
Abstract
Alzheimer’s disease (AD) is the most common form of dementia, characterized by amyloid-β peptide (Aβ) aggregates, phosphorylated tau protein (p-tau), and progressive neurodegeneration. Amyloid-β peptide 42 (Aβ42) is considered an early trigger of AD pathogenesis. We have previously reported that Aβ N-terminus monoclonal antibody (mAb) A8 alleviated cognitive dysfunction and reduced the abundance of soluble Aβ in the brains of the senescence-accelerated mouse prone 8 (SAMP8) mouse model. To confirm the efficacy of mAb A8 in the double-transgenic APPswe/PS1ΔE9 (APP/PS1) mice, here we reported the related findings. The Morris water maze (MWM) data showed that the A8 treatment group had a shorter escape latency than the control groups in the place navigation test and the probe trial (p < 0.05). Moreover, immunohistochemistry showed decreased levels of both Aβ and p-tau in the brains of APP/PS1 mice. Regarding Aβ levels, western blot results showed that Aβ42 oligomer (p < 0.01) but not Aβ40 levels were diminished in brains of A8-treated APP/PS1 mice. Western blot results showed that phospho-tau (pSer231) (p < 0.01) but not tau levels were reduced in A8-treated mouse brains. Furthermore, transmission electron microscopy images indicated ultrastructural improvements, including an increased (p < 0.01) density of synapses and a reduction of abnormally enlarged mitochondria (p < 0.01), in the brains of A8-treated mice. Taken together, our data showed that mAb A8 is highly efficacious in APP/PS1 mice as a treatment for AD, and the underlying mechanism may target synaptic pathology by inhibiting the amyloid cascade.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that threatens aging societies. AD is characterized by deposition of amyloid-β (Aβ) [1], formation of neurofibrillary tangles (NFTs) [1], and neuronal loss with dementia. It is estimated that more than 46.8 million people worldwide had dementia as of 2015, and this number is expected to double every 20 years to 131.5 million in 2050. In the past few decades, many efforts have been made to find the ultimate treatment for AD. All current Food and Drug Administration (FDA) approved drugs are palliative, whereas passive immunotherapy using an antibody has been shown as one promising approach for AD treatment.
Several mAbs against Aβ have been developed and tested in clinical trials to decrease the neurodegeneration caused by Aβ. These mAbs include the following: (1) Bapineuzumab (AAB-01; Pfizer Inc., New York, NY, and Janssen Pharmaceuticals, Inc., Raritan, NJ), a humanized 3D6 mAb (IgG1) targeting the N-terminus of Aβ, became the first mAb to undergo clinical trials for AD [2]. However, amyloid-related imaging abnormalities with edema/effusion (ARIA-E) and the lack of a definite curative effect resulted in the termination of bapineuzumab in phase III of clinical trials. (2) The humanized m266 mAb (IgG1) solanezumab (LY2062430; Eli Lilly and Company, Indianapolis, IN), which target the central region of Aβ, has greater affinity for monomers. It failed to slow down cognitive decline in AD in phase III clinical trials. One of the reasons for the failure is that the antibody was not able to reach therapeutic concentrations in the brain; another reason was the occurrence of ARIA-E [3]. However, Panza et al. [4] reported the potential of solanezumab and gantenerumab to prevent AD in people with inherited mutations that cause its early onset. (3) Gantenerumab [5] (Hoffman-La Roche, Basel, Switzerland) was the first fully human mAb against a conformational epitope on Aβ fibrils. The antibody was found to be safe in phase I clinical trials, and it has been tested in a phase II/III study to determine whether it mitigates cognitive dysfunction in AD patients. (4) Aducanumab (BIIB037; Biogen, Inc., Cambridge, MA) [6], a human IgG1 developed from a B-cell library from healthy aged people, binds to the Aβ N-terminal region, recognizing oligomers and fibrils. Mitigation of cognitive decline has been observed in a phase Ib clinical trial. Therapeutic treatment is desperately required to slow the progression of dementia. In clinical data, most anti-Aβ mAbs against pathogenic aggregates or conformational epitopes succeeded in curing cognitive deficits, whereas mAbs with greater affinity for Aβ monomers failed to do so. Therefore, numerous anti-Aβ antibodies of the former type should be screened and tested for AD immunotherapy.
The reasons for the different effects of the antibodies in AD treatment may stem from the different mechanisms of Aβ clearance. Rodent experiments have indicated that 0.11% of the circulating antibodies enter the central nervous system [7], where they act through the following mechanisms: (1) Macrophage phagocytosis and complement activation [8]. Immunotherapy works together with the human immune system to counteract the process of Aβ aggregation. (2) Formation of a peripheral Aβ sink [9] by circulating antibodies. Moreover, antibodies in the brain ventricular fluid might bind Aβ and act as a sink in the central nervous system. (3) Catalytic modification of Aβ conformation [10]. (4) Neonatal Fc receptor (FcRn)-mediated efflux of intracranial antibody–antigen complexes across the blood–brain barrier. The mechanisms of antibody-based Aβ immunotherapy differ depending on the type of antibody. Regardless, however, Aβ clearance or balance of Aβ production and clearance is the main aim of antibody-based AD immunotherapy.
