
Abstract
Background:
Rivastigmine is a cholinesterase inhibitor, approved for the treatment of mild-to-moderate dementia of Alzheimer’s type.
Objective:
To explore the efficacy and safety of the maximal tolerated dose of rivastigmine capsules in Chinese patients with mild-to-moderate Alzheimer’s disease (AD).
Methods:
The study was a multicenter, open-label, single-arm, phase IV clinical study in mild-to-moderate drug-naïve AD patients treated with rivastigmine capsules. The primary endpoint was the changes in the total scores of Alzheimer’s Disease Assessment Scale-Cognitive subscale (ADAS-Cog) from baseline to week 16. Secondary endpoints included changes in the scores of the following assessment scales and safety: Alzheimer’s Disease Cooperative Study; Activities of Daily Living; Mini-Mental Status Examination (MMSE); Neuropsychiatry Index (NPI), and Caregiver Burden Inventory.
Results:
222 patients were enrolled. Of these, 136 (75.1%) patients received and maintained the effective dose (≥6 mg/d) of rivastigmine for at least 4 weeks. The ADAS-Cog scale score improved in rivastigmine-treated patients at week 16 compared with baseline (p < 0.001) by 2.0 (95% CI: –3.0 to –1.1) points, which met the pre-defined superiority criteria. NPI-10 and NPI-12 scores improved by 3.6 and 4.0 points at week 16 (p = 0.001, p < 0.001), respectively. A total of 107 patients (59.1%) experienced adverse effects (AEs) during the study; common AEs included nausea (20.5%), vomiting (16.6%), anorexia (7.8%), dizziness (7.7%), and diarrhea (7.2%).
Conclusion:
This was the first phase IV study on rivastigmine in mainland China. The study preliminarily demonstrated that rivastigmine capsules showed good tolerability and efficacy in mild-to-moderate AD patients with the maximal tolerated dose.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease, and the main clinical manifestations include cognitive impairment, linguistic incompetence, visuospatial dysfunction, disabilities in social activities and behavior, and psychological symptoms [1]. The incidence of AD tends to increase in older subjects [2]. Epidemiologic data shows that there are about 8 million people affected by AD in China, thus causing a heavy burden of AD costs [3]. Etiologically, it is hypothesized that AD involves abnormal amyloid-β (Aβ) protein metabolism, tau protein hyperphosphorylation [4], and glutamate excitotoxicity [5]; the primary pathological changes involve cholinergic nervous pathways radiating from the protocerebral basilar part to the cerebral cortex and hippocampus [6, 7]. Cholinergic pathways are relevant to concentration, memory, learning ability, and other cognitive processes. Cholinesterase inhibitors are the mainstay symptomatic treatments for AD recommended by the Federation of European Neuroscience Societies (FENS) and the National Institute for Health and Clinical Excellence (NICE) [8, 9]. Multiple studies have revealed that cholinesterase inhibitors can improve cognitive symptoms, neuropsychiatric symptoms, and activities of daily living [10–13].
Rivastigmine capsules were approved in 2000 by the NMPA (National Medical Products Administration) for treating mild-to-moderate AD [14, 15]. Rivastigmine is a pseudo-irreversible cholinesterase inhibitor with dual specificity for acetylcholinesterase and butyrylcholinesterase [7, 11]. It is metabolized to an inactive metabolite (NAP-226-90) with little or no involvement of the hepatic cytochrome P450 system [16]. The lack of involvement of the cytochrome P450 system means that rivastigmine has fewer clinically relevant drug-drug interactions in the elderly population likely to be receiving multiple concomitant medications for numerous comorbidities [15, 16].
Clinical data has confirmed that patients treated with rivastigmine benefit more with higher dosages [15, 17]. One retrospective analysis of four randomized, 26-week, double-blind, placebo-controlled trials assessed the tolerability, safety and efficacy of oral rivastigmine (capsules) in patients with mild-to-moderately severe AD [18]. The study involved 878, 1,053, and 863 patients, who were randomized to 1 to 4 mg/d rivastigmine, 6 to 12 mg/d rivastigmine, and placebo groups, respectively. Rivastigmine-treated groups had greater changes on total ADAS-cog and memory domain scores (p = 0.0001) compared to placebo groups; patients treated with rivastigmine at doses of 6-12 mg/d had clinically relevant and statistically significant improvements in cognitive and global assessments, as well as in activities of daily living [18]. In the long-term efficacy study, where patients were treated with rivastigmine for 2 years, the mean Mini-Mental State Examination (MMSE) score declined by 1.84 points. In patients with mild-to-moderate AD who received no active medical treatment, the MMSE scores declined by 2–4 points each year [19], thus suggesting that rivastigmine may delay cognitive decline by at least 1 year [19].
