
Abstract
Alzheimer’s disease (AD) is a currently incurable neurodegenerative disorder being the major form of dementia worldwide. AD pathology is initiated by cerebral aggregation of amyloid-β (Aβ) peptides in the form of amyloid plaques; however, the mechanism how Aβ peptide aggregates participate in the disease progression and neurodegeneration is still under debate. Human neuroblastoma cell line SH-SY5Y is a convenient cellular model, which is widely used in biochemical and toxicological studies of neurodegenerative diseases. This model can be further improved by differentiation of the cells toward more neuron-like culture using different protocols. In the current study, dbcAMP, retinoic acid with TPA, or BDNF were used for differentiation of SH-SY5Y cells, and the resulting cultures were tested for the toxicity toward the Aβ42 peptide. The toxicity of Aβ42 peptide depended on the type of differentiated cells: RA and TPA- differentiated cells were most resistant, whereas dbcAMP and RA/BDNF- differentiated cells were more sensitive to Aβ toxicity as compared with non-differentiated cells. The differentiated cultures provide more appropriate cellular models of human origin that can be used for studies of the mechanism of Aβ pathogenesis and for a screening of compounds antagonistic to the toxicity of Aβ peptides.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that is responsible for up to 75% of all dementia cases [1]. Brains of AD patients are characterized by the presence of amyloid plaques, consisting of amyloid-β (Aβ) peptides, and neurofibrillary tangles consisting of hyperphosphorylated tau proteins. Aβ peptide aggregates accumulate in the brain long before the onset of neurological symptoms and play a significant role in the neurodegeneration. Toxicity of Aβ is intensively studied in cellular models, which can shed light on the mechanisms of disease progression at cellular level. In these studies, a variety of in vitro cellular models, which include primary rodent neuronal cultures, neuroblastoma cell lines (mouse Neuro-2A, rat PC12, human SH-SY5Y cells), and human induced pluripotent stem cells (iPSCs), are widely used [2, 3]. Each model has its own advantages and disadvantages; however, human origin, price advantage, and accessibility make human neuroblastoma cell line SH-SY5Y the most popular cellular model in toxicological and biochemical studies of AD. Unfortunately, SH-SY5Y cells in the non-differentiated state lack several essential features of neurons specifically affected in AD. Most importantly, they expose several morphological subtypes with rounded cell bodies and only few short neurites [4] and moreover, they proliferate rapidly. Cancerous cell line-based models can be further improved by application of specific agents, which induce differentiation toward neuron-like morphology, expression of neuron-specific proteins, and inhibition of proliferation.
It is known that differentiation of SH-SY5Y cells can be induced by various agents including retinoic acid (RA), brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), tetradecanoylphorbol acetate (TPA), 17 beta-Estradiol (E2), 3β-hydroxy-5-cholestene (cholesterol), N(6),2′-O-dibutyryladenosine 3′:5′ cyclic monophosphate (dbcAMP), insulin-like growth factor 1 (IGF-1), and several others [5–9].
RA treatment enhances the outgrowth of neurites [10] and leads to increase of the survival of SH-SY5Y cells through the upregulation of BCL-2 protein [11] and changes in cellular sodium homeostasis [12]. Treatment with RA makes SH-SY5Y cells responsive to neurotrophins including BDNF and sensitive to further differentiation. Sequential treatment of SH-SY5Y cells with RA and BDNF leads to outgrowth of neurites, synaptogenesis, and modulates the cellular survival. At the molecular level, treatment leads to increased expression of VAChT and ChAT, and to increased activity of AChE, suggesting differentiation toward the cholinergic neuronal phenotype [13, 14].
Differentiation of SH-SY5Y cells with dbcAMP also induces morphological changes in cells toward a neuron-like phenotype with neurite outgrowth and branching, exposing noradrenergic phenotype [9]. DbcAMP is degraded by intracellular esterases to butyrate and monobutyryl cAMP (mbcAMP), which activates protein kinase A (PKA). It was shown that both the butyrate and the activation of PKA play an important role in dbcAMP mediated differentiation of SH-SY5Y cells [9]. Several studies have demonstrated that dbcAMP increases the expression of amyloid-β protein precursor (AβPP), the precursor molecule for Aβ peptides [15, 16].
