
Abstract
Hereditary cerebral amyloid angiopathies (CAA) are rare disorders of early onset and severe course. We describe a 47-year-old patient with Iowa-type amyloid precursor protein (APP) mutation-related hereditary CAA that manifested with concomitant lobar hemorrhage and venous sinus thrombosis. To analyze the cerebral amyloid-β burden, an amyloid-PET was performed, demonstrating low cortical retention except for the calcarine cortex. High amyloid retention was also found in the thalamus and pallidum. The co-occurrence of CAA and venous thrombosis has not been previously reported in Iowa CAA and its mechanism is yet to be elucidated. Low cortical florbetapir-PET uptake does not rule out CAA in young patients, who may benefit from genetic testing to reach diagnosis when suspicion is strong.
INTRODUCTION
Cerebral amyloid angiopathy (CAA) is a group of disorders that share deposition of amyloid fibrils in the walls of cortical and leptomeningeal capillaries and arterioles. It is associated with lobar hemorrhages, cortical microhemorrhages, microinfarcts, leukoencephalopathy, and cognitive decline [1, 2]. While sporadic CAA is common among the elderly, hereditary CAA comprises rare entities presenting as autosomal dominant disorders of earlier onset and more severe clinical manifestations [3]. Mutations of the amyloid precursor protein (APP) gene, located in the coding region of the amyloid-β (Aβ) peptide, are primarily associated with hereditary CAA [4].
CAA definitive diagnosis requires full postmortem histological examination according to Boston criteria [5]. These criteria can provide in vivo diagnosis with varying degrees of certainty, attending to imaging features. In recent years, amyloid-PET imaging has emerged as a tool to quantify the burden and distribution of cerebrovascular Aβ deposition in CAA [6, 7]. It has proven good ability to differentiate intracerebral hematoma (ICH) related to CAA or to arterial hypertension, as well as CAA from Alzheimer’s disease (AD) and non-CAA microangiopathies [8].
We describe a case of Iowa-type APP mutation-related hereditary CAA that manifested with symptomatic ICH and cerebral venous thrombosis (CVT). We analyzed the brain Aβ burden with florbetapir-PET finding unexpected results.
MATERIAL AND METHODS
A previously healthy 47-year-old man first attended the emergency department due to sudden mild motor aphasia preceded by a 2 day-long worsening headache. An urgent computed tomography (CT) scan revealed a large left parieto-temporal ICH. Vascular imaging techniques (CT angiography and conventional arteriography) did not show vascular abnormalities. Further magnetic resonance imaging (MRI) disclosed a left sigmoid sinus thrombosis adjacent to the hematoma which extended to the origin of the internal jugular vein, as well as many subcortical microhemorrhages and superficial siderosis in both cerebral hemispheres (Fig. 1a-c). No cerebral calcifications were found. A study was conducted to determine possible conditions associated with CVT. Autoimmune disorders and hereditary thrombophilias were ruled out. There was no history of head trauma, previous venous thrombotic events or other risk factors for CVT identifiable with laboratory tests such as anemia, liver disease, kidney disease, and inflammatory conditions.
