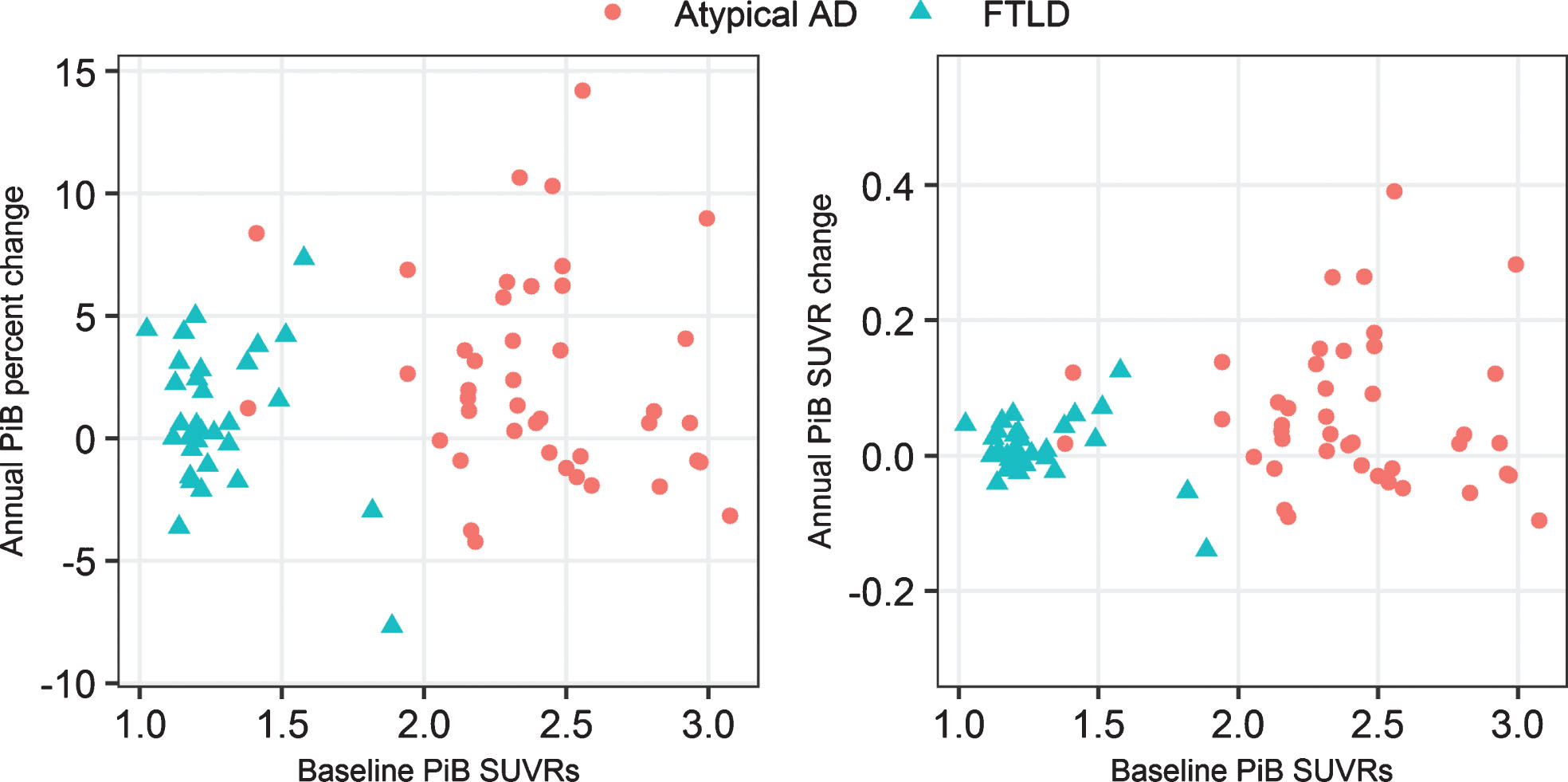
Plots of baseline Aβ SUVRs versus rate of Aβ accumulation (SUVR change and percentage change) by diagnosis.
The results of the mixed models showing the association of demographic factors to baseline Aβ SUVR are shown in Table 2 and Fig. 3. The results showing associations to rate of Aβ accumulation are shown in Table 3 and Fig. 3. When the cohorts were dichotomized by age, older age (>70 years) was associated with higher baseline Aβ SUVR and faster rates of Aβ accumulation in FTLD (Fig. 4). The effect of age was significantly different in atypical AD compared to FTLD. The two atypical AD age groups had more comparable baseline SUVRs but, if anything, the younger atypical AD group had faster accumulation. Rates of accumulation in the younger atypical AD group were significantly greater than in the younger FTLD group.