However, gastrointestinal reactions including nausea, vomiting, diarrhea, and anorexia were common adverse effects (AEs) of cholinesterase inhibitors, particularly during the dose escalation period, which could impede treatment with higher dosages [11]. There is also a lack of post-market evidence in the Chinese population that demonstrates the efficacy and safety of rivastigmine capsules.
To address this issue, the current study was designed to investigate the clinical efficacy and safety of the individualized maximal tolerated dose of rivastigmine capsules in Chinese patients with mild-to-moderate AD. The study aimed to provide evidence of the clinical efficacy of rivastigmine capsules, i.e., the improvement in the cognitive score based on Alzheimer’s disease assessment scale-cognitive subscale (ADAS-cog), and to explore its tolerability and safety in the target population.
METHODS
Patients
Between August 2014 and September 2015, 222 patients with probable mild-to-moderate AD were screened, and 187 patients were enrolled from 13 study centers, including Xuanwu Hospital, Capital Medical University, China-Japan Friendship Hospital, Peking University Third Hospital, and other Chinese hospitals. Patients aged 50–85 years with probable AD (according to the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s disease and Related Disorders Association [NINCDS/ADRDA] criteria). The primary inclusion criteria were as follows: 50–85 years old (inclusive); male or female; MMSE scores of 10–26 points (inclusive); diagnosed with AD according to the criteria of the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV); clinically diagnosed with probable AD according to NINCDS-ADRDA standards; patients who had never received cholinesterase inhibitors (ChEI) or memantine; patients for whom there had been head scans (CT or MRI) imaging evidence supporting the diagnosis of AD, and these head scans were performed in the past one year or before enrollment; patients whose disease status was stable; informed consent form signed by the patient or his/her legal guardian.
The main exclusion criteria included patients with severe AD; dementia symptoms caused by other diseases such as cerebrovascular disease or vitamin B12 or folate deficiency, hypothyroidism, normal pressure hydrocephalus, severe traumatic brain injury, alcoholic encephalopathy, or other neurodegenerative diseases (Parkinson’s disease, frontotemporal dementia, Huntington’s disease, etc.); a modified Hachinski ischemic score (MHIS)>4; and severe disability making the patient incapable of meeting the clinical research requirements (e.g., blindness, severe deafness leading to an inability to communicate, or severe language disorders rendering the patient unable to communicate). In addition, patients who had previously received ChEI or memantine were excluded from the study.
All patients signed an informed consent form which was approved by the Institutional Review Board (IRB)/Independent Ethics Committee (IEC)/Research Ethics Committee (REC) before inclusion in the study. The design, performance, and reporting of the study conformed to the ethical principles specified in the ICH-GCP (International Conference on Harmonization of Good Clinical Practice) and the Declaration of Helsinki.
Study design
This was a 16-week multicenter open-label single-arm study (International Clinical Trial Registration Number: NCT01948791) evaluating the clinical effectiveness of rivastigmine capsules with a maximal tolerated dose for the treatment of mild-to-moderate AD. After a 2-week screening period, patients who met the eligibility criteria were treated according to Table 1. Rivastigmine capsules were initiated at a dose of 1.5 mg bid and gradually up-titrated every 4 weeks in increments of 1.5 mg bid, up to a maximum dose of 6 mg bid by week 12. During the up-titration, 4-weekly visits were conducted. The maximal tolerated dose of capsules was maintained from week 12 to 16 (maintenance phase). In the case of safety or tolerability issues, dose adjustments (interruptions or down-titrations) were implemented. The maintenance dose was defined as the target dose assigned to the treatment group or the maximal tolerated dose for each individual patient. At the end of week 12 (V5) and at the end of week 16 (V6), patients returned to the site for efficacy assessments. At the end of weeks 4, 8, 12, and 16 (V3, V4, V5, and V6), patients returned to receive safety assessments. For the detailed study protocol, see the Supplementary Material.
Dose titration
DL, dose level *The study drug was first administered at the baseline visit; **The maintenance dose is defined as the target dose of 12 mg/d or the maximal tolerated dose of each patient.
