
Abstract
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease in the adult population. There is evidence of an inverse epidemiological relationship between AD and cancer, another prevalent age-related disease. This has led to hypothesize that there could be a common biological mechanism, deregulated in opposite directions that might explain the phenomenon of mutual protection. The immunological system and its regulatory checkpoints are good candidates to explain why having survived a cancer could protect from developing AD. During cancerous growth, the neoplastic cells induce immune tolerance to block the host’s immunity system that would prevent tumor growth. This has led to the development of drugs that block distinct immune checkpoints, such as Programmed Death 1 (PD-1) and its major ligand PD-L1, that have shown great promise in treating diverse types of cancer. We propose that in those individuals who survived a cancer, the immune system is left in a state of diminished tolerance or proinflammatory systemic milieu, after its successful attempt to fight the cancer, that protects them from developing AD.
THE INVERSE RELATIONSHIP BETWEEN ALZHEIMER’S DISEASE AND CANCER
Alzheimer’s disease (AD) is a brain degenerative disease, characterized by early failure in episodic memory and gradually worsening of cognitive and motor symptoms. It constitutes the most frequent dementia in worldwide population, corresponding to the 70% of all diagnosed dementias, with an estimated 44.4 million cases accounted in 2016 [1] and predicted to double its prevalence every 20 years [2]. AD is a frequent cause of disability in the elderly population [3], with consequences not only for individuals and families but also for health and social welfare systems [4], since it does not yet have an effective treatment to reverse or stop its progression.
On the other hand, cancer is one of the leading causes of death before age 70 years in 91 out of 172 countries, according to estimates of the World Health Organization (WHO) in 2015 [5], and, just as AD, it is more frequent in the elderly. Despite the emergence of new therapies every year, cancer remains the leading cause of death in the highest-income countries [6].
Interestingly, there is epidemiological evidence of an inverse relationship between cancer and AD, observed in various epidemiological studies: patients with a history of cancer in the past have less risk of developing AD and conversely, patients suffering from AD show a lower risk of cancer in the future [7, 8]. This finding has been corroborated in several other studies [9–16] and has been observed for all types of cancer (except prostate) [10].
Deregulation in opposite directions of biological mechanisms involved in both AD and cancer might explain the inverse protection observed between these two disorders. As the role of the immune system in the development of age-related diseases has become a topic of emerging importance, especially in cancer [17], reviewing the control it may exert on the ontogenesis and upfolding of AD, has become of utmost relevance.
Regarding the biological mechanisms that might explain the inverse association between cancer and AD, it has been hypothesized that there could be a common biological mechanism, deregulated in opposite directions [18, 19] that might explain the phenomenon of mutual protection: for example, cell susceptibility to oxidative damage [18]. In studies with cell cultures from patients, it has been observed that cell death induced by oxidative damage is increased in lymphocytes from AD and mild cognitive impairment (MCI) patients when exposed to H2O2, compared to lymphocytes from healthy control donors of similar age, whereas lymphocytes from patients who had survived a cancer in the past showed an increased resistance to cell death [20]. In addition, this higher susceptibility to cell death correlates with the severity of the dementia [21–24]. The type of cell death observed in AD patients’ lymphocytes under H2O2 exposure was markedly protected by inhibition of Poly [ADP-ribose] polymerase 1 (PARP1), and not by caspase inhibition, the ultimate mark of apoptotic death, suggesting that it corresponds to a PARP1-dependent, caspase-independent type of apoptosis [25] that has been named Parthanatos [26]. In physiological conditions, PARP1 is an enzyme involved in DNA repair, but under severe DNA damage it can lead to non-apoptotic cell death, by consumption of large amounts of cellular NAD+ and ATP. In addition, we have found evidence that p53, a tumor suppressor protein that is altered in 50% of cancers [27], and has been shown increased in the brain of AD patients [28], might have a role in the different susceptibility to H2O2-induced death of lymphocytes in AD and cancer patients [22], suggesting that the p53 pathway might be already activated in AD and MCI patients compared to healthy controls.