It has been widely confirmed, at least in mouse models of AD pathogenesis, that the Aβ load in the brain can be reduced and the accompanying cognitive decline can be inhibited through immunotherapy. However, the effects of monoclonal antibody treatment on cognition-associated synaptic dysfunction and mitochondrial lesions have not been reported in preclinical studies or clinical trials of AD immunotherapy. Synapse loss is the major correlate of cognitive impairment, and soluble Aβ aggregates impair synaptic and network activity [11]. Wang Z and colleagues [11] reported that human brain-derived Aβ oligomers bind to synapses and disrupt synaptic activity. Furthermore, Henstridge et al. [12] reported that synapse loss was significantly increased in cognitively impaired cases among amyotrophic lateral sclerosis patients and was not due to cortical atrophy.
Mitochondrial dysfunction [13] has been closely linked to the pathogenesis of AD, but the relationship between mitochondrial pathology and neuronal damage is poorly understood. Morphological alterations of mitochondria may play an important role in the pathogenesis of AD, which has been associated with oxidative stress [14] and Aβ-peptide-induced toxicity [15]. Therefore, evaluation and detection of synaptic pathology and mitochondrial damage should be considered in studies of AD immunotherapy.
We previously reported [16] that A8, a mAb targeting the N-terminal amino acid of Aβ, inhibited Aβ fibril formation and had a potential effect in the SAMP8 mouse model of AD. Afterwards, A8-derived single-chain variable-fragment (scFv) antibodies were expressed in baculovirus and used as regulators of Aβ assembly and disaggregation. Our previous data [17] showed that anti-Aβ scFv expressed in baculovirus blocked Aβ fibril elongation and promoted its disaggregation. However, the efficacy and mechanisms of action of A8 on cognitive dysfunction in APPswe/PS1ΔE9 (APP/PS1) mice were not reported.
In the present study, the in vivo component demonstrated that neutralization of pathologic Aβ by intraperitoneal administration of A8 mAb in APP/PS1 mice mitigated synaptic pathology, improved spatial memory, and increased Aβ clearance in the brain. Our study first showed that the increased synaptic density and decreased mitochondrial lesions may be the underlying mechanisms of passive immunotherapy using the monoclonal antibody A8, which has potential as a therapeutic agent for AD. Furthermore, our results suggest that behavioral impairment might be reduced via the upregulation of synaptic density and the recovery of mitochondrial morphology.
MATERIALS AND METHODS
Chemicals and antibodies
The monoclonal antibody A8 [16] was generated previously and maintained by our group. Mouse anti-Aβ (1–42) (12F4), mouse anti-Aβ (17–24) (4G8), and mouse anti-Aβ (1–16) (6E10) were purchased from Covance (Emeryville, CA, USA). Rabbit anti-Aβ (1–40) was purchased from abcam (Cambridge, MA, USA). Mouse anti-phospho-tau (pSer231) was from Signalway Antibody LLC (SAB) (College Park, MD, USA). Anti-β-actin and horseradish peroxidase-conjugated secondary antibody were from Zhongshangjinqiao Inc. (Beijing, China). A broad-range protein marker was purchased from New England Biolabs Inc. (Ipswich, MA, USA).
Animals and treatment
APPswe/PSΔE9 (APP/PS1) transgenic AD model mice (C57BL/6J background) and wild-type (WT) C57BL/6J mice, all of them 4-month-old females with a body mass of 20 g ± 2 g, were obtained from the Institute of Experimental Animals of the Chinese Academy of Medical Science [18] and maintained at the Department of Laboratory Animal Science, Peking University Health Science Center. This mouse strain shows spatial memory deficits at 3 months of age. Mice were housed in standard mouse feeding cages (318 × 202 × 135) with the controlled conditions (temperature, ∼24°C, 12 h light/dark cycle, and humidity (35∼40%). Mice were free to get food and water throughout the study. Protocols involving experiments with animal subjects were designed and carried out in compliance with the guide for care and use of laboratory animals from the Peking University Research Council and were approved by the Animal Care and Use Committee of Peking University. Mice were randomly allocated into different groups according to the results of the table of random numbers: (1) APP/PS1 control group, (2) IgG injected group, (3) A8 mAb treated group, and (4) WT control group. The total number of the mice was 64, and there were 16 mice in each group. To determine whether A8 mAb treatment delays the learning and memory defects that are characteristic of transgenic AD model mice, 4-month-old APP/PS1 mice were injected intraperitoneally once a week with A8 antibody (20 mg/kg) or nonspecific mouse IgG (Sigma-Aldrich, St. Louis, MO, USA) for 8 weeks. At the end of this treatment, mice were subjected to object recognition testing. The time-line diagram of the study design is shown in the Supplementary Material.
Behavioral analysis
To determine whether administration of A8 mAb improves the cognitive performance of an AD mouse model, we intraperitoneally injected 4-month-old APP/PS1 mice with 400 μg of purified A8 mAb (20 mg/kg) once a week for 8 weeks. At the same time, APP/PS1 mice treated with nonspecific mouse IgG (Sigma-Aldrich), untreated APP/PS1 mice and wild-type C57BL/6 mice were used as the control groups.