Efficacy endpoints
The primary objective of this study was to analyze changes during the period from baseline to week 16 in the total ADAS-Cog score in patients treated with rivastigmine capsules at the maximal tolerated dose. The primary study endpoint (ADAS-Cog scale) is used to evaluate the cognitive function of AD patients. The scale comprises 11 items, namely word recall, following commands, constructional praxis, naming objects/finger, ideational praxis, orientation, word recognition, recall of test instructions, spoken language ability, word-finding difficulty, and comprehension of spoken language. The maximum score is 70 points, and a higher score indicates more severe cognitive impairment. Thus, if the score decreases in points after rivastigmine treatment, there is an improved cognitive function.
Secondary endpoints were the change in the following parameters between baseline and week 16: MMSE, AD Cooperative Study-Activities of Daily Living scale (ADCS-ADL), Neuropsychiatry Index (NPI), and Caregiver Burden Inventory (CBI). The MMSE scale has a maximum score of 30 points, and is used to reflect the overall cognitive function; a lower score indicates more severe cognitive damage. Therefore, an increase in the MMSE score after rivastigmine treatment indicates an improved condition. The ADCS-ADL scale is used by caregivers to evaluate the patients’ ability to perform six basic activities of daily living (BADL) and eight instrumental activities of daily living (IADL). These activities include using the telephone, shopping, doing housework, and writing. The maximum total score on this scale is 78, and a higher score indicates a stronger function; a positive change therefore indicates an improved condition after rivastigmine treatment. The NPI is used to evaluate the neuropsychiatric behavioral symptoms of AD. This index includes 12 neuropsychiatric behavioral symptoms that are common in AD, including delusions, hallucinations, depression, anxiety, euphoria, and anomalous behavior. The NPI is used to evaluate the occurrence of neuropsychiatric behavioral symptoms of patients during a period of approximately 1 month, and the target score (0–12 points) is the product of frequency (0–4 points) and severity (0–3 points) of each item. A maximum score of 12 is given to each symptom, with an overall scale of 0–144 points; a higher score indicates more serious neuropsychiatric behavioral symptoms. Therefore, a reduction in the NPI score indicates an improved condition after rivastigmine treatment. The CBI contains 24 items and is used to evaluate the caregiver burden. Each item is given a score of 0–4 points according to the severity of the burden. The maximum total score is 96 points, and a higher score indicates a heavier caregiver burden; thus, a negative change in the CBI indicates an improved condition after rivastigmine treatment.
The safety endpoints were the percentage of patients reaching and maintaining the effective dose (≥6 mg/d) of rivastigmine for at least 4 weeks; the percentage of patients with AEs or serious AEs (SAEs); dropout rates; any change in weight from the baseline value; pulse rate; systolic and diastolic blood pressure; hematology, hematochemistry, urine and other laboratory examinations.
Statistical analysis
Primary analysis was based on the per-protocol set (PPS), which included patients who had received the study drug at least once and had undergone a baseline evaluation, with at least one evaluation of the primary efficacy variables at week 16. All patients in this set were without major protocol deviations. The intent-to-treat set (ITT) included patients who had received the study drug at least once and had undergone a baseline evaluation and at least one evaluation after treatment to investigate the primary efficacy variables. The safety set (SS) included all patients who had received the study drug at least once and had undergone at least one safety evaluation after baseline evaluation. Statistical analyses were performed using SAS® Version 9.2. For analyses of missing scores/visits based on the ITT population, Last Observation Carried Forward (LOCF) approach was used to impute the missing (total) scores. In addition, changes in the scores from baseline to week 12 were also subjected to statistical analyses. The hypothesis was to test the non-inferiority of post-baseline changes in ADAS-Cog from baseline. If the upper limit of the 95% confidence interval (CI), calculated based on the mean difference between the baseline score and post-baseline score, fell on the left of 1.40, post-baseline non-inferiority could be concluded. If the upper limit of the 95% confidence interval, calculated based on the mean difference between the baseline score and post-baseline score, fell on the left of 0, then superiority could be concluded. The sample size estimation was based on the non-inferiority hypothesis on one sample. Assuming a standard deviation (SD) of 5.38 and a clinical non-inferiority margin of 1.40, 158 patients were needed for enrollment to obtain a 90% power, as estimated at the one-sided significance level of 0.025. Assuming a dropout rate of 15%, the final sample size for this study was 186 subjects. The primary population used for statistical analyses was based on the PPS without imputation of missing data, and it was repeated on the ITT population to examine the sensitivity of the conclusions drawn from the primary analysis. One sample t-tests were used to investigate the changes from baseline to confirm the treatment effect; in addition, the 95% CI of the difference of the mean of treatment were calculated. Safety data were analyzed descriptively based on the SS population. Safety variables were AEs (including SAEs), vital signs, and electrocardiography (ECG). AEs were coded using MedDRA 18.0 terminology. All the AEs experienced on or after the first day of drug administration were analyzed using descriptive statistics, while the other AEs were only flagged and listed instead of being included in the summary tables. All analyses were defined as bilateral tests and the significance level was 0.05 (equivalent to unilateral tests with a 0.025 significance level), which allowed a p value and a bilateral 95% CI to be calculated. Subgroup variables from baseline to post-baseline were compared by a one-sample t-test, after that ANCOVA analysis was used to detect significant differences between the groups.