IMMUNITY AND CANCER
Studies on the relationship between the immune system and cancer began formally in the late twentieth century, when the association between pathogen-driven inflammation and its effect against tumors was established [29]. Twenty years ago, it was demonstrated that endogenously produced interferon-γ (IFN-γ) was able to protect the host against tumor growth [30, 31].
Evidence shows that cancer patients can develop T cells able to specifically recognize and destroy tumors in vitro [32, 33]. T cells are able to proliferate in response to stimulation with autologous tumor cells by secreting cytokines such as interleukin-2 (IL-2), IFN-γ, granulocyte-macrophage colony stimulating factor (G-MCSF), and tumor necrosis factor-α (TNF-α) [34]. These observations have resulted in the identification and characterization of tumor antigens recognized by human T cells [35, 36]. Ample evidence supports the involvement of cytokines in events leading to the initiation, promotion, invasion, and metastasis of cancer [37]. In chronic inflammatory processes, cytokines such as TNF-α and IL-6 induce the generation of free radicals that can damage DNA, potentially causing mutations that lead to tumor initiation [38]. Tumor growth can be also favored by proinflammatory cytokines, such as IFN-γ, that stimulate cell proliferation; while anti-inflammatory cytokines, such as IL-10 and TGF-β, can contribute to tumor immune evasion. Moreover, chemokines such as IL-8 might have an important role in cell migration to other tissues, promoting metastasis [38].
The pro or antitumor roles of cytokines depend on the balance between different inflammatory mediators and the stage of tumor development [38]. The presence of tumor-infiltrating lymphocytes (TILs) in cancer tissues is indicative of an active host immune response against cancer cells. TILs contain various immune cell subsets that can suppress or promote tumor progression. Among these, CD4+ and CD8+ T cells are the main components of tumor specific, cellular adaptive immunity.
CD8+ cytotoxic T lymphocytes (CTLs) recognize major histocompatibility complex (MHC) class I molecules bearing cognate antigenic peptides derived from endogenous proteins. As the majority of tumor cells express MHC class I, CD8+ T cells can recognize and directly lyse them by releasing perforins and granzymes. Indeed, several studies have shown that a high presence of tumor-infiltrating CD8+ T cells is associated with favorable prognosis in colorectal [39], ovarian [40], breast [41], pancreatic [42], and biliary tract cancer [43]; although this does not apply to all kinds of tumors [44, 45].
Similarly, CD4+ T cells are essential for regulating immune responses, a role exercised mainly through the secretion of different cytokines. During the activation of T cell receptors (TCR) in a local cytokine milieu, CD4+ T cells can differentiate into several lineages of effector T cells or T regulatory cells (Tregs), as defined by cytokine expression and cell function patterns. Upon antigen-specific stimulation, effector CD4+ T helper 1 (Th1) cells release cytokines involved in antitumor and antiviral immunity, such as IL-2 and IFN-γ [46, 47]. On the other hand, Tregs can inhibit effector T cell functions in physiological and diseased states, through cell-cell contact mechanisms or secretion of regulatory cytokines, such as IL-10 and TGF-β [48]. One Treg specific marker and master regulator of Treg development and functioning is Foxp3 [49]. Considering that Tregs within the tumor microenvironment might significantly suppress local antitumor immune responses [50], increased Foxp3+ T cells in peripheral blood or tumor tissues have been associated with negative prognosis for various cancers [51–53].