The Morris water maze (MWM) was used as described previously [16] to assess changes in the learning and memory ability of mice in each group. In brief, the MWM consists of a metal pool (diameter 130 cm, height 47 cm) filled with water tinted with milk (to a depth of 30 cm) at 23–26°C. This test requires the animal to find a hidden platform (diameter 9 cm, submerged 1 cm below the surface) through the memory of visual cues around the room. During the test, a flag with a geometric design was positioned above the wall of the pool in a constant location for the mice to use as a navigation cue. A video camera above the pool recorded the swimming track of every mouse, and the data were stored on a computer connected to the camera. The process includes three parts: (1) the visual platform test, (2) the hidden platform test (or place navigation test), and (3) the probe trial test. The mice were wiped with a dry towel and dried with a heater after each swim. The escape latency, travel distance and swimming speed of each mouse were recorded.
In the visual platform test, the mice were put in the water to swim in the presence of the cue flag. In this stage, the mice were required to find the platform, which was elevated above the surface of the water, in no more than 60 s and then stand on the platform for 20 s. If a mouse could not climb onto the platform, it was guided to the platform and was allowed to stand on it for 20 s.
The place navigation test measured the spatial learning and memory of APP/PS1 mice. A round platform on which the mice could stand to escape the water was positioned in a specific location in the pool. For each trial, the mouse was released into the pool from a random direction, with its nose against the pool wall. The trial ended when a mouse climbed up and stand on the platform. After finding and climbing onto the platform, each mouse was allowed to remain there for 20 s and was then returned to its cage. Animals that did not locate the platform within 60 s were guided to it and allowed to remain there for 20 s before being returned to their cages. Before the place navigation test, each mouse was allowed to swim freely for 2 min in the water maze in order to familiarize them with the environment. The latency to reach a submerged platform was measured over 4 days (4 consecutive trials per day). Escape latency was defined as the time taken to find the platform.
In the probe trial, the platform was removed, and the mice were allowed to search for the former platform location for 90 s. The latency to find the previous location of the platform was measured. Data were recorded through an overhead camera linked to a computer to quantify latency. Mice were tested in a blinded manner, and differences in latency and swimming speed were analyzed. The experimenters were unware of the animal’s groups and medicine injected. The analysis and experimental group assignment were also performed according to the blinding rule.
Immunohistochemistry (IHC)
Three days after MWM testing, mice were anesthetized with 2% pentobarbital sodium (45 mg/kg) (the duration of anesthesia, analgesia, and muscle relaxation met the experimental requirements) and perfused with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) before IHC staining [16, 19]. Tissue blocks from APP/PS1 mice were fixed by immersion in 4% w/v buffered (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.2) paraformaldehyde. Samples were dehydrated in graded ethanol solutions followed by xylene, then embedded in paraffin. Mouse anti-Aβ (17–24) (4G8) or mouse anti-Aβ42 (12F4) (1 : 150 dilution) and anti-phospho-tau (pSer231) (SAB) (1 : 100 dilution) were used as primary antibodies. Sections (4 μm) were stained with a standard avidin-biotin-peroxidase method using 3,3′-diaminobenzidine substrate. The experimenters were unware of the tissue block groups and antibodies used. The analysis and experimental group assignment were also performed in a blinded manner. We used the following criteria to determine whether the Aβ or p-tau staining was positive: (1) the background color was light or no staining, while the positive markers were clear; (2) the positive signal location was clear; (3) no antigen expression was found in the negative control. For counting, 3 animals per group, 3 sections per animal, and 3 visual fields per section were observed. One hundred cells in each visual field were counted, and the number of positively signals was recorded; the results were expressed as means ± standard error of means (SEM).
Western blot assays
Brain tissues were homogenized in cold RIPA buffer (0.05 M Tris, 0.9% NaCl, 5 mM EDTA, pH = 7.4) [20] with a protease inhibitor (Complete Protease Inhibitor Mixture; Roche Molecular Biochemicals, Mannheim, Germany) to extract total brain protein, and the total protein concentrations were determined by BCA (bicinchoninic acid) protein assays (Pierce Chemical Co., Rockford, IL). For Aβ detection, the RIPA buffer contained 8 M urea. So the 8 M urea-treated Aβ aggregates can be detected by Aβ40 or Aβ42 specific antibody. Aliquots (25 μg) of transgenic mouse brain extracts (soluble samples) were separated by 15% SDS-PAGE and transferred to nitrocellulose membranes. The sample loading buffer contained SDS, and the samples were boiled for 5 min before being added to the gel.
As for the study of specificity of A8 mAb, the Aβ42 peptides (DAEFRH DSGYEV HHQKLV FFAEDV GSNKGA IIGLMV GGVVIA) and the Aβ40 peptides (DAEFRH DSGYEV HHQKLV FFAEDV GSNKGA IIGLMV GGVV) obtained from the manufacturer (Shanghai Sangon Biological Engineering & Technology and Service Co. Ltd., Shanghai, China) were > 95% pure. Aβ oligomer was prepared according to the methods described previously [21]. Equal amounts of Aβ oligomer sample (5 μg) were boiled, loaded, and separated by 15% SDS-PAGE and transferred to nitrocellulose membranes.
Membranes were blocked for 1 h, then rinsed and incubated with anti-beta amyloid 1–42 (12F4) (1 : 1000), anti-beta amyloid 1–40 (ab17295) (1 : 1000), A8 mAb (1 : 1000), anti-beta amyloid 1–16 (6E10) (1 : 1000), or anti-phospho-Thr231 (1 : 500) antibodies overnight at 4°C. After being rinsed, the blots were incubated for 1 h with a horseradish peroxidase (HRP)-conjugated sheep anti-mouse IgG secondary antibody (1 : 10,000, Santa Cruz Biotechnology). The blots were imaged on X-ray film with SuperSignal® West Pico Chemiluminescent Substrate (Pierce-Thermo Scientific, Rockford, IL, USA). The intensities of the bands were measured and analyzed by densitometry with Quantity One 4.6.2 (Molecular Dynamics, Inc., Sunnyvale, CA). Western blots were repeated at least three times. The experiments and analysis were also performed according to the blinding rule.