RESULTS
Patient population
This study recruited 222 subjects from 13 study centers; 187 patients passed the screening, and 151 subjects (68%) completed the study. Of all enrolled patients, 36 (16.2%) subjects discontinued the study before its planned end, including 12 subjects (5.4%) who discontinued due to safety reasons (AEs) and 3 subjects (1.4%) who discontinued due to efficacy dissatisfaction. Details of all the subjects who discontinued the study are shown in Fig. 1 A total of 165 patients received treatment with the study drug at least once and underwent at least one evaluation of the primary efficacy variable (ADAS-Cog) from baseline; these patients were included in the ITT. A total of 181 patients were treated with the study drug at least once and underwent a safety evaluation; these patients were included in the SS. Finally, 151 patients were included in the PPS (Fig. 1).
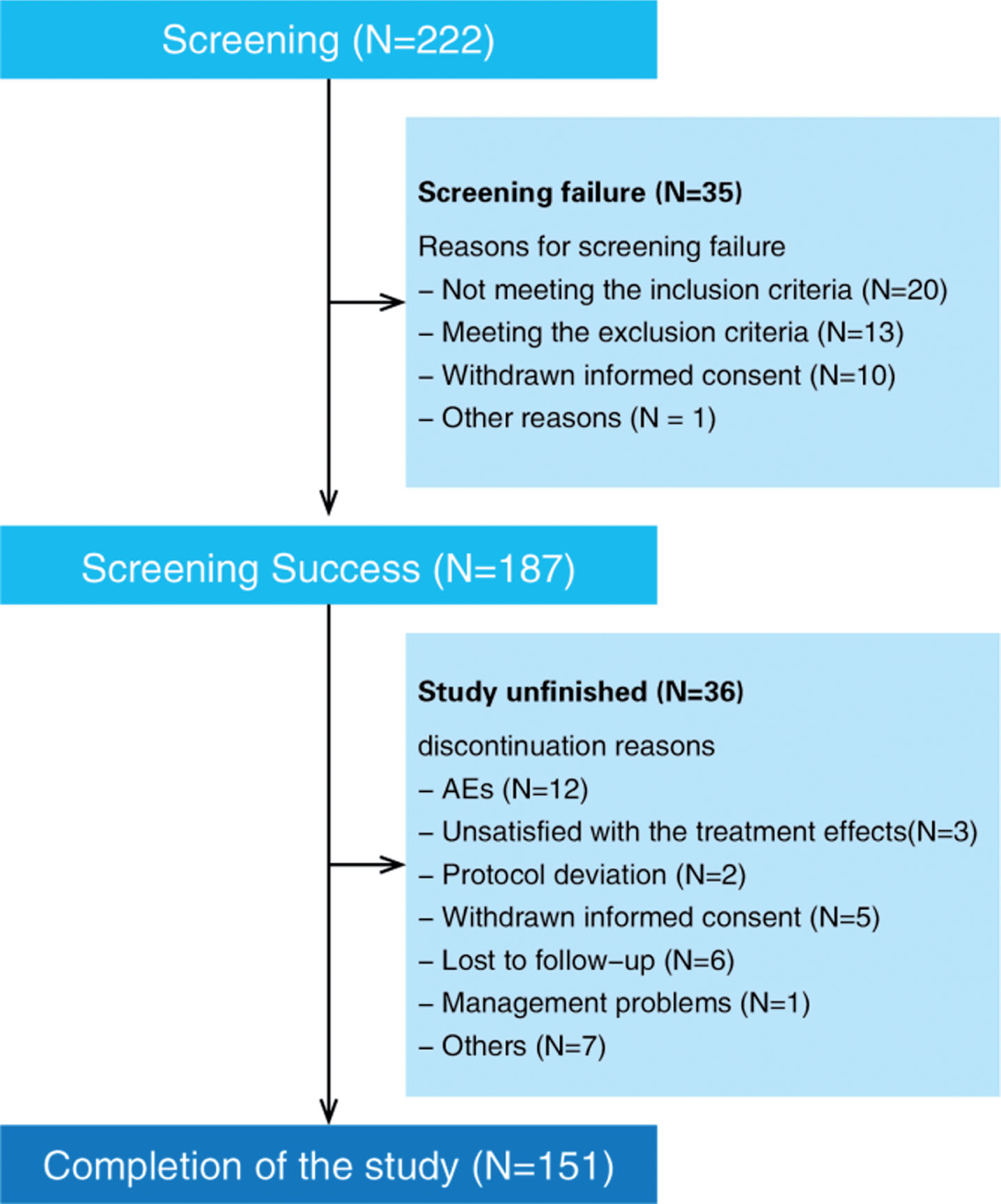
Distribution of study patients. AEs, adverse effects.