IMMUNE CHECKPOINT INHIBITION IN CANCER TREATMENT
The amplitude and quality of the immune response, which is initiated through antigen recognition by the T cell receptor, is regulated by a balance between co-stimulatory and inhibitory signals (named immune checkpoints). Under normal physiological conditions, these immune checkpoints are crucial for the maintenance of self-tolerance to limit collateral tissue damage during anti-microbial immune responses, playing a major role in maintaining the homeostasis of the immune system. Also, immune checkpoints can be co-opted by cancer to evade immune destruction [54, 55]. The observation that blocking the classical immune checkpoint receptor cytotoxic T lymphocyte antigen 4 (CTLA-4) could mediate tumor regression in murine models led to the clinical development and approval of anti-CTLA-4 as a treatment for patients with metastatic melanoma [56]. Subsequently, drugs blocking distinct checkpoints, such as Programmed Death 1 (PD-1) and its major ligand PD-L1, have shown great promise in treating many diverse types of cancer. This has revealed new treatment options for patients and has revolutionized the approach to cancer therapy [57, 58]. For this novel treatment for cancer, the Nobel Prize in Physiology and Medicine was awarded to James P. Allison and Tasuku Honjo in 2018.
Immune checkpoint inhibition can be accomplished using antibodies against CTLA-4 or the PD-1 pathway, either alone or in combination [58]. Antibodies against CTLA-4 (ipilimumab and tremelimumab) were the first to be tested in clinical trials in patients with advanced melanoma in 2000, and ipilimumab was approved in 2010. CTLA-4 is an intracellular protein that translocates to the surface after the binding of the T cell receptor to its antigen, where it displaces CD28 from its binding to the receptors CD80 and CD86 that are needed for the cytotoxic T cell response. Therefore, antibodies targeting CTLA-4 unblock the inhibition of the immune response, promoting it. Reports show that some patients have been cancer free for as long as 10 years. Nevertheless, it seems that not all patients respond [59, 60]; also, severe side effects have been reported [61].
In a similar way, the PD-1 receptor present in immune cells engages with its ligand PD-L1, expressed by tumors and many somatic cells upon exposure to proinflammatory cytokines, downregulating the anti-tumor T cell effector function [58]. T cells infiltrating the tumor bind to tumor antigens triggering the expression of PD-1 in T cells and releasing IFN-γ, which is the most important stimulator of PD-L1 expression in cancer resident cells [62]. Blockade of the PD-1-pathway is now considered one of the most important advances in the treatment of cancer, more specific and less toxic than the anti CTLA-4 treatment [58]. Currently there are five anti-PD-1 agents and/or anti-PD-L1 antibodies approved by the FDA for the treatment of eleven types of cancer: nivolumab, pembrolizumab, avelumab, durvalumab, atezolizumab [58]. They are particularly favorable in cancers induced by carcinogens or driven by viral infections (Hodgkin’s lymphoma, Merkel cell carcinoma of the skin, microsatellite instability-high cancers, and desmoplastic melanoma), with response rates around 50–90%. The more common melanoma variants show response rates of 35–40%, and cancers related to smoking (non-small cell lung cancer, bladder and urothelial, head and neck, and gastro-esophageal) show rates of 15–25%. The majority of patients receiving anti PD-1 or PD-L1 antibodies have toxicities similar to placebo; however, endocrinopathies can develop (thyroid, hypophysis adrenal, and type I diabetes) [58]. Several other immune checkpoint inhibitors are being examined, and combination therapies between two checkpoint inhibitors, or a checkpoint inhibitor plus traditional cancer treatments, are being tested with good results [54, 58]. In addition, combinations of PD1 and Lymphocyte-Associated Gene 3 (LAG3), a new immune checkpoint inhibitor, are currently being tested [63].
IMMUNITY AND INFLAMMATION IN ALZHEIMER’S DISEASE
The role of neuroinflammation in AD has been debated for a long time and until now it is uncertain whether inflammation is a cause, contributor, or secondary phenomenon in the disorder [64–68]. Activated microglia, the main innate immunity cells in the central nervous system, were reported in the original description of Alois Alzheimer in 1907 [69]; nevertheless, for a long time it was considered merely an epiphenomenon present only in late stages of the disease [64, 67]. Nowadays the immune system, including both innate and adaptive, is considered a key factor in AD that is not restricted only within the brain, since the peripheral immune system is clearly involved in the pathogenesis of AD [64, 70–74]. In addition, increased percentages of lymphocytes both CD8+ and CD4+ have been described in the brains of AD patients [75].