Transmission electron microscopy (TEM)
The sample preparation and TEM methods were described previously [22, 23] with some modification. Mice were perfused with 4% paraformaldehyde (PFA), and brain samples were fixed overnight in 4% PFA/2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 (Beijing Zhongjingkeyi Technology Co., Ltd., Beijing, China). The samples were then postfixed in 1% OsO4. Subsequently, the samples were dehydrated through graded (50% 10 min, 70% 10 min, 80% 10 min, 90% 10 min, and 100% 10 min ×2) ethanol, stained with uranyl acetate, and embedded in Epon 812 resin (Beijing Zhongjingkeyi Technology Co., Ltd., Beijing, China). Thin (900 nm) sections were stained with toluidine blue, and ultrathin (80 nm) sections from matching areas of experimental and control tissue blocks were cut and stained with 2% uranyl acetate for 20 min and 0.2% lead citrate for 2 min. The ultrathin sections were visualized using an electron microscope (H-7650 or JEM 1400) at 80 kV. Electron micrographs from at least three mice in each group were analyzed. The thin section preparation, TEM image observation and analysis were also performed in a blinded manner.
Statistical analyses
Experiments were analyzed with one-way or two-way ANOVA and the chi-squared test. A α level of 0.05 was used for all statistical significance tests. SPSS 17.0 was used to analyze the collected data. Data were expressed as the mean ± SEM. To analyze the results of the place navigation test, we first used Mauchly’s test of sphericity to judge the appropriateness of repeated-measures analysis of variance. If the assumption of sphericity was met, statistical comparisons were made by one-way repeated-measures analysis of variance and Tukey post hoc pairwise comparisons, with significance defined as p < 0.05. To analyze the probe trial, we used single-factor analysis of variance and Student’s t-test, with significance defined as p < 0.05. For western blots, data were expressed as the mean ± SEM. We used single-factor analysis of variance and Student’s t-test, with significance defined as p < 0.05.
RESULTS
Spatial learning and memory ability in APP/PS1 mice was improved by A8 treatment
To investigate the therapeutic effect of A8 on cognitive dysfunction in transgenic AD mice, we treated 4-month-old APP/PS1 mice with A8 (n = 16) or nonspecific mouse IgG (n = 16). At the same time, untreated APP/PS1 (n = 16) and wild type (WT, C57BL/6J) (n = 16) mice were used as the negative and positive control groups. One week after the last injection, the spatial learning and memory capacity of the mice was investigated using the MWM test. The place navigation test was performed from the first to the fourth day, and the spatial probe trial was carried out on the seventh day (Fig. 1A). In the place navigation test, as shown in the recorded motion traces, the mice in the A8 treatment group and the wild-type group searched for the platform, while the APP/PS1 and IgG groups swam close to the pool wall and did not approach the platform (Fig. 1B, those four figures are representative for the four different groups). The escape latency decreased in the A8 treatment group and the wild-type group but not in the other two groups (Fig. 1C). The difference was statistically significant on the 3rd and 4th day of this test (p < 0.05) (Fig. 1D). In the probe trial, A8-treated APP/PS1 mice showed significantly shorter latency than IgG-treated mice (p < 0.05) or APP/PS1 control mice (p < 0.05) (Fig. 1E), and A8-treated APP/PS1 mice also showed significantly higher swimming speed than IgG-treated mice (p < 0.05) or APP/PS1 control mice (p < 0.05) (Fig. 1F). Therefore, passive immunotherapy with A8 ameliorated behavioral deficits in APP/PS1 mice as indicated by the MWM test.