12-O-tetradecanoylphorbol-13-acetate (TPA) is a biologically active phorbol ester affecting cell growth and differentiation via protein kinase C (PKC) [13]. During differentiation by TPA, SH-SY5Y cells undergo morphological changes, discontinue replication, and reach a stable cell population. It has been previously shown that sequential exposure of SH-SY5Y cells to RA and TPA induces 3-fold increase in TH, 4-fold increase in DAT, 3-fold increase in D2 and 6-fold increase in D3 receptor levels as compared to undifferentiated cells, characteristic for a dopaminergic cellular phenotype [7, 17].
We have differentiated SH-SY5Y cells by using three protocols: RA/BDNF, RA/TPA, and dbcAMP, which all induced differentiation toward more neuron-like phenotype characterized by dense neurite network. The cultures were tested against toxicity of Aβ42 peptide by using different viability tests as well as microscopy and immunocytochemistry. After 48 h exposure to Aβ42, all differentiated cells and neurites were covered with Aβ layer and neurite fragmentation was observed. However, cells exhibited different sensitivity toward Aβ42 peptide in viability tests: dbcAMP treatment and RA/BDNF treatment increased the sensitivity whereas RA/TPA- differentiated cells were the most resistant toward Aβ toxicity.
MATERIALS AND METHODS
Cell culture
SH-SY5Y human neuroblastoma cells (ATCC, Europe) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 50 U/ml penicillin, 50 μg/ml streptomycin solution (Gibco) in an incubator at 37°C and 5% CO2. The medium was changed every 2-3 days and cells were split using Trypsin-EDTA solution (Gibco). Cells were seeded in poly-L-lysine (PLL) (Sigma) coated (dbcAMP) or uncoated (RA/TPA and RA/BDNF) white clear bottom 96-well plates (Greiner Bio-One) for toxicity experiments or 24-well plates (Greiner Bio-One) for microscopy experiments. Prior to the application of differentiating agents, cells were allowed to adhere for 24 h.
Obtained cells were treated according to the following differentiation protocols: cells were pre-differentiated with RA (10 μM; Sigma Aldrich) in full media for 4 days and then treated with BDNF (50 ng/mL; Alomone Labs) in serum free media for 2 days or with 80 nM TPA (Sigma Aldrich) in serum free media for 3 days; dbcAMP (Sigma Aldrich) treatment was conducted with 2 mM dbcAMP in full media for 2 days and subsequently in serum-free media containing 2 mM dbcAMP for an additional day.
Cells were visualized with Zeiss Axiovert 200 M, Axiocam MRc5 with 20x A-Plan with Ph2 and 20x Plan NeoFluo objectives.
Cell viability measured by WST-1
The effects of Aβ42 on the viability of cells were determined using the cell viability assay WST-1 (Roche). WST-1 allows colorimetric measurement of cell viability due to reduction of tetrazolium salts to water-soluble formazan by viable cells. The amount of formed formazan dye correlates with the number of viable cells. The measurements were completed 48 h after cells treatment with Aβ42. 1 μM staurosporine (Santa Cruz) added to serum-free control group was used as a positive control to induce apoptosis during 48 h. The experiments with HEPES buffer were used as a negative control. 5 μl/well of WST-1 reagent was added to 100 μl cell culture medium, incubated at 37°C for 2 h and absorbance was measured at 450 nm using TECAN Genios Pro microplate reader.
Propidium iodide assay
Propidium iodide (PI) is a red-fluorescent DNA-binding dye used to detect nonviable cells with disrupted cell membranes as it cannot cross intact cell membranes. 0.5 mM PI in phosphate buffered saline (PBS, Sigma) was added to 100 μl cell culture 0.5 μl/well and incubated for 10 min at 37°C. Fluorescence was measured using TECAN Genios Pro microplate reader (excitation 540 nm, emission 612 nm). Results are presented as the fold increase from control islets.
Peptide preparation
Lyophilized amyloid-β 1–42 peptide (Aβ42) (rPeptide) was dissolved in 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP) (Sigma) to disaggregate the peptide oligomeric and fibrillar assemblies, vortexed and incubated for 1 h at room temperature, divided into aliquots and dried overnight in a vacuum desiccator. The aliquots were stored at –80°C. One day before experiments, the aliquots were treated with HFIP again as described before. Defibrillized Aβ42 aliquot was dissolved in 10 mM NaOH and incubated on ice for 10 min. Next, equal amount of 40 mM HEPES buffer containing 200 mM NaCl (pH 7.3) was added to a final peptide concentration of 400 μM. Prepared peptide solution was immediately applied to the serum-free differentiated cell culture at final concentration of 10 μM and 20 μM.