Retrospective epidemiological studies show that the use of nonsteroidal anti-inflammatory drugs (NSAIDs) is associated with reduced prevalence of AD [76]. Data in animal models also show that inhibition of inflammation, by the administration of anti-inflammatory drugs, is beneficial for AD [77]. However, newer large-scale clinical trials of anti-inflammatories including NSAIDs, aspirin, and prednisone have not corroborated these findings in patients [78, 79]. One explanation might be that different inflammatory mechanisms take place throughout different stages of the disease. This hypothesis is supported by recent data showing that NSAIDs have a beneficial role when administered preclinically for 2-3 years, but an adverse effect at later stages of AD pathogenesis [80].
There is also genetic and bioinformatic data that support a role for increased inflammation in AD. Key microglial genes, such as CD33 (an immune receptor that inhibits microglial phagocytosis) and TREM2 (triggering receptor expressed on myeloid cells 2), also involved in phagocytosis, are associated with increased risk of AD [81–83]. Also, an accelerated cognitive decline and increased risk of AD is observed in patients that suffer chronic infections such as periodontitis, and other non-transmissible diseases consider to be chronically pro inflammatory [84].
INFLAMMATION VERSUS ANTI-INFLAMMATION IN AD
The presence of Aβ peptides acting as pathogen-associated molecular patterns (PAMPs) may impede resolution of inflammation and has led to the assumption that an increased inflammatory activity is present in AD. Microglia are proposed to be chronically activated in an autotoxic loop [70, 85]. Heneka et al. speculate that misfolded Aβ peptides represent a conserved molecular pattern for which the innate immune system has developed immunological receptors [64]. This has been shown in animal models, after the injection of oligomers of Aβ into the ventricles or hippocampus of wild type mice, which was associated with memory impairment and accompanied by gliosis and expression of proinflammatory cytokines such as IL-1, TNF-a, IL-6, and IL-10, supporting the hypothesis that these forms of Aβ promote inflammation [86].
However, in the long run, the long-term chronic stimulation of microglia is known to make them ineffective (“frustrated or degenerating” microglia), and this could attract regulatory T cells, Tregs [87, 88]. Astrocytes are also capable of secreting immunoreactive mediators and can induce the proliferation of Tregs. Accordingly, there are reports of increased immune mediators such as inflammatory cytokines and chemokines in the tissues and body fluids of AD patients even at early stages of the disease [89, 90].
Despite this evidence in favor of a chronic inflammatory state in AD, there are also signs of anti-inflammatory responses, and even signs of an immunosuppressed milieu, along the course of the disease. Microglial cells in transgenic AD mice are normally in a low inflammation state and hypo responsive to Aβ peptide [91, 92]. Also, blockade of the anti-inflammatory interleukins IL-4 or IL-10, or a strong proinflammatory stimulus, activates the phagocytic activity of microglia and leads to plaque clearance in AD models and in in vitro studies [93, 94]. In accordance, proteins related to the IL-10 pathway have been reported increased in the AD brain [94, 95]. In the APP/PS1 transgenic mouse model of AD conditionally expressing IL-1β, a proinflammatory cytokine, Cherry et al. demonstrated that inflammation decreased Aβ deposition, in opposition to what has been observed in vitro, in which inflammatory cytokines impair Aβ clearance [96]. They propose that the discordance between the in vivo and in vitro results might be explained because in vitro studies occur in a closed environment in which microglia are the only cells present. In vivo, instead, other cells are recruited to the site of inflammation, initially proinflammatory but later on anti-inflammatory, that secrete T helper 2 (Th2) and other anti-inflammatory cytokines. The discordant results obtained in AD studies, showing evidence in favor of increased but also of decreased inflammation, might be explained by the presence of different states of inflammation along the course of the disease.