The demographic and baseline characteristics of the ITT were as follows: the mean age of the study population was 69.6 years, and 61.2% were female. Overall, 62.4% of patients had comorbidity, including hypertension (33.3%), diabetes (12.1%), lacunar infarction (7.3%), hyperlipidemia (4.8%), and coronary artery disease (4.8%) (Table 2); 21.2% of the patients received concomitant medication during the trial. There were no obvious differences in the demographic data or baseline characteristics between the PPS and the ITT.
Patient demographic data and baseline characteristics (ITT, n = 165)
ITT, intent-to-treat set; SD, standard deviation; MHIS, Hachinski ischemic score; ADAS-Cog, Alzheimer’s disease assessment scale-Cognitive; ADCS-ADL, Alzheimer’s Disease Co-operative Study-Activities of Daily Living Inventory; MMSE, Mini-Mental Status Examination; NPI, Neuropsychiatry Index; CBI, Caregiver Burden Index.
Drug efficacy
In the PPS, there was a statistically significant difference in the change in total ADAS-Cog scale score at week 16 after treatment compared with baseline (p < 0.001). Rivastigmine-treated patients improved by 2.0 (95% CI: –3.0 to –1.1) points from baseline according to the ADAS-Cog scale. The upper limit of the CI was less than both the non-inferiority criteria of 1.4 and the superiority criteria of 0. Therefore, both the pre-defined non-inferiority criteria and the pre-defined superiority criteria were met. For the ITT population, the change in total ADAS-Cog scale score at week 16 after treatment compared to baseline was similar to that in the PPS (Table 3).
Changes in ADAS-Cog, MMSE, ADCS-ADL, NPI-10, NPI-12, CBI scores with rivastigmine treatment from baseline to week 16 and week 12
ADAS-Cog, Alzheimer’s disease assessment scale-Cognitive; ADCS-ADL, Alzheimer’s Disease Co-operative Study-Activities of Daily Living Inventory; MMSE, Mini-Mental Status Examination; NPI, Neuropsychiatry Index; CBI, Caregiver Burden Index; PPS, per-protocol set; ITT-LOCF, intention to treat-last observation carried forward, single sample t-test. aADAS-Cog, NPI-10, NPI-12, and CBI: a negative value indicates a significant improvement in patient performance after treatment compared with baseline; bMMSE and ADCS-ADL: a positive value indicates a significant improvement of patient performance after treatment compared with baseline; *indicates that test results are statistically significant at α=0.05 significance level.
In the PPS, the total MMSE score at week 12 after treatment with rivastigmine improved by 0.6 points (95% CI: 0.1 to 1.0; p = 0.015) compared with baseline, and improved by 0.3 points at week 16 (95% CI: –0.1 to 0.8; p = 0.175). The total ADCS-ADL score numerically improved by 0.4 points (95% CI: –0.8 to 1.5; p = 0.530) and 0.9 points (95% CI: –0.5 to 2.3; p = 0.228) at week 12 and 16 after treatment, respectively. There were no statistically significant differences in the ADCS-ADL score before and after treatment. The change in total NPI-10 score between baseline and week 12 and week 16 after treatment was improved by 2.6 (95% CI: –4.1 to –1.0; p = 0.001) and 3.6 (95% CI: –5.3 to –1.9; p < 0.001), respectively. The change in total NPI-12 score at week 12 and week 16 after treatment from baseline was improved by 2.9 (95% CI: –4.8 to –1.0; p = 0.003) and 4.0 (95% CI: –6.2 to –1.9; p < 0.001), respectively. The change in total CBI from baseline to week 12 and 16 after treatment was improved by 1.2 (95% CI: –2.7 to 0.3; p = 0.114) and 1.4 (95% CI: –3.3 to 0.5; p = 0.140), respectively, but these changes were not statistically significant. The results of the ITT population were similar to those described above (Table 3).