Immunocytochemistry
Cells were seeded on glass coverslips in 24-well plates. After differentiation, culture media was removed and cells were washed twice with PBS. Cells were fixed using 4% PFA (for TUJ antibody - methanol for 15 min at 4°C) for 20 min at room temperature, washed twice with PBS and blocked with 3% goat serum diluted in PBS for 20 min at room temperature to avoid non-specific staining. Cells were washed twice with PBS and primary antibodies diluted in 0.2% Tween solution were applied for incubation overnight at 4°C. Primary antibodies - anti-Synaptophysin 1 (1:2000) (Synaptic Systems), anti-βIII tubulin (1:2000), anti-AβPP/β-Amyloid (1:2000) NAB228 (antigen is 1–11aa from Aβ) were used (Cell Signaling Technology).
Samples were washed three times for 5 min with PBS and incubated with secondary antibodies diluted in PBS for 1 h at room temperature in dark. Secondary antibodies, Alexa Fluor 488 goat anti-mouse and Alexa Fluor 568 goat anti guinea pig (1:2000) (Thermo Fisher Scientific), were used.
Samples were washed three times for 5 min each with PBS, and nuclei were counterstained with DAPI for 5 min. Coverslips were rinsed once with PBS and once with water and applied onto the microscope slides mounted with a drop of ProLong® Diamond antifade reagent (Life Technologies). Cells were visualized using a confocal microscope Zeiss Duo 510 META with 63X oil immersion objective.
Statistical analysis
Statistical analysis was performed by using one-way analysis of variance (ANOVA) with the post hoc Dunnett’s multiple comparison test. The graphs represent data from at least three independent experiments, all performed in triplicates as mean±standard error of the mean (SEM). In the cell viability assay, positive cells were normalized to 100% in negative control (HEPES buffer). In the PI assay, results are presented as the fold increase from control islets. Statistical significance of p < 0.05 is represented as *, p < 0.002 as **, p < 0.0002 as ***, p < 0.0001 as ****. Statistical analyses were performed with GraphPad Prism 7.