BREAKING IMMUNOSUPPRESSIVE PATHWAYS IN AD
The group of Schwartz et al. [97–99] propose that systemic immunity should be boosted rather than suppressed in neurodegenerative disorders, to induce an immune response to repair the damage, similar to what is seen in immunotherapy for cancer. They propose that the entrance of reparatory peripheral monocytes into the brain through the brain barriers is impaired in AD, and that stimulation of inflammation could have a protective role in AD [66, 101]. The proposal of Schwartz et al. is that in AD there is chronic inflammation in the brain, and at the same time, decreased entrance of protective immune cells from the periphery due to elevated levels of peripheral Tregs. They have shown that Tregs increase in the spleen of 5xFAD mice as they grow older [97, 98]. The elevated levels of peripheral Tregs in turn promote immune tolerance and impair the entrance of beneficial leukocytes into the CNS through the cerebrospinal fluid (CSF)-blood barrier—the choroid plexus (CP)—which is the best adapted gateway for the passage of immune cells to the CNS. The entrance of leukocytes through the CP is dependent on the INF-γ dependent expression of passage proteins in the CP, such as ICAM and VCAM [66, 100].
Interestingly, Baruch et al. showed that either a transient depletion of Foxp3+ Tregs or its pharmacological inhibition, induced breakage of immune tolerance and allowed passage of reparatory leukocytes into the brain, mitigating AD pathology in the 5xFAD transgenic mouse model of AD [97]. Monocyte-derived macrophages (CD45high/CD11bhigh) were recruited around Aβ plaques in the brain and there was Aβ plaque clearance, mitigation of the brain inflammatory response and reversal of cognitive decline [97]. Furthermore, increasing Treg activity with all-trans retinoic acid (ATRA) worsened the outcome [97]. Interestingly, in a follow up study, Baruch et al. [98] showed that immune checkpoint inhibition may be beneficial for AD. They demonstrated that anti PD-1 treatment in 5xFAD mice provoked an INF-γ-dependent systemic immune response, which was followed by the recruitment of monocyte derived macrophages into the brain, clearance of cerebral Aβ plaques and improved cognitive performance [98]. However, this result was not replicated in another study using other mouse models [102], nor in a model of prion disease lacking PD-1 [103], maybe due to different experimental settings (see Table 1).
Immune tolerance modulation in Alzheimer’s disease’s mouse models
*Tregs, T regulatory cells; ATRA, all-trans-retinoic-acid; Aβ, amyloid-beta; KO, knock-out.
The studies in animal models of AD suggest that the beneficial effect of lowering Tregs seems to have an optimum point in time. Dansokho et al. showed that depletion of Tregs at very early stages of the disease (4-5 weeks), using anti CD25 antibody, worsened cognitive deficit in an APP/PS1 model [104]. They did not find changes in plaque load but there was reduced recruitment of microglia around plaques [98]. Also, in another study using the 3xTg-AD mouse model, Baek et al. [105] demonstrated that depletion of Tregs at early stages of the disease (4 weeks of age) was accompanied by an accelerated onset of cognitive deficit, increased Aβ plaque burden and microglia/macrophage inflammatory response in hippocampal CA1 and CA3 regions [105]. These two studies suggest that a certain degree of tolerance might be beneficial at very early stages of the disease to avoid excessive inflammation in animal models of AD. However, at later stages of the disease, in 6-month-old AβPPswe/PS1dE9 transgenic mice, Yang et al. demonstrated that systemic transplantation of Tregs educated with human umbilical cord derived mesenchymal stem cells improved impaired cognition [106]. In all, these experiments in transgenic AD mouse models show that the role of Tregs seems to be different depending on the stage of the disease (see Table 1). At early stages, lowering immune tolerance by the depletion of Tregs worsens AD pathology, suggesting a beneficial role of Tregs in reducing an excessive inflammatory state. On the contrary, at later stages, the presence of Tregs inducing immune tolerance would have a detrimental role by interfering with the passage of immune cells to the brain to help eliminate Aβ damage.