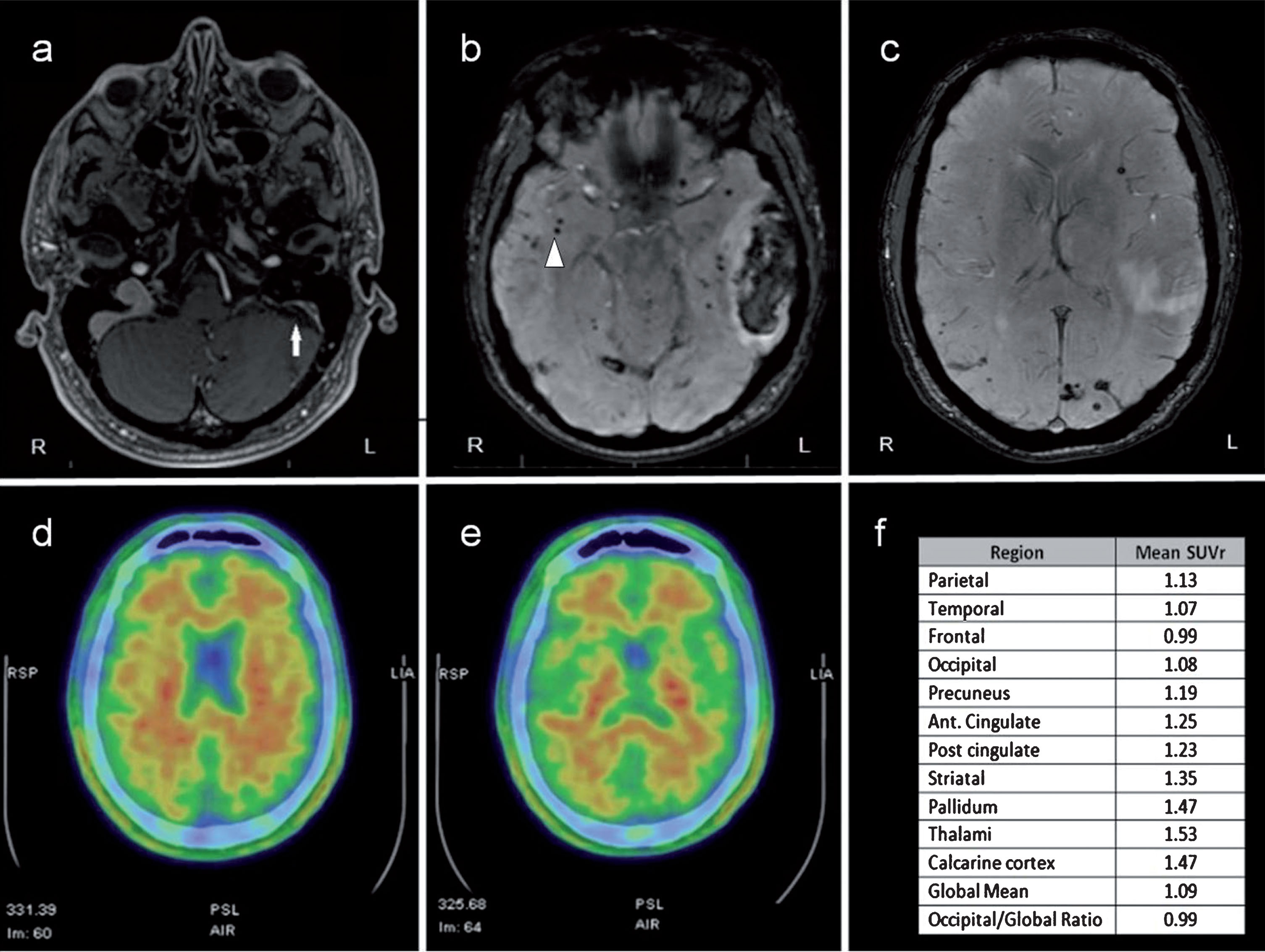
Neuroimaging characteristics of the patient. a) Contrast-enhanced T1-weighted MRI showed left sigmoid sinus and jugular vein thrombosis (white arrow). b, c) SWI MRI showed multiple subcortical microhemorrhages (white arrowhead) in both cerebral hemispheres and left parieto-temporal hematoma. d, e) 18F-florbetapir PET study informed as negative after binary visual reading analysis. f) 18F-florbetapir PET regional quantification. Data are shown as the mean of left and right hemispheres. SUVr, standard uptake value ratio.
Detailed family history was obtained (Fig. 2). His father had suffered many cerebrovascular events of unknown mechanism and imprecise timing, and he was diagnosed in the 7th decade of life with mixed degenerative-vascular dementia. His MRI showed diffuse leukoencephalopathy and features that suggested the presence of CAA. He died at age 75 due to cerebral hemorrhage. His elder brother underwent an MRI study at age 47, which showed leukoencephalopathy. No susceptibility-weighted nor gradient-echo sequences were obtained. His sister’s history was unremarkable. Cases of cerebrovascular events of unknown mechanism in his father’s family were reported, but additional information could not be retrieved. Finally, there was no evidence of venous thrombotic events in other family members.