RESULTS
Morphology of differentiated SH-SY5Y cells
Treatment of SH-SY5Y cells with differentiating agents used induces neurite outgrowth with elongated neurites that form connections with surrounding cells comparing to non-differentiated cells (Fig. 1). RA/BDNF resulted in almost homogeneous cell population whereas small populations of undifferentiated cells were present in dbcAMP and RA/TPA differentiated cell cultures. The success of differentiation was also visualized using immunostaining with an early neuronal marker beta III tubulin (Fig. 1, right column).
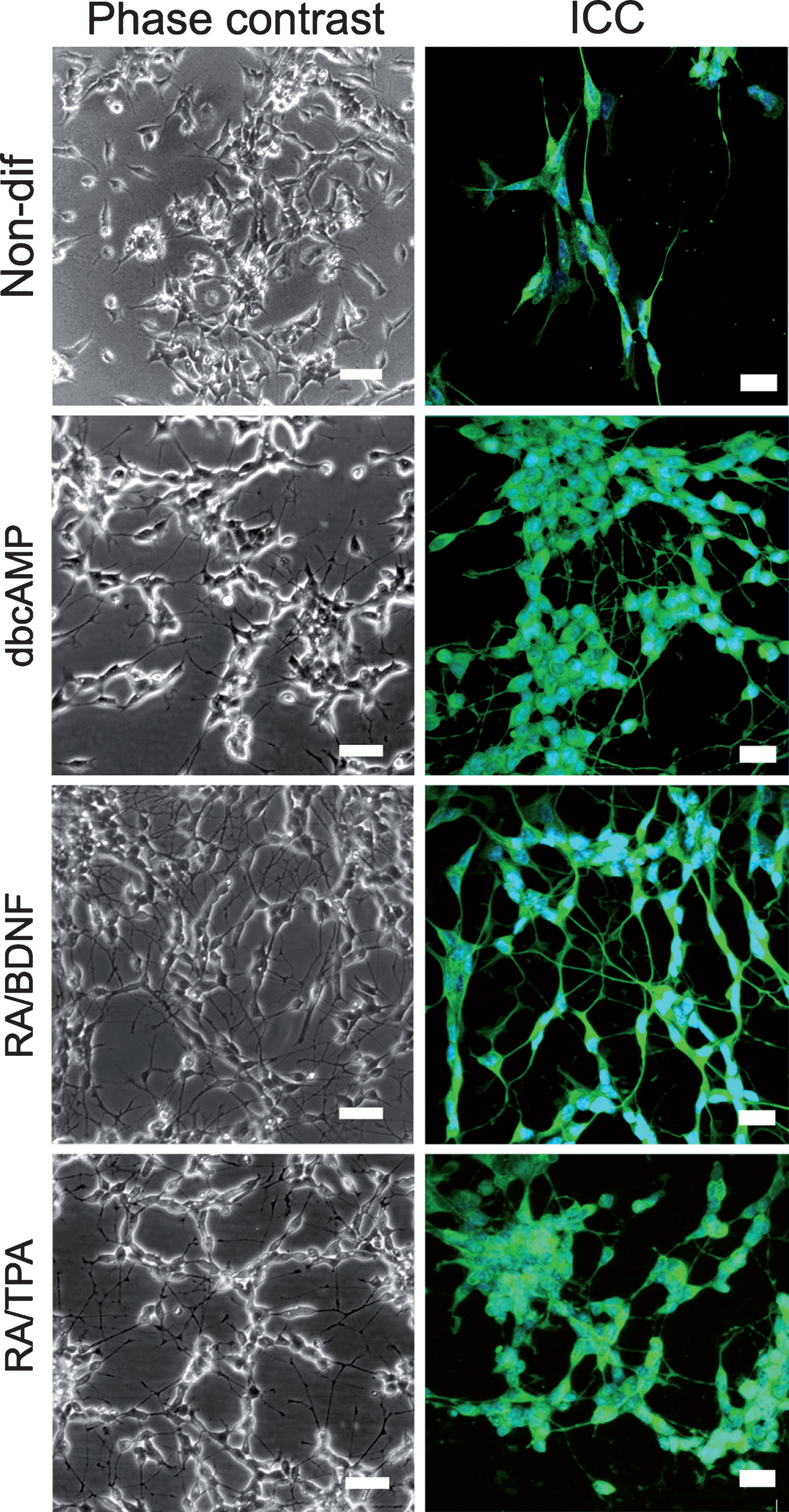
Visualization of the differentiated SH-SY5Y cells. Phase contrast (left column) images obtained with Zeiss Axiovert 200 M, Axiocam MRc5 with 20x A-Plan with Ph2. Scale bar 50 μm. Immunohistochemistry for TUJ (right column) with counterstained nuclei (DAPI) were visualized with the confocal microscope Zeiss Duo 510 META and 63x oil immersion objective. Stack layers with z-interval of 320 nm are presented in sum intensity projection. Brightness and contrast were increased for clarity. Images were edited by using Fiji software. Scale bar 20 μm.
Toxicity of Aβ42 on non-differentiated SH-SY5Y cells
The toxicity of Aβ on non-differentiated SH-SY5Y cells was determined, which served as a reference for comparison of the effects of differentiation (Fig. 2A, B). The results showed that after 48 h, 10 μM Aβ42 was not toxic in viability (WST-1) test (100.2±3.4% compared with negative control) as well as in cell permeability (PI) tests (1.1±0.1 fold increase over control). In contrast, 20 μM Aβ42 exposed significant toxicity by reducing viability to 57.4±8.7%, and increasing PI uptake was 1.8±0.2 fold over control.