It is difficult to extrapolate these results in animal models of AD—which have a high genetic load of AD mutations—to the reality of patients, who develop AD slowly along many decades. Tregs have been reported elevated in the elderly and in patients with AD [107]. Another study found higher levels of Tregs in MCI, but not in mild AD patients [108]. Still, another study reported that MCI patients had higher expression of the more suppressive (cytoplasmic) form of Tregs compared to AD [109]. Recently, Oberstein et al. [110] compared Tregs and Th17 cells in patients with MCI due to AD compared to non-AD MCI and healthy controls. They reported that Th17 cells were significantly increased in MCI due to AD patients. Peripheral Tregs (CD4 + CD127lowCD25+), although not significantly different between groups, were positively correlated with the levels of CSF total and phosphorylated-tau in subjects with AD. The mechanism of action of Tregs to suppress the immune response is unknown, but probably involves IL-10 and TGF-β production.
In all, the role of inflammation and immune response in AD is still controversial. As in other neurodegenerative disorders, the abnormal folding of proteins leads to a chronic form of neuroinflammation accompanied by microglial activation and increased release of cytokines and chemokines in the brain and also in peripheral fluids [101]. Later on, microglial phagocytosis becomes compromised complicating Aβ clearance [87, 88]. In addition, an immunosuppressive milieu is established due to an alteration of the passage of beneficial immune cells into the brain that prevents resolution of the damage, as evidenced by the beneficial effect of unlocking immune checkpoint in the 5xFAD mouse model of AD [97, 98]. This beneficial effect of checkpoint inhibition is similar to the successful treatment shown by unleashing immune tolerance in cancer therapy [54, 55].
IMMUNE-MEDIATED INVERSE RELATIONSHIP: PRESENT AND FUTURE
We propose that deregulation of immune mechanisms might be the basis of the mutual protection observed between AD and cancer. The chronic neuroinflammation present in AD might represent a state of permanent inflammation that protects against the development of cancers in the body. On the other hand, the development of a cancer and surviving it might leave a state of an unlocked immune tolerance that protects against the progression or development of AD (see Fig. 1). In the future, it will be interesting to further explore whether having had cancer in the past may protect against AD in the future. Since the development of cancer induces immune tolerance (blocking the host’s immunity to be able to continue its growth), it is possible to assume that in those individuals who survived a cancer, the immune system would grant protection against the development of AD, after being left in a state of diminished tolerance or proinflammatory state in its successful attempt to fight the cancer.