Several items of ADAS-Cog scores in the PPS at week 16 were numerically improved after treatment compared with baseline, specifically naming objects/finger improved by 0.1 (95% CI: –0.2 to –0.0), p = 0.040; recall of test instructions improved by 0.2 (–0.5, –0.0), p = 0.040; word-finding difficulty improved by 0.2 (–0.3, –0.0), p = 0.007; and comprehension of spoken language improved by 0.3 (–0.4, –0.1), p < 0.001. There were few differences in ADCS-ADL items between baseline and treatment, except for two items [patients talking about the latest things (p = 0.010) and reading magazines, newspapers, or books (p = 0.004)], which indicated an improved condition after rivastigmine treatment at week 16 compared with baseline. In the NPI, the score of several items improved at both weeks 12 and 16 compared with baseline, including delusions (p = 0.028), agitation/aggression (p = 0.002), depression (p = 0.024), apathy/indifference (p = 0.002), irritability (p = 0.012), and abnormal behavior (p < 0.001).
Safety evaluation
Drug exposure
Among the 187 patients who passed the screening assessment, 181 patients received treatment with rivastigmine according to the provided protocol, while data concerning drug treatment was unavailable in the remaining 6 patients because of loss of follow-up. A total of 136 (75.1%) patients received and maintained the effective dose (≥6 mg/d) of rivastigmine for at least 4 weeks, and 105 (58.1%) patients received the maximal tolerated dose DL4 (6 mg/dose, bid) during the maintenance period.
Adverse events
In the trial, 109 patients (62.2%) experienced at least one AE; 107 (59.1%) patients experienced AEs during the treatment period, and 86 (47.5%) AEs were related to drugs. Most AEs were mild-to-moderate gastrointestinal reactions, such as nausea (20.5%), vomiting (16.6%), anorexia (7.8%), dizziness (7.7%), and diarrhea (7.2%) (Fig. 2). Overall, 7 (3.9%) patients experienced at least one SAE, which all occurred during the treatment period and no deaths occurred. The investigator judged that only one SAE (dizziness) could have been rivastigmine-related. A total of 12 patients (6.6%) dropped out of the study due to AEs, half of which were due to gastrointestinal reactions. Laboratory examinations, vital signs, ECG, physical examinations, and neurological examinations revealed no obvious safety problems.
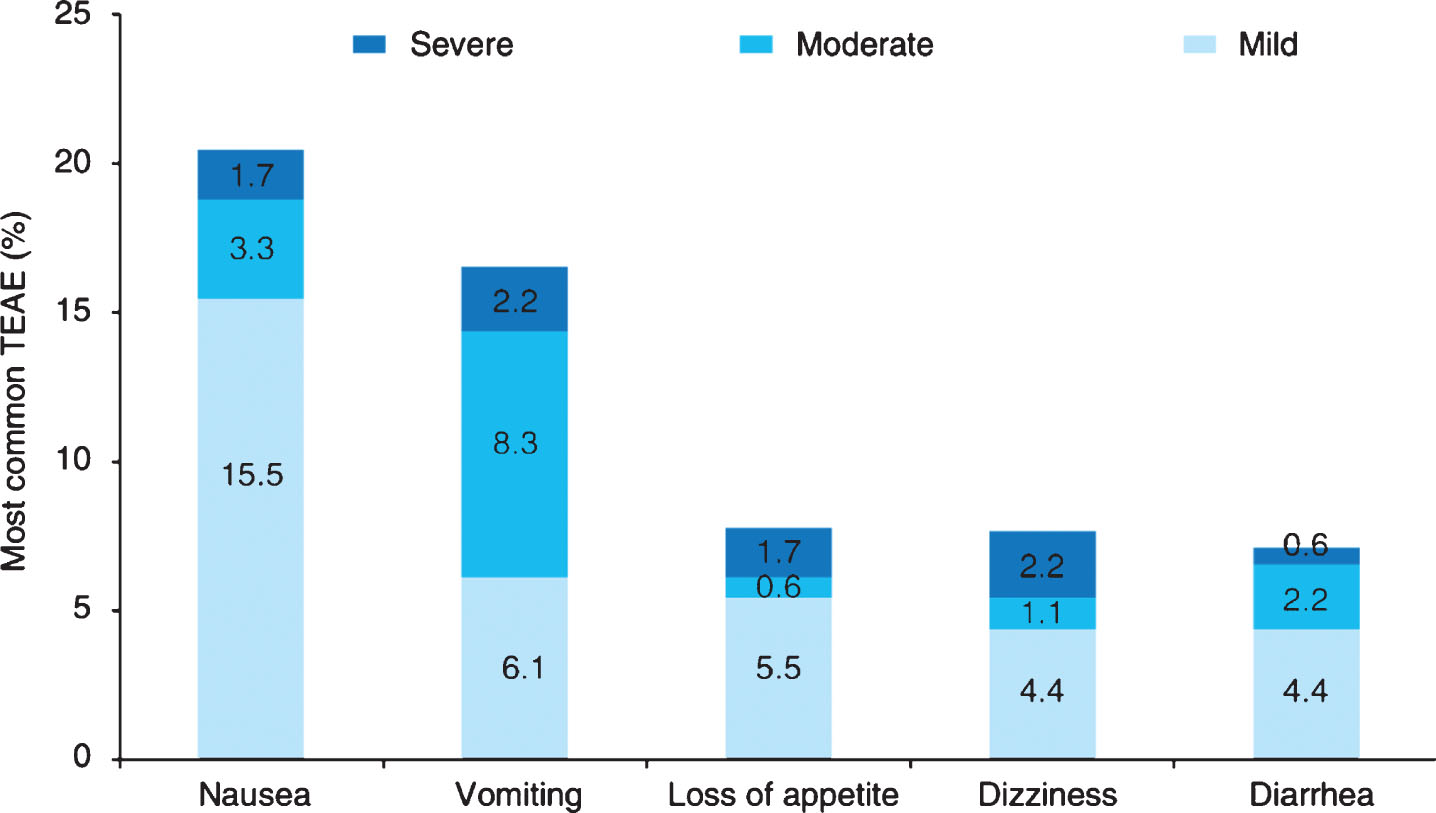
Overall incidence of treatment emergent adverse event (TEAE) in the Safety Set (n = 181).
DISCUSSION