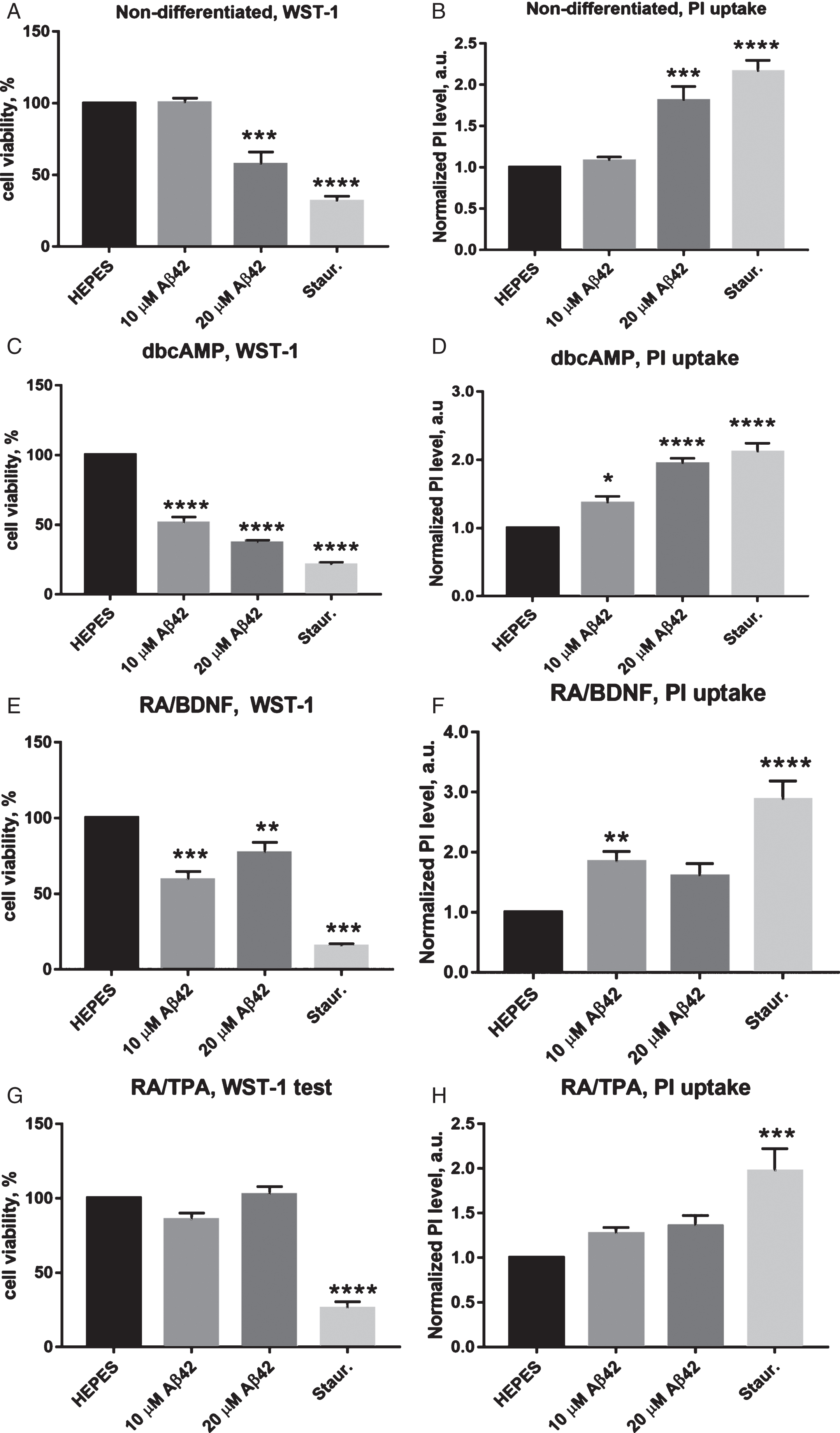
Quantitation of viable and dead cells after 48 h incubation with Aβ42. WST-1 viability test results are on panels A, C, E and G. PI uptake estimation results are on panels B, D, F and H. Negative control: HEPES buffer; positive control: staurosporine. The figure displays the mean±SEM; n = 4 for non-differentiated cells; n = 4 for dbcAMP cells; n = 10 for RA/BDNF cells; n = 6 for RA/TPA cells. ****p < 0.0001; ***p < 0.0002; **p < 0.0021; *p < 0.0489. One-way ANOVA followed by a Dunnett’s multiple comparisons test at the 0.05 level was used for statistical analysis.
Toxicity of Aβ42 on dbcAMP differentiated cells
Incubation with Aβ42 had a statistically significant effect on the viability of dbcAMP differentiated cells after 48 h incubation period as measured with WST-1 assay (Fig. 2C). 10 μM and 20 μM Aβ42 induced respectively a 51.3±4.2% and 37.2±1.7% decrease in the cell viability, which was in both cases statistically significant (p < 0.0001).
PI assay revealed that 20 μM Aβ42 induced a significant (p < 0.0002), almost 2-fold increase in number of permeabilized cells (1.94±0.1 fold increase over control) (Fig. 2D), incubation with 10 μM Aβ42 caused 1.37±0.1 fold increase, which was also statistically significant (p < 0.04).
Toxicity of Aβ42 on RA/BDNF differentiated cells
RA/BDNF differentiated cells were also susceptible to Aβ toxicity; however, 10 μM Aβ42 led to a slightly lower relative viability (59.4±5.3 %, p < 0.0002) as compared to the effect of 20 μM Aβ42 (77.2±6.8%, p < 0.0021) after 48 h incubation (Fig. 2E).