Effects of age, APOE ɛ4, disease duration, and sex on baseline Aβ SUVR
Effects of age, APOE ɛ4, disease duration, and sex on rates of Aβ accumulation expressed as either percent change or SUVR change
All models were age adjusted except model with age as predictor.

Plots illustrate the association between demographic features (age, APOE genotype, disease duration and gender) and both baseline Aβ SUVR and rate of Aβ accumulation (expressed as annualized SUVR change and annualized percentage change). Plots for APOE, disease duration, and gender were age corrected.
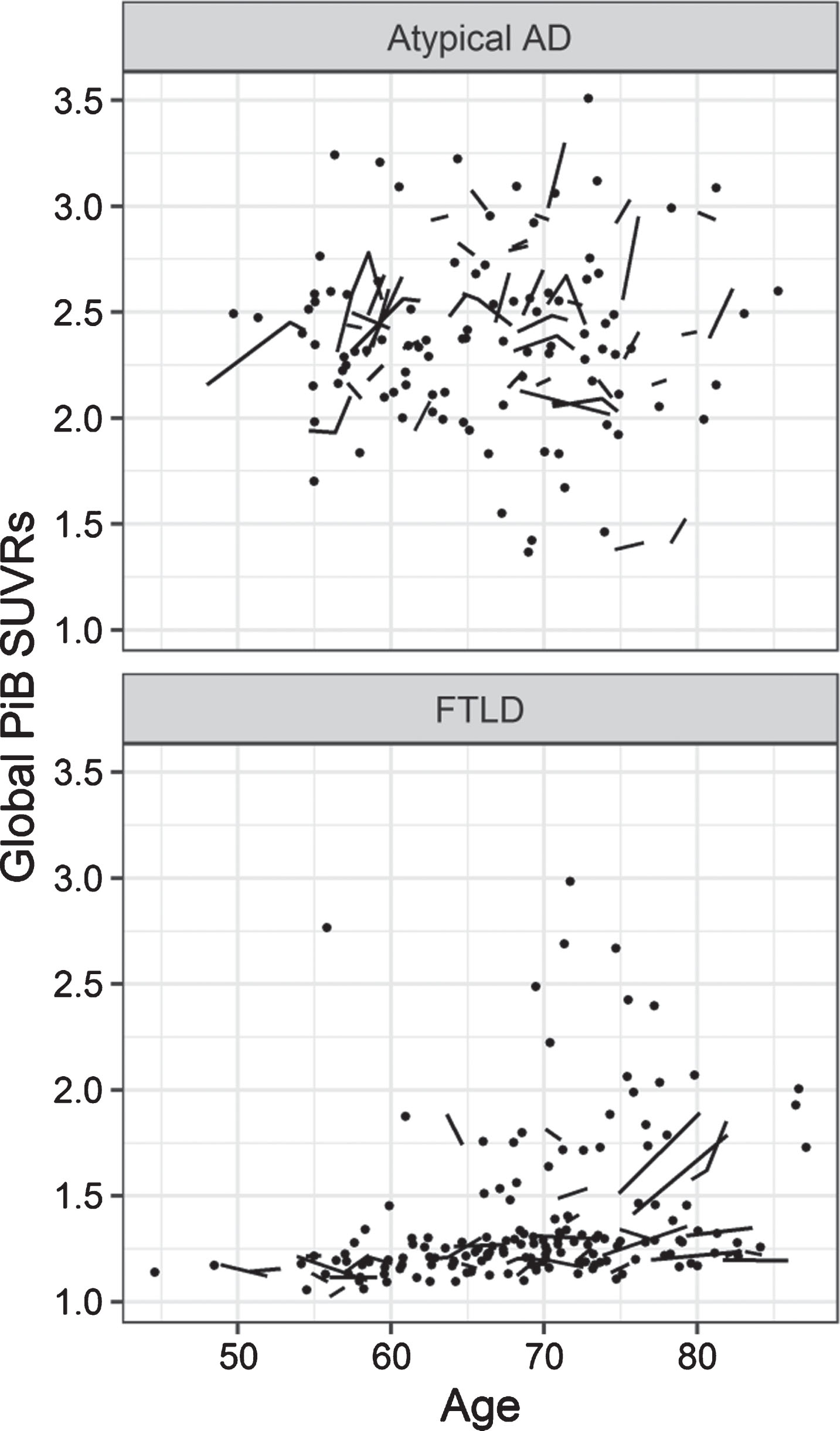
Plots of global Aβ SUVRs by age in atypical AD and FTLD.