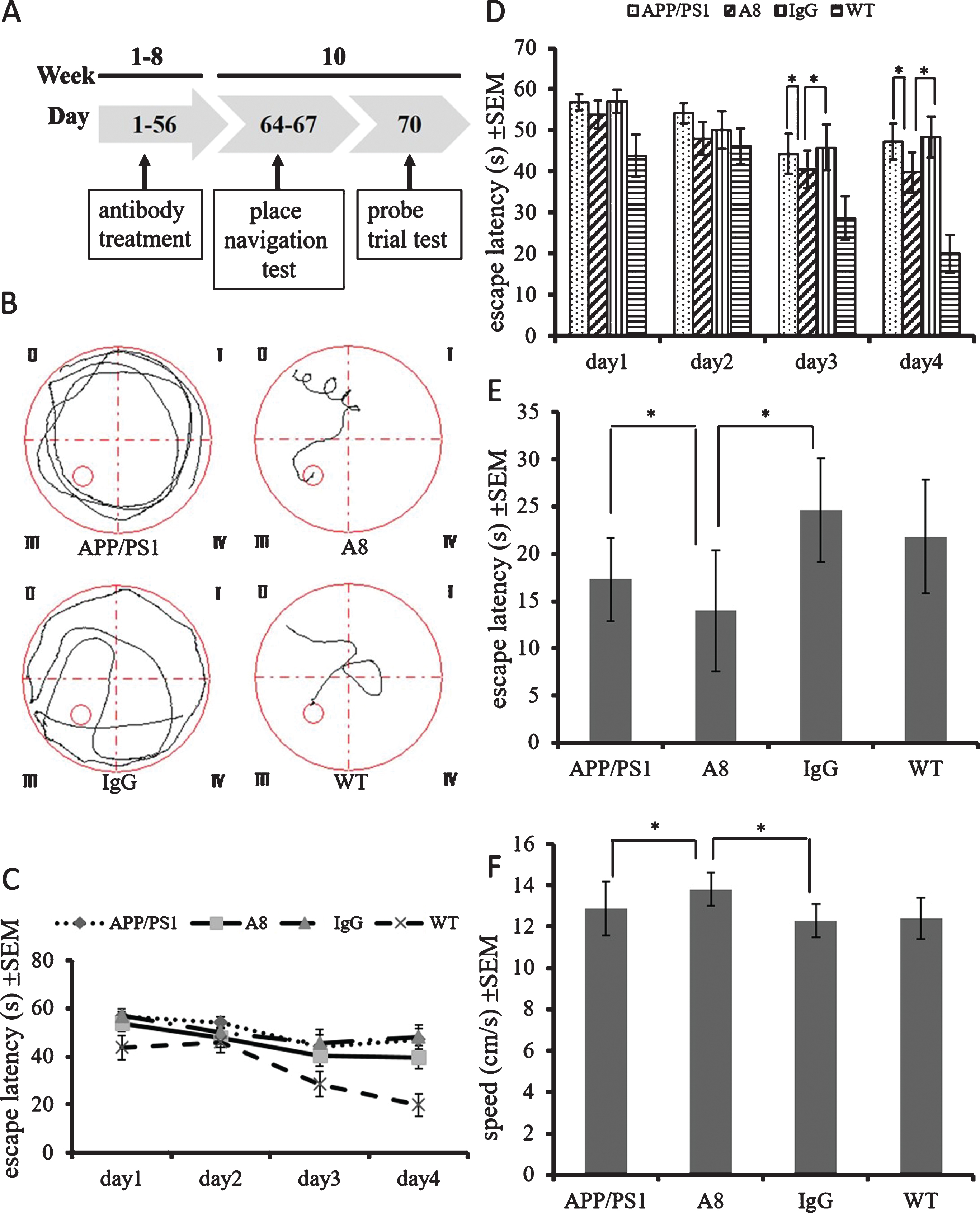
Memory impairment in APP/PS1 mice was alleviated by A8 treatment. Four-month-old APP/PS1 mice were intraperitoneally injected with a dose of 400 μg/week of A8 or nonspecific mouse IgG for 8 weeks. A) During the seven days following the administration of antibodies, a place navigation test (day 1 to day 4) and a probe trial (day 7) were performed in the Morris water maze (MWM). B) The motion traces of mice in each group were recorded by a digital camera. Those four figures (indicated as APP/PS1, A8, IgG, and WT) are representative for the four different groups (APP/PS1 untreated, A8-treated APP/PS1, IgG-treated APP/PS1, and WT untreated). C, D) The escape latency (Y scales) of mice in each group to find the platform in the place navigation test. There is a significant difference between the A8-treated group (n = 16) and the IgG control group or APP/PS1 control group (n = 16) on day 3 and day 4 (Mauchly’s test of sphericity, one-way repeated-measures analysis of variance and Tukey post hoc pairwise comparisons. n = 16 per group. Error bars represent SEM, *p < 0.05). E) The escape latency (Y scales) of mice in the probe trial. A8-treated APP/PS1 mice reached the platform site in a significantly shorter time than IgG control mice or APP/PS1 control mice did (One-way ANOVA and Student’s t-test, n = 16 per group. Error bars represent SEM, *p < 0.05). F) The swimming speed (Y scales) of mice in the probe trial (One-way ANOVA and Student’s t-test, n = 16 per group. Error bars represent SEM, *p < 0.05).