Similarly, treated cells exhibited also increase in amount of permeabilized cells, measured by PI test (Fig. 2F). In case of 10 μM Aβ42, there was a 1.8±0.2 fold increase over control, whereas 20 μM Aβ42 displayed lower 1.6±0.2 fold increase over control.
Toxicity of Aβ42 on RA/TPA differentiated cells
RA/TPA differentiated cells were not sensitive to Aβ42 toxicity. In the WST-1 assay, both peptide concentrations induced only minor changes in relative viability of the cells (Fig. 2G), being 85.7±3.7% in the case of 10 μM Aβ42 and 102.6±5.3% in the case of 20 μM Aβ42. Viability of staurosporine-treated cells as a positive control was decreased significantly to 26.4±4.1% (p < 0.0001) from control.
Quantification of PI-positive cells also did not expose statistically significant toxic effects of Aβ42 on RA/TPA differentiated cells after 48 h incubation (Fig. 2H). In case of 10 μM Aβ42, the number of permeabilized cells was increased 1.3±0.1 fold over control, and in the case of 20 μM Aβ42, a 1.4±0.1 fold increase was observed. The positive control, staurosporine, induced nearly two-fold increase in PI-positive cell count (2.0±0.2).
Pathological changes in cell culture and coverage with Aβ peptide
Pathological changes were detected in all differentiated cultures after 48 h incubation with 10 μM Aβ42 peptide; however, the signs of pathology were completely different from the effects of staurosporine (Supplementary Figure 1). DbcAMP differentiated cells lost all neurites after 48 h incubation even with 10 μM Aβ42 (Supplementary Figure 1F, J). In the case of RA/TPA differentiated cells, which were most resistant to amyloid, we observed a relatively small increase in the amount of dead cells after 48 h incubation even with 20 μM Aβ42; regardless, neurite fragmentation was seen in the case of both 10 and 20 μM Aβ42 as compared with the negative control (Supplementary Figure 1H, L). Both effects were substantially larger in the case of RA/BDNF differentiated cells (Supplementary Figure 1G, K). It is known that Aβ interacts with the plasma membrane and there is also evidence for localization of Aβ42 on the cell membranes in the brain of AD patients [18]. As distinct differentiation methods revealed different sensitivities to Aβ42, the next step was to evaluate the differences in the distribution of the Aβ42 peptide in the cell cultures. For these experiments, cells were stained for AβPP/Aβ and synaptophysin 1 after the treatment with 10 μM Aβ42 for 24 h. Synaptophysin 1 is a major synaptic vesicle membrane protein, whose expression is a broad-range marker of neural and neuroendocrine cells including neuroblastoma cells [19]. By means of ICC, we detected that in case of all cell types studied (non-differentiated cells and three differentiated cell cultures), the cell bodies and neurites were too large extent covered with the Aβ42 peptide aggregates (Fig. 3), whereas Aβ aggregates were also present in the intracellular space (Fig. 3B, G, L, Q, white arrows).