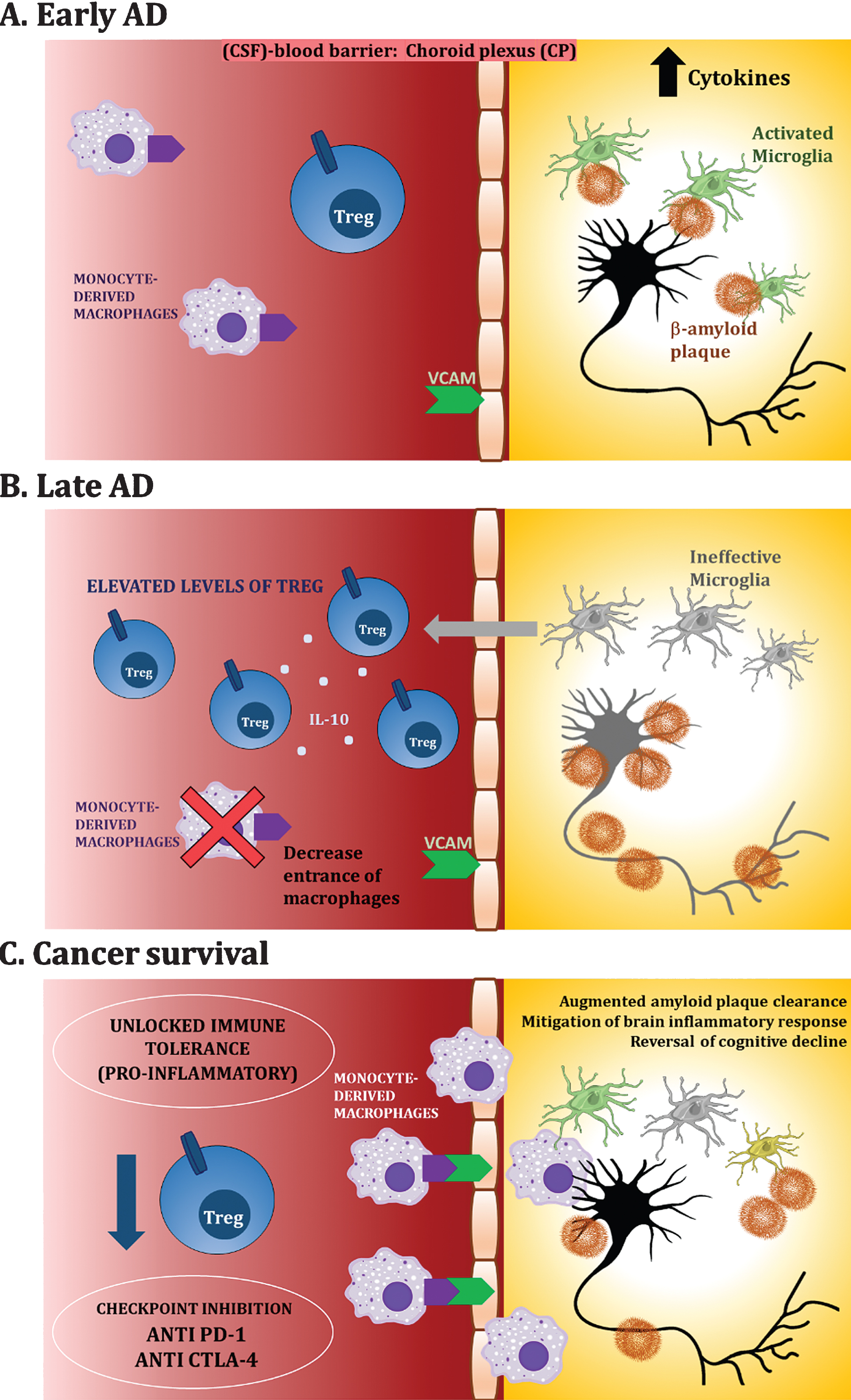
Schematic diagram of proposed dysregulation of immune mechanisms and unlocked immune tolerance in cancer and AD patients. A) Early AD. In vulnerable brains, neuroinflammatory processes take place in which activated microglia surround Aβ plaques, promoting a pro-inflammatory milieu. B) Late AD. Long-term chronic stimulation of microglia makes them ineffective (“frustrated or degenerating” microglia), and this in turn, attracts Tregs. C) Cancer survival. Having survived a cancer or through the use of checkpoint inhibitors such as anti PD-1, unlocked immune tolerance allows recruitment of macrophages to the brain promoting amyloid clearance and other neuroprotective processes.
Previous studies have reported a role of the immune system on the inverse comorbidity between AD and cancer. Kong et al. [111] compared microarray gene expression data through fast independent and network component analysis and found 10 transcriptional factors closely related to cytokines that were inversely associated between AD and breast cancer. In another study, Aramillo Irizar et al. [112] comparing gene expression data from four vertebrate species across four different tissues and along different points in time, showed an increased expression of immune genes in AD and Parkinson’s disease, beyond those observed with aging, while the reverse was seen in cancer. On the other hand, Sanchez-Valle et al. [113] identified a significant number of genes of the immune system and of a chronic inflammation state that were deregulated in the same direction in AD and glioblastoma, which might explain the direct co-morbidity that has been observed between these two diseases [114].
CONCLUDING REMARKS
Although a radical proposal, there seems to be enough epidemiological and cellular evidence to promote the study of immune checkpoints as a novel mechanism to explain the inverse association between AD and cancer. As new immune-mediated processes are being described in AD, it is of utmost importance to review the basic models, since these could present a unique opportunity to understand the mechanisms and help find successful treatments for two of the most challenging diseases of the last two centuries.