Aβ42 oligomer and plaque but not Aβ40 levels were diminished in the brains of A8-treated APP/PS1 mice
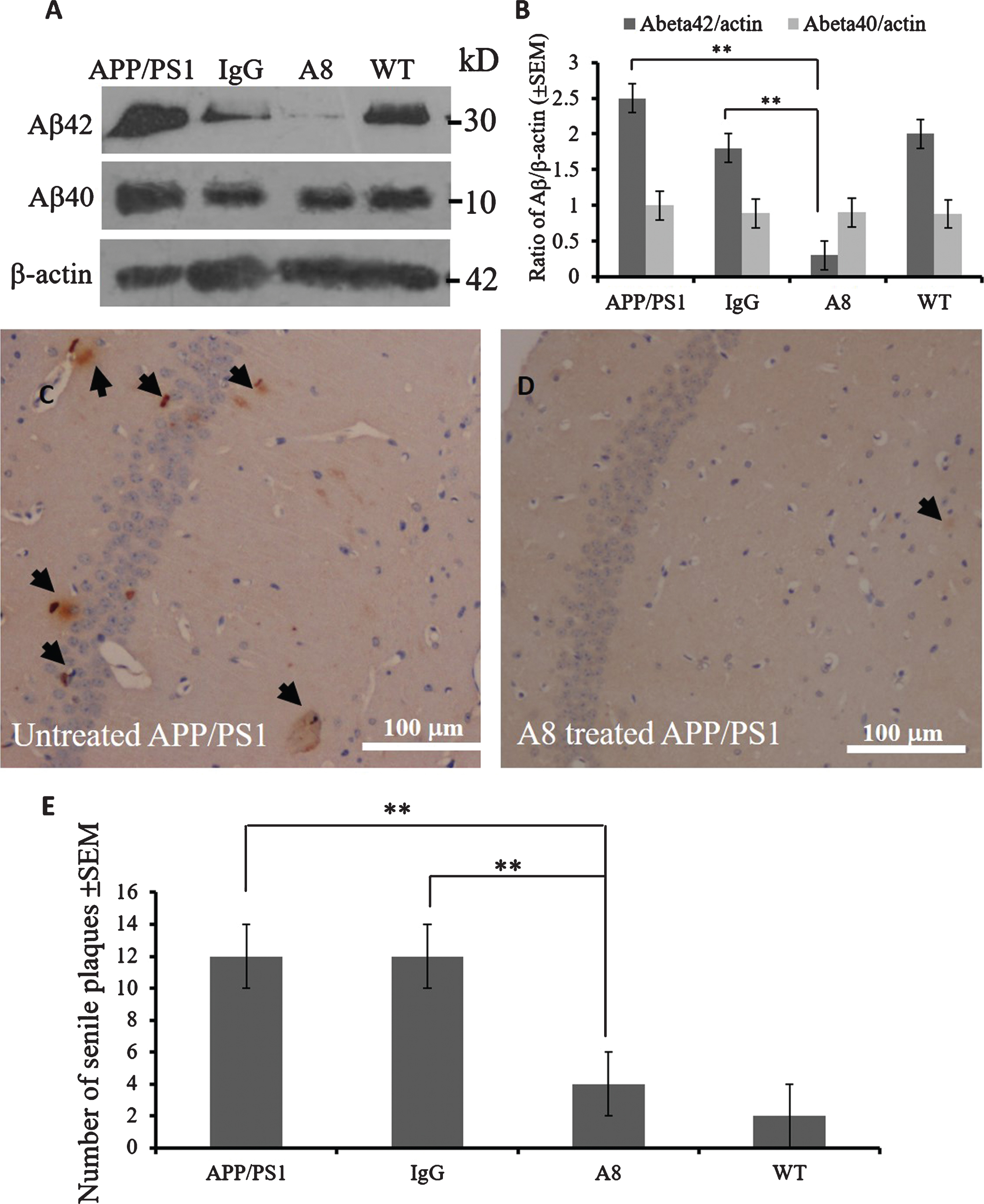
To investigate changes in Aβ load after A8 mAb treatment, we sacrificed each group of mice three days after the MWM test. Aβ42 oligomer and Aβ40 levels were detected via western blot assays, and the Aβ plaques were visualized by immunohistochemistry. The western blot results showed that the expression levels of Aβ42 oligomers (about 30 kD) but not Aβ40 aggregate (about 10 kD) in the brains of the A8-treated group were lower (p < 0.01) than those of the IgG-treated APP/PS1 mouse group or APP/PS1 control mice (Fig. 2A, B), while there was no significant difference in the level of either form between the APP/PS1 and IgG groups.
Aβ
The results in Figure 2A and Figure 2B suggested that A8 mAb mainly targeted Aβ42 oligomers rather than Aβ42. Specificity of A8 mAb to Aβ42 is shown in Supplementary Figure 1. Aβ42 or Aβ40 peptides were aggregated in the same conditions, and A8 or 6E10 mAb were used to detect the aggregates. The results in Supplementary Figure 1 show that A8 recognized Aβ42 oligomers (Lane 2) but not Aβ40 aggregates (Lane 1), while 6E10 recognized both Aβ42 and Aβ40 (data not shown).
Brain samples were fixed in 4% paraformaldehyde. Brain sections, 4 μm thick, were analyzed by IHC with anti-Aβ (4G8) or anti-Aβ42 (12F4) staining. The brown plaques in the hippocampal region, indicating positive Aβ signal, were counted in three fields of view on each section for three mice per group. The numbers of senile plaques in each group were indicated as average values ± standard error. No significant differences in the Aβ levels in brain sections were found between the IgG-treated and APP/PS1 mouse control groups (p > 0.05) (data not shown). However, compared with the IgG group or APP/PS1 control mice (Fig. 2C), the A8-treated group (Fig. 2D) had a significantly decreased number of Aβ plaques in the hippocampal region (p < 0.01, Fig. 2E).
Phospho-tau (pSer231) but not tau levels were reduced
To investigate the effects of A8 administration on tau pathology, we analyzed the expression levels of phospho-tau (p-tau) and tau by western blotting. As shown in Figure 3A, the p-tau231 level in the cortex was lower in the A8 group than in the IgG group or APP/PS1 control mice (p < 0.01, Fig. 3B). IHC confirmed that the level of phospho-tau (pSer231) was reduced significantly in the A8-treated group (Fig. 3D) compared with the IgG-treated control group or APP/PS1 control mice (Fig. 3C) (p < 0.01, Fig. 3E). The staining in the cortex (Fig. 3C) of IgG-treated or APP/PS1 control groups are called neuritic plaques, which is p-tau positively stained in the Aβ plaques. However, no significant differences were found in the expression level of tau among these groups (data not shown).