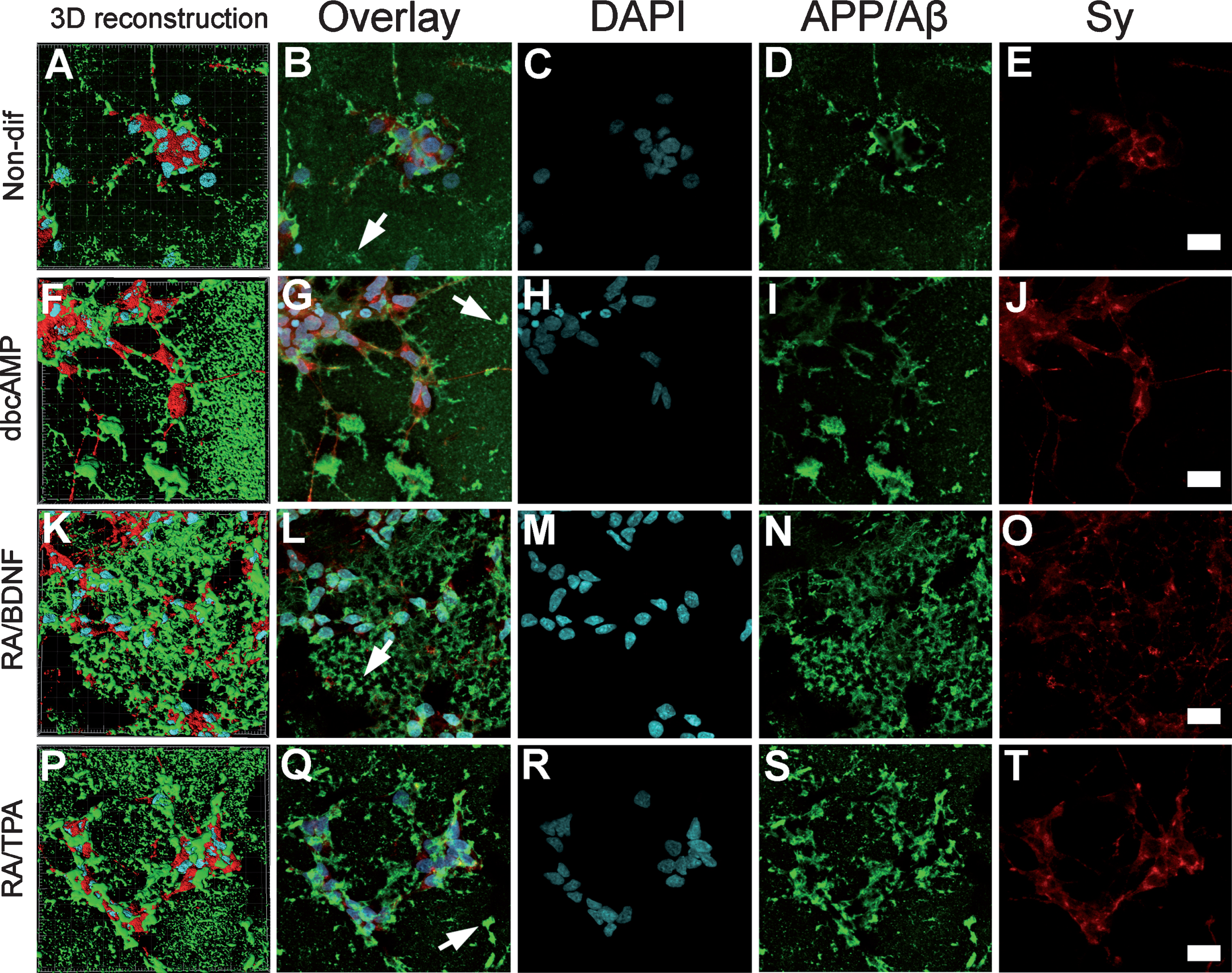
Distribution of Aβ in cell cultures after 24 h incubation. Immunocytochemistry of 10 μM Aβ42-treated SH-SY5Y cells for APP/Aβ (panels D, I, N, and S) and Sy (panels E, J, O, and T) with DAPI counterstained nuclei (panels C, H, M, and R) visualized with the confocal microscope Zeiss Duo 510 META and 63x oil immersion objective. White arrows (panels B, G, L, and Q) indicate the aggregates in the intracellular space. Stack layers with z-interval of 320 nm presented in sum intensity projection. Three-dimensional reconstruction of stacks (panels A, F, K, and P) generated with Imaris software. Brightness and contrast were increased for clarity. Images were edited by using Fiji software. Scale bar 20 μm.
DISCUSSION
To expand our understanding of the neurotoxicity of amyloid peptides in AD, we compared the effects of Aβ42 on human neuroblastoma cell line SH-SY5Y differentiated toward more neuron-like morphology, exposing different biochemical phenotypes [20]. Three different treatments included: 1) dbcAMP, 2) RA followed by BDNF, and 3) RA followed by TPA, which supposedly induce noradrenergic [9], cholinergic [14], and dopaminergic [7] phenotypes, respectively. In all cases, differentiation induced the outgrowth of βIII-tubulin positive networked neurites.
One of the most important factors in Aβ toxicity studies is peptide pre-treatment and preparation for cell culture experiments. It has been shown that the origin of the Aβ peptide [21] as well as the protocol of the preparation of the peptide before applying it to the cells can affect the results [22, 23]. Therefore, comparative studies of Aβ toxicity should be performed using strictly standardized protocols of peptide preparation. In the current study, we used freshly HFIP- treated and alkali-solubilized recombinant Aβ42, which minimizes the amount of preformed fibrillary seeds present in the initial solution. In our previous study with RA/BDNF differentiated SH-SY5Y cells, we used a slightly different Aβ dissolving protocol solubilizing the HFIP-pretreated peptide at neutral pH and observed a lower toxic effect, especially in PI assay [24]. In many studies, amyloid peptide is pre-treated with organic solvents before toxicity experiments in order to get oligomers [25, 26] or protofibrils [27, 28] with enhanced toxicity. However, since an increasing amount of evidence points to the decisive role of intermediate fibrillization species in the cellular toxicity [22], we suggest that toxicity of the peptide in the state of active fibrillization can be better model for the in vivo toxicity of the peptide than artificially generated formulations.