This is the first phase IV clinical study on the cholinesterase inhibitor rivastigmine performed in order to explore the efficacy and safety of the maximal tolerated dose in a Chinese population. The initial dose used was 3 mg/d, which was then gradually up-titrated to the target dose of 12 mg/d according to patient tolerance. Results demonstrated that rivastigmine capsules showed good tolerability and efficacy with the maximal tolerated dose for treating mild-to-moderate AD patients.
Rivastigmine treatment was well-tolerated and improved the cognitive function and neuropsychiatric behavior of patients. For the primary endpoints in the PPS, the total ADAS-Cog scale score at week 16 improved by two points compared with baseline (p < 0.001); the total ADAS-Cog score also improved after week 16 of treatment in the ITT-LOCF. In the PPS, improvements in naming objects/finger, recall of test instructions, word-finding difficulty, and comprehension of spoken language were statistically significantly different compared with baseline. In 2004, one real-world study showed that AD patients treated with rivastigmine for 6 months exhibited a decline in total ADAS-Cog score by 1.29 points from baseline, which meant that cognition had improved significantly [20]. Other studies reported that AD patients who responded inadequately to treatment with cholinesterase inhibitors could benefit from treatment with the AChE-BuChE inhibitor rivastigmine [21, 22].
Common neuropsychiatric behavioral symptoms of AD patients include depression, apathy, agitation and aggression, anxiety, sleep disturbance, irritability, appetite changes, aberrant motor behavior, delusions, disinhibition, hallucinations, and euphoria [23]. It is hypothesized that high expression levels of BuChE, which are mainly distributed in the amygdala and hippocampus in AD patients could lead to the decline of acetylcholine, which subsequently affects cognition and memory [7, 24–26]. Furthermore, BuChE in the frontoparietal region of AD patients may cause hallucinations and symptoms of agitation via acetylcholine hydrolysis [7, 26–28]. Therefore, inhibiting BuChE expression could improve symptoms of hallucinations and agitation in AD patients. In the current study, the total NPI scale (NPI-10 plus NPI-12) score at weeks 12 and 16 after treatment in the PPS decreased significantly compared with baseline. In addition, the scores of individual NPI items (delusions, agitation/aggression, depression, apathy/indifference, irritability, abnormal behavior) were also significantly lower at week 16 than baseline. This suggests that neuropsychiatric behavioral symptoms of AD patients were improved by treatment with rivastigmine.