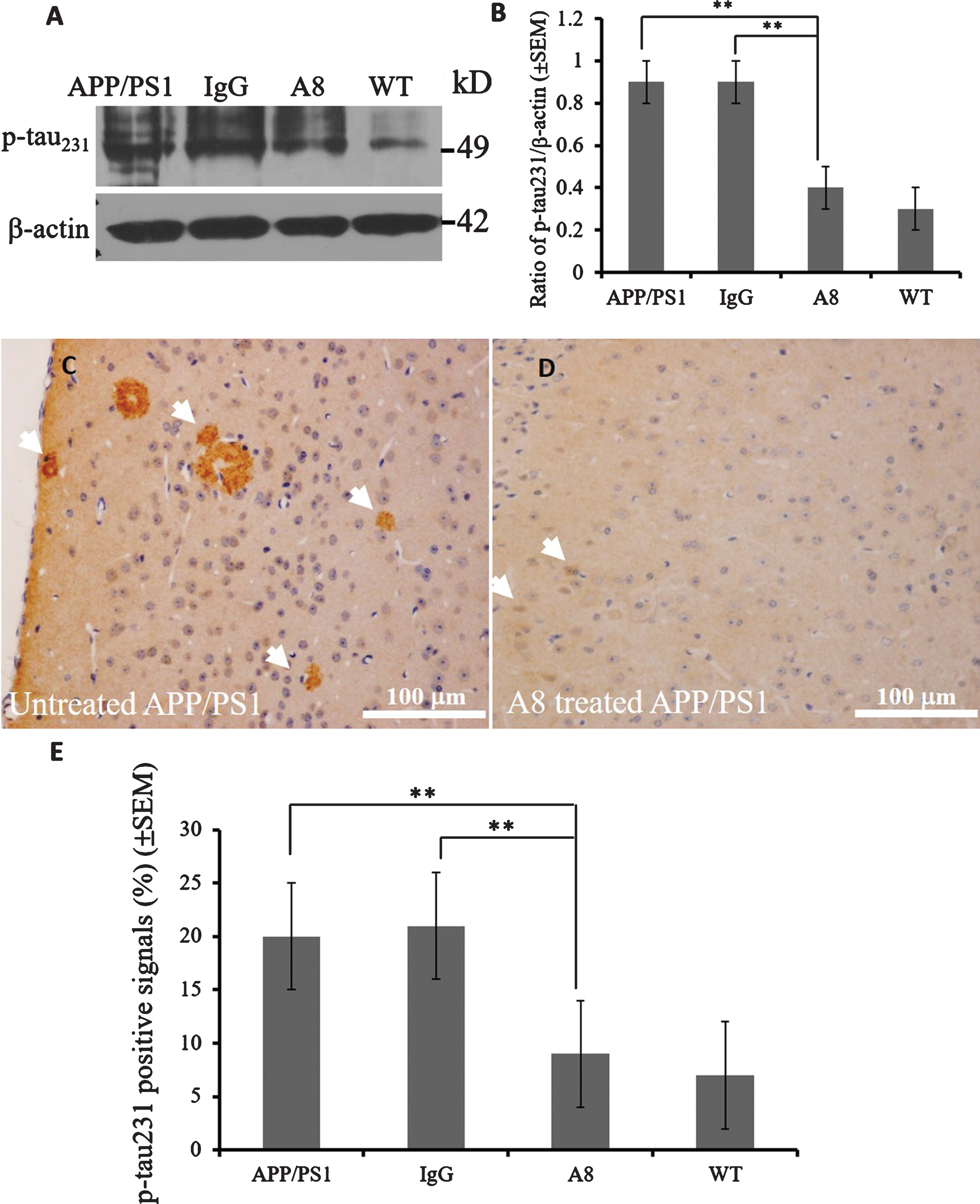
Phospho-tau expression levels were reduced by A8 treatment. A, B) A reduced level of phospho-tau231 can be detected by a western blot; line 1, control group; line 2, IgG-treated group; line 3, A8-treated group; line 4, wild-type mice. In the quantitative graph of the western blot data, the ratio of phospho-tau231 to β-actin was decreased in the A8-treated group (n = 3 mice) compared with the IgG control group or APP/PS1 control mice (n = 3 mice) (One-way ANOVA and Student’s t-test, n = 3 per group, error bars represent SEM, **p < 0.01). C) Phospho-tau231 signal in the cortex (arrow) appeared as brown-stained neuritic plaques in IgG control group. D) The signal of phospho-tau decreased after A8 treatment compared with control treatment (scale bar = 100 μm). E) Compared with the IgG control group (n = 3 mice), phospho-tau231 levels measured by IHC were reduced significantly (One-way ANOVA and Student’s t-test, n = 3 per group, error bars represent SEM, **p < 0.01) in the cortex of A8-treated mice (n = 3 mice), as shown by the quantitative graph.
Density of synapses increased in the brains of A8-treated mice
To investigate whether synaptic loss could be alleviated by A8 treatment in APP/PS1 mice, the synapses in the hippocampus of each group were counted in TEM images. The micrographs showed that the density of synapses was decreased in APP/PS1 mice (Fig. 4A, C) compared with A8-treated mice (Fig. 4, 4D) (p < 0.01, Fig. 4E). The results suggest that A8 mAb has the potential ability to protect synapses from toxicity caused by Aβ accumulation.