By comparing the toxicity of similarly pre-treated Aβ42 peptide toward differently prepared SH-SY5Y cells, we established that dbcAMP treatment toward noradrenergic phenotype and RA/BDNF treatment toward cholinergic phenotype increase the susceptibility of cells to the toxicity of Aβ42 (Tables 1 and 2). In the study with rat primary neuronal cell cultures, it has been demonstrated that Aβ is more toxic to noradrenergic neurons in comparison with cholinergic neurons [29], similarly to our results with human derived cell line. It can also be speculated that the higher susceptibility to Aβ42 peptide might be caused by the dbcAMP-induced overexpression of AβPP [15], which proteolytic processing yields Aβ peptides and may enhance the toxic effects endogenously. Surprisingly, in the case of RA/BDNF differentiated cells the Aβ42 peptide at 10 μM concentration was more toxic than at 20 μM level. The reason for this unordinary concentration dependence is not clear; however, it can be hypothesized that as 20 μM peptide fibrillizes faster, then the exposure time of cells to the toxic intermediate species crucial for Aβ-induced neurotoxicity [22, 24] is shorter. Significant sensitivity of RA/BDNF cells toward amyloid can also be enhanced by increased expression of AβPP, showed by Holback et al [30], similarly to dbcAMP- differentiated cells; however, the irregular concentration effect on this type of cells need further investigation. RA/TPA differentiated dopaminergic SH-SY5Y cells were almost resistant to Aβ42 toxicity. Such cells are also more resistant to other toxic compounds like 6-OHDA [17], which is used to destroy dopaminergic neurons by different mechanisms as compared to Aβ. Moreover, TPA treatment had protective effect against Aβ toxicity also in case of hippocampal primary neurons, which is assumingly mediated by activation of protein kinase C [31], known to support cell survival [32]. Thus, RA/TPA differentiated SH-SY5Y culture is characterized by high resistance against Aβ-mediated toxicity applicable for studies of the underlying mechanisms of tolerance.
Comparative table of cell viability WST-1 test results
The table displays the mean±SEM; n = 4 for non-differentiated cells; n = 4 for dbcAMP cells; n = 10 for RA/BDNF cells; n = 6 for RA/TPA cells.
Comparative table of PI test results
The table displays the mean±SEM; n = 4 for non-differentiated cells; n = 4 for dbcAMP cells; n = 10 for RA/BDNF cells; n = 6 for RA/TPA cells.
In parallel with toxicological studies, we also monitored distribution of extracellularly applied Aβ42 in cell cultures by ICC. The visualized pattern of peptide distribution on cell bodies and neurites in different cultures was generally similar. We expected that the increased resistance of RA/TPA-differentiated cells toward Aβ might be associated with decreased peptide association with cell membranes, which composition might differ from other cells studied. It is known that the composition of cell membranes is crucial for seeding of Aβ peptide aggregation [33, 34] and involved also in cell death signaling [35]. However, our assumption was not confirmed, as we did not find any correlations between the extent of Aβ distribution in the studied cell cultures and the peptide toxicity, which indicates that the reasons of RA/TPA-differentiated cell resistance toward Aβ toxicity need further clarification.
To conclude, we have generated three neuron-like cellular models of human origin by differentiation of SH-SY5Y cells according to different prescribed protocols, which possess several advantages over non-differentiated cell line model, and compared the toxic effects of Aβ42 peptide on these systems. We have found that RA/TPA differentiated cells SH-SY5Y cells were almost resistant against toxicity of Aβ. DbcAMP-differentiated and RA-BDNF-differentiated SH-SH5Y cells might be useful for further studies of the mechanisms of Aβ toxicity as well as in the screening of compounds protecting the neuronal cells from the toxic effects of Aβ. DbcAMP differentiation should be preferred because of the normal concentration dependence; RA/TPA differentiated cells can be used for unravelling the mechanisms of Aβ tolerance, which might contribute to better understanding of the neurodegeneration and neuroprotection in case of AD.