All of the following results were corrected for age.
DISCUSSION
Despite the fact that atypical AD and FTLD showed very different baseline levels of Aβ SUVR, rates of Aβ accumulation were similar in the two cohorts overall. However, demographic factors had different relationships to Aβ burden and rate of accumulation in the two diseases, and differences between atypical AD and FTLD emerged in rates of Aβ accumulation when stratifying by age and disease duration.
As would be expected, and by design, baseline Aβ SUVR was higher in atypical AD than in FTLD, reflecting the fact that the atypical AD patients all had biomarker evidence for AD. However, we observed more similar results across the two groups regarding rate of Aβ accumulation. There was a tendency for atypical AD to show higher rates of accumulation than FTLD, although a large degree of variability was observed in both cohorts and the rate measures were not significantly different overall. In fact, a large proportion of patients in both groups showed stable Aβ SUVR over time. We did not find any relationship between rate of Aβ accumulation and baseline Aβ SUVR in either group or across the entire cohort, suggesting that patients’ Aβ SUVR will not help to predict the rate that they will subsequently accumulate Aβ over time. Studies assessing the normal-to-typical Alzheimer’s dementia spectrum have found that rate of Aβ accumulation increases with baseline Aβ SUVR up until an SUVR of approximately 2.0 and then starts to plateau as rates decrease again [4, 6]. Our FTLD cohort resembles the cognitively normal patients in these models, showing low Aβ SUVR and low rate of change. Our atypical AD group nearly all had baseline Aβ SUVR over 2.0, and hence Aβ may already be saturated and rates may have plateaued in this cohort. We did, however, have a couple of patients with very high Aβ SUVR up to 3.0 that were still showing high rates of accumulation.
A number of demographic features influenced baseline Aβ SUVR and rate of Aβ accumulation in our analysis, although we typically observed different effects in atypical AD and FTLD. One striking difference was that age was only related to Aβ in FTLD and not in atypical AD. Within the FTLD cohort, older age was associated with greater baseline Aβ SUVR, as we and others have noted [20–22], but also greater rates of Aβ accumulation. Hence, Aβ deposition is strongly age-related in FTLD, concurring with findings from an autopsy study of Aβ in FTLD [24]. This suggests that the development and progression of AD continuum pathology is more common in older patients. Of four FTLD patients that showed Aβ uptake on PiB-PET in one study, 100% showed Aβ deposition on autopsy and three had sufficient tau pathology to meet pathological criteria for AD [24]. Similarly, age has been shown to be related to Aβ SUVR [36] and rate of Aβ accumulation [5] in clinically normal people. The fact that age was not associated with baseline or accumulation of Aβ in atypical AD concurs with previous cross-sectional studies in atypical AD syndromes [22] and longitudinal studies in typical AD [2]. In fact, although not significant, the rate of Aβ accumulation actually appears a little higher in younger versus older atypical AD patients, and in patients under age 70 atypical AD had significantly faster rates of Aβ accumulation than FTLD. This difference between diagnostic groups was hidden when all ages were merged in the main analysis. Interestingly, we have also recently shown that younger atypical AD patients have faster rates of accumulation of tau measured by [18F]flortaucipir PET [37], suggesting a more aggressive disease course in young atypical AD.
The influence of the APOE ɛ4 genotype was also only observed in FTLD, with greater baseline Aβ burden observed in APOE ɛ4 carriers. This finding that APOE ɛ4 increases the risk of Aβ deposition has previously been noted in other autopsy and cross-sectional studies that have assessed FTLD phenotypes [20, 38]. We did not, however, find any evidence that APOE ɛ4 influences rate of Aβ accumulation in FTLD. Hence, APOE ɛ4 appears to increase the risk of developing Aβ but does not play a role in the rate that the Aβ spreads through the brain. This concurs with previous longitudinal findings from one study in clinically normal individuals [5], although others did find a relationship between APOE ɛ4 genotype and faster rates of Aβ accumulation in clinically normal individuals [10, 36]. Of note, the FTLD APOE ɛ4 carriers still had a very low Aβ SUVR and atypical AD showed higher baseline Aβ than FTLD both in APOE ɛ4 carriers and non-carriers. APOE ɛ4 genotype did not influence the Aβ SUVR or rate of accumulation of Aβ in atypical AD. While APOE ɛ4 may have played a role in the original development of Aβ in these patients, there may, therefore, be another unknown factor, or factors, which play a role in determining Aβ burden and rate once Aβ is present. There has been mixed results concerning whether APOE ɛ4 influences rate of Aβ accumulation in AD, with some finding an association [2] and others not [6].