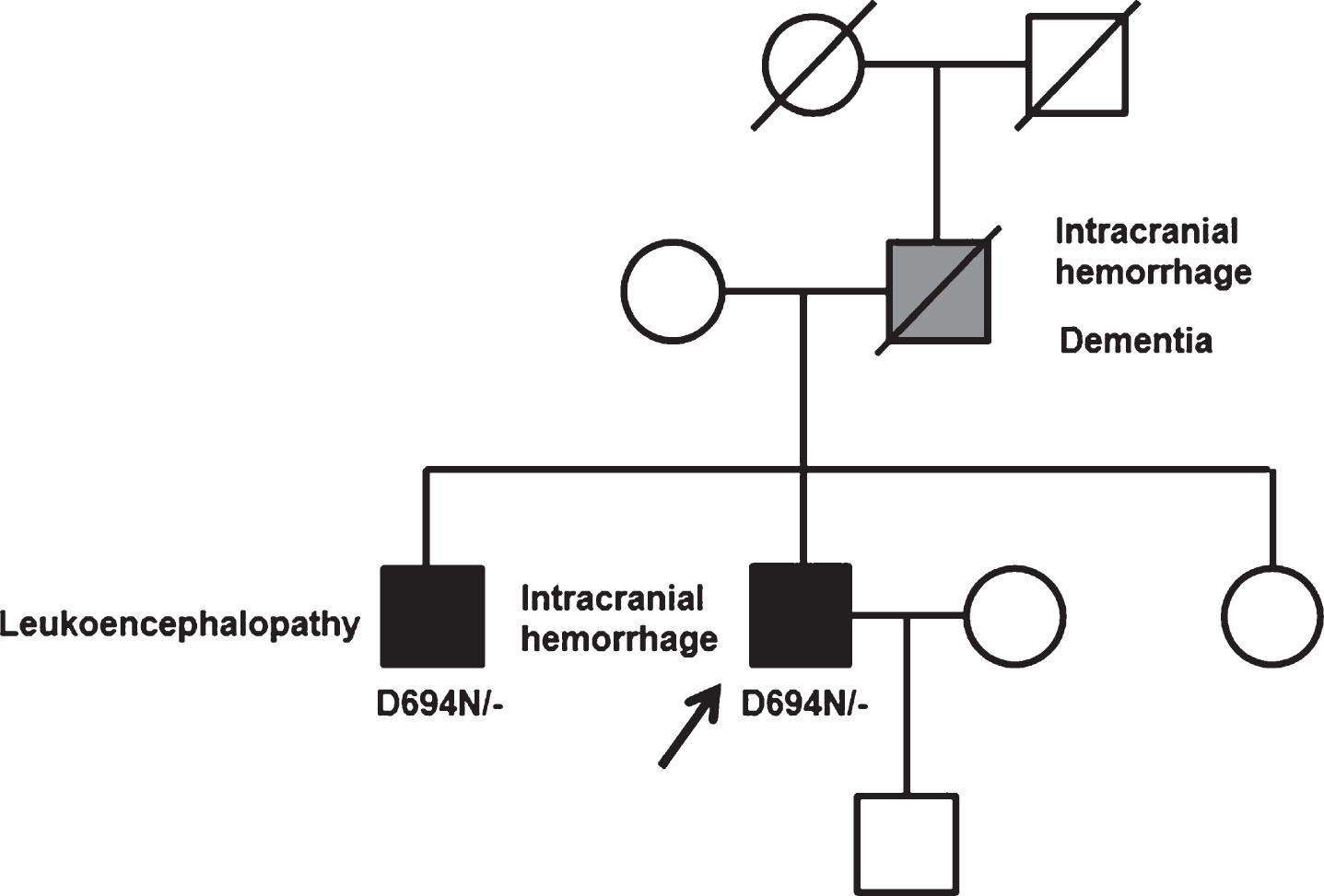
Pedigree of the family carrying Iowa APP mutation. Pedigree showing as black symbols the two definitely affected subjects, and in grey symbol, the probably affected subject. Proband is indicated with an arrow.
Genetic testing from a blood sample was performed addressing the strong suspicion of hereditary CAA. Several genes related to CAA included in a commercial genetic panel were sequenced (APOA1, APE, APP, CST3, GSN, ITM2B, PRNP, PSEN1). All coding exons of interest were sequenced using NextSeq500 sequencing system (Illumina) and analyzed with HD Genome One Research Edition software.