In a previous study performed in patients who were untreated or treated with placebo, the ADCS-ADL score declined by 11.5 points every year [29]. By contrast, the total ADCS-ADL scores of patients in the PPS increased by 0.4 and 0.9 points at weeks 12 and 16, respectively, after rivastigmine treatment compared with baseline in the current study. There was also a significant difference in two items of the ADCS-ADL (patients talking about the latest things and patients reading magazines, newspapers, or books) at week 16 compared with baseline. This suggests that rivastigmine could delay the decline in some of the daily living activities in AD patients.
In the current study, the safety results suggested that rivastigmine doses above 6 mg/d and up to the maximum approved dose (12 mg/d) were well-tolerated in general. The dose was modified according to the subject’s safety and tolerability profile in the study, and 75.1% of the patients in this study reached and maintained the effective dose of rivastigmine (≥6 mg/d) for at least 4 weeks, which was higher than the dose commonly used in most patients in mainland China (1–3 mg/d). At week 16 of rivastigmine treatment, the treatment-emergent AEs in the current study were primarily mild-to-moderate gastrointestinal adverse reactions, such as nausea (20.5%), vomiting (16.6%), and anorexia (7.8%), most of which required no additional treatment. A prospective, randomized, open-label clinical trial in Italy showed after 12-week treatment of rivastigmine, most of adverse events, such as nausea (6.5%), vomiting (6.5%), headache (2.2%), anorexia (0), were transient and mild-to-moderate intensity, and resolved spontaneously without intervention [30]. Compared with the results in this study, the adverse event rate in Caucasians appeared lower than that in Chinese. The post-hoc analysis of a double-blind trial in patients with AD showed there was an association between a higher adverse event rate and low body weight among patients receiving rivastigmine capsules; however, among patients receiving rivastigmine patch, lower body weight was not associated with a higher adverse event rate [31]. Given that the body weight in Chinese is generally lower than that of Caucasians, it seemed reasonable to explain that the adverse event rate in Chinese treated with rivastigmine capsules was higher than that in Caucasians. The incidence of gastrointestinal side effects with rivastigmine has been associated with high maximum plasma concentrations (C max) and short times to C max (T max). Studies have shown that compared with rivastigmine capsules, rivastigmine transdermal patches have a low C max and a long T max. During the administration period of these patches, the patient’s plasma concentration has been shown to be stable, with no first-pass effects and less adverse reactions [30]. The emergence of rivastigmine transdermal patches may improve the tolerability of rivastigmine and enable more patients to receive higher dosages [32].
There are several limitations in the design of this study. It was a single-arm study with no blinding method or placebo control, which may cause bias in the results. Since the study was performed for only 16 weeks, the short study period may cause heterogeneity and prevent patients from achieving higher tolerated dosages. Moreover, due to the development of the upgraded rivastigmine transdermal patches, which have improved tolerability, it would be worthwhile to investigate the real-world effectiveness with maximal tolerated dose. Further limitations are the low sample size and reduced statistical power, and the risk of credence of statistical analysis of single items in various domains that could limit the reproducibility and significance of these analyses. Thus, a more well-designed long-term study may be needed to observe further effects.
Conclusions
In conclusion, this is the first phase IV clinical study on rivastigmine in patients from mainland China. The results showed that treatment with rivastigmine capsules for 16 weeks significantly improved the cognitive symptoms and neuropsychiatric behavior of Chinese patients with mild-to-moderate AD, and the treatment was well-tolerated even up to the dosage of 12 mg per day. Developing the rivastigmine transdermal patch may compensate for the weaknesses of the safety profile of rivastigmine capsules, and could be a new treatment option in China.
Footnotes
ACKNOWLEDGMENTS
This study was sponsored by Novartis Pharmaceutical China. All the authors performed the trial. A writing committee prepared an initial draft of the manuscript, based on a report provided by Novartis; JPJ edited, read and approved the final manuscript, and all authors contributed to its finalization by an interactive review. In addition, the authors had full access to all of the data and that the decision on what to publish was made independent of sponsor.