An unexpected finding was that disease duration was strongly associated with rate of Aβ accumulation in FTLD, with faster rates in patients with disease duration under four years. This finding does not appear to be driven by the distribution of the specific FTLD syndromes, since disease duration did not differ across the syndromic groups (p = 0.88). In addition, disease duration was not correlated with age (Spearman correlation = –0.14, p = 0.47) or scan interval (Spearman correlation = –0.07, p = 0.70) in FTLD, and age was accounted for in the analysis. These results could suggest that patients with shorter disease duration may have more rapidly progressing illnesses, and the spread of AD continuum pathology may be in some way contributing to, or be associated with, worse clinical outcomes that lead the patient to present to a clinician earlier in the disease course; it is possible they may even die sooner than patients with FTLD in the absence of AD. There could also be bias in our cohort, whereby patients with both FTLD and AD continuum pathology are less likely to return to be assessed at longer disease durations. Given this uncertainty, the finding will need to be confirmed in independent samples. Rate of Aβ accumulation did not differ by disease duration in atypical AD, although baseline Aβ SUVR was higher in patients with longer disease duration. This finding makes sense given that we know Aβ accumulates over time in atypical AD, and hence, the longer a patient has been affected then the higher the Aβ burden.
The only demographic factor that influenced rates of Aβ accumulation in atypical AD was sex, with faster rates observed in women, even after correcting for age. We did not observe any sex differences in Aβ burden at baseline. The reason for this sex difference is unclear. Sex differences have been observed in relation to Aβ in patients in the clinically-normal-AD spectrum, although results are mixed. Consistent with the fact that we did not observe any sex differences in baseline Aβ, a pathological study found no differences in senile plaque burden by sex in cases with advanced neurofibrillary tangle stage, and a PET study did not identify differences in global Aβ uptake by sex [39]. One study suggested that women have early neural resistance to Aβ [40]. However, other studies found that men had greater Aβ burden than women [41, 42]. One longitudinal PET study found no differences in rate of Aβ accumulation by sex [5], although another found that rates were greater in men than women, although this effect went away after correcting for baseline Aβ burden [4]. More work will, therefore, be needed to confirm our finding and to determine how these findings relate to disease progression in atypical AD. Of note, our cohort had a high proportion (67%) of women, as is typical of other posterior cortical atrophy and logopenic aphasia cohorts [12, 23].
An important aspect of our study was that we analyzed relationships separately between atypical AD and FTLD, allowing us to identify different demographic determinants of Aβ accumulation in the different cohorts. Evaluating them together would have masked these associations and potentially driven other misleading associations. We also analyzed rate of Aβ accumulation as an annualized change in Aβ SUVR, as well as annualized percent change in SUVR, and the results were almost identical using the two methods. Our cohorts were large and consisted of a number of different atypical AD and FTLD syndromes which should increase generalizability of the findings. However, we acknowledge that the results may not generalize to other syndromes that were not included or to cohorts with different syndromic breakdowns. We also lacked autopsy findings to confirm pathological diagnosis which is most important in FTLD since we lack neuroimaging biomarkers for the underlying FTLD proteinopathies. However, previous studies have shown good concordance between the syndromes we assessed and underlying FTLD pathology [16, 43], particularly between primary progressive apraxia of speech [44], progressive supranuclear palsy [43, 45] and agrammatic primary progressive aphasia [43, 46] and underlying FTLD tauopathies; and between semantic dementia and underlying TDP-43 deposition [46–48]. We did not include behavioral variant frontotemporal dementia or corticobasal syndrome in our FTLD cohort since the pathology of these syndromes is notoriously heterogeneous and often includes AD [43, 50].
The findings from this study increase our knowledge of Aβ pathology in neurodegenerative diseases, and provide demographic risk factors for Aβ accumulation, including age and disease duration in FTLD and sex in atypical AD. This will be important to help predict the behavior of Aβ PET in patients with these neurodegenerative diseases and aid in the interpretation of Aβ PET findings. The results also highlight interesting associations that may lead to a better understanding of the underlying pathophysiological mechanisms in these diseases.