Amyloid imaging with 18F-florbetapir PET (Amyvid, Eli Lilly & Co. and Avid Radiopharmaceuticals Inc.) was performed 6 months later (Fig. 1d-f). Binary visual analyses were made with the Syngo PET MMWP-VE61B (Siemens Healthcare) software using the interpretation criteria for Amyvid [9]. The quantitative analysis was developed using SPM12 with Matlab. The images were normalized in the MNI space, using the anatomical atlas neuromorphometrics for segmentation by regions. Amyloid retention was expressed as standard uptake value ratio (SUVr), using the cerebellum as reference region. PET imaging was evaluated by two nuclear medicine physicians.
RESULTS
Genetic testing revealed a c.2080 G>A (p.D694N) Iowa heterozygous mutation of the APP gene. The APOE genotype was ɛ4/ɛ3. Genetic testing of his brother was later performed, showing the same c.2080 G>A APP heterozygous mutation.
With regards to the 18F-florbetapir PET, visual analysis was informed as negative for amyloid deposition (Fig. 1d, e). Quantification of global florbetapir retention was low (SUVr <1.10), indicating scarce cortical Aβ burden (Fig. 1f) and supporting previous visual analysis. Interestingly, SUVr were borderline in precuneus and cingulate when analyzing cortical areas separately. The occipital-to-global amyloid-PET uptake ratio was below 1. Deeper analysis demonstrated certain cerebral subregions with elevated amyloid retention (SUVr >1.40; Fig. 1f) such as calcarine cortex, thalamus, and pallidum bilaterally, as well as right parietal operculum and left precentral gyrus.
Due to the high risk of hemorrhage, the patient did not receive anticoagulation therapy for venous thrombosis. Another MRI study performed 9 months later showed similar findings. He made a good clinical recovery, which allowed him to resume his previous employment.
DISCUSSION
To our knowledge, this is the seventh known family with confirmed hereditary CAA related to Iowa APP mutation [10–16].
Although our patient’s clinical features are similar to those of previously reported hereditary cases, this is the first one associated with CVT. A comprehensive study to identify conditions associated with CVT was negative and no identifiable risk factors were determined. Few cases of sporadic CAA and CVT have been described in the literature [17, 18]. The precise mechanism of this association is unknown. It has been suggested that amyloid deposits in cerebral venous vessels could contribute to the development of venous thrombosis in the setting of CAA [18]. We also propose that altered venous flow secondary to hemorrhage and inflammatory response may have played a role. Therefore, our case provides one more record of the concurrent appearance of these two conditions.
Amyloid-PET imaging in Iowa-type hereditary CAA with Pittsburgh Compound B (PiB) was previously reported by Greenberg et al. [19]. This study showed an elevated PiB retention in bilateral occipital cortex with lower global amyloid burden than it is observed in AD. Conversely, our patient’s study was informed as negative after two different readings methods which included regional amyloid burden analysis.
It has been described that whole-cortex tracer uptake in CAA shows an intermediate pattern between controls and positive AD cases, and a predilection for occipital regions [8]. However, no occipital cortex retention was seen in our patient. Interestingly, certain subregions including calcarine cortex showed an increase in amyloid retention (SUVr >1.4). Therefore, standardized analysis in cortical regions of interest may identify a mild tracer uptake in specific subregions in CAA that can be overlooked in visual analysis.
Postmortem brain examination of patients with Iowa APP mutation show severe CAA with Aβ-positive immunostaining and variable amount of neurofibrillary tangles and neuritic plaques [11, 15]. Although no pathological examination is available in our patient, it seems that amyloid pathology has not reached clearly detectable levels by florbetapir-PET.
It should be noted that a negative amyloid-PET result does not necessarily exclude CAA in certain clinical contexts. That is particularly relevant in early-onset hereditary CAA, in which the diagnosis relies on careful clinical suspicion and genetic testing. Additional clinical, imaging, and neuropathological reports about Iowa mutation-related CAA are needed to help clarify uncertainties about these questions and to provide further information about this entity.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0800r2).
ETHICAL APROVAL
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1975 Helsinki declaration.