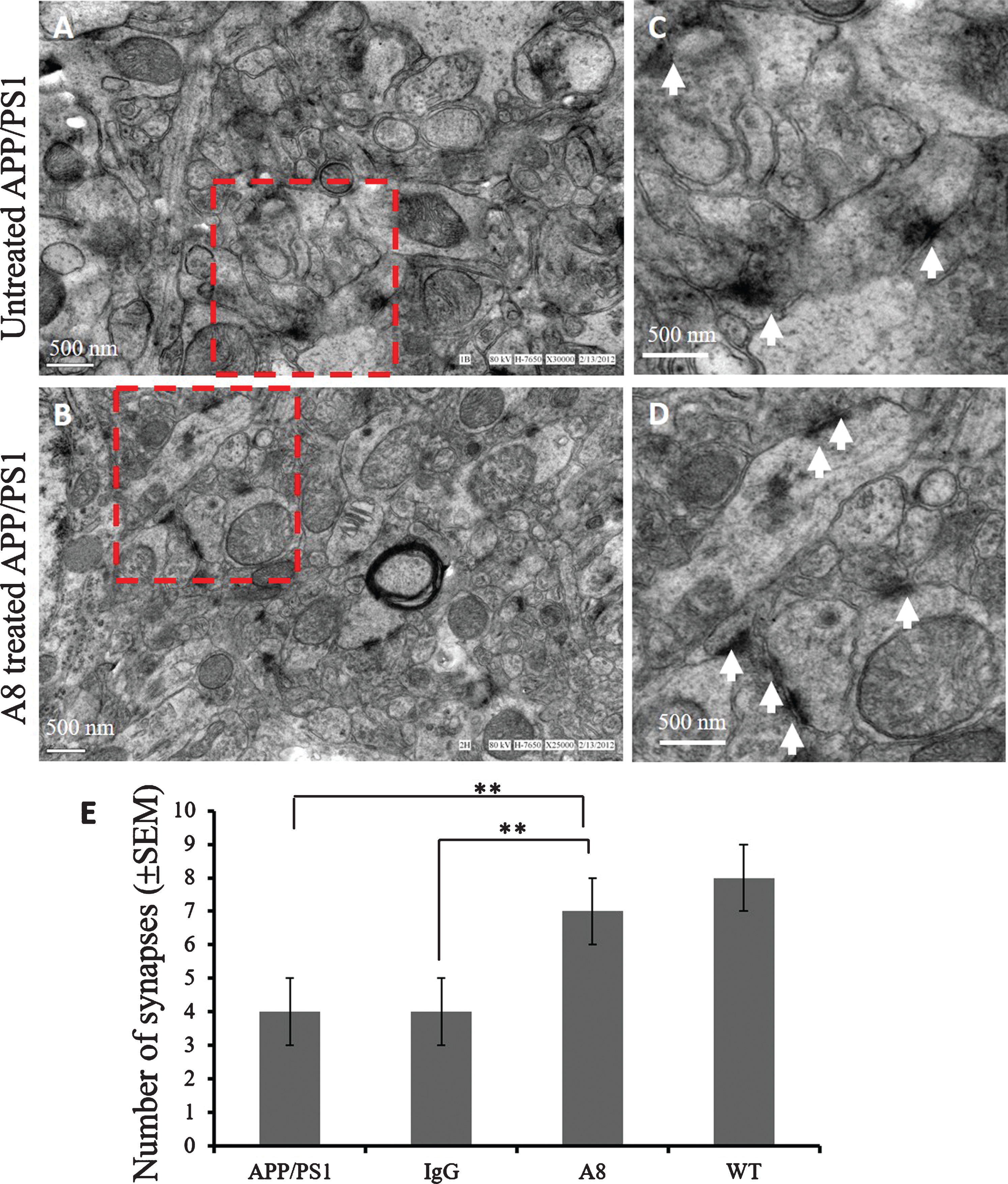
Loss of synapses in the hippocampus was inhibited by A8 treatment. Hippocampal tissue from each group was isolated and fixed in 4% PFA/2.5% glutaraldehyde and then postfixed in 1% OsO4. The stained ultrathin sections were visualized under a TEM. The methods of sampling and observing synaptic images under electron microscope are as follows: under the condition of 20,000–30,000 times magnification, the characteristic synaptic structures including presynaptic membrane, synaptic space and postsynaptic membrane were observed under the transmission electron microscope. The numbers of synapses in equal areas were counted in three fields of view on each grid, and three grids from different mice were analyzed in each group. A) EM images of the hippocampus in the APP/PS1 control group. B) EM images from the mAb A8 treatment group. C, D) The higher magnified image of dotted line frame in A and B (scale bar = 500 nm). E) Quantitative graph of the numbers of synapses in equal areas. The numbers of synapses were counted in three fields of view on each ultrathin section, and three ultrathin sections from different mice were analyzed in each group (One-way ANOVA and Student’s t-test, n = 3 per group, error bars represent SEM,**p < 0.01).
The number of mitochondria with abnormal morphology decreased in the brains of A8-treated mice
To confirm the ultrastructural changes accompanying the increase in synapse density, we examined the mitochondrial pathology. TEM images showed that subcellular structural abnormalities including very large mitochondria (Fig. 5A, arrow), mitochondrial degradation and mitochondrial dissolution (Fig. 5A, C, arrowhead) appeared in the hippocampus CA3 of APP/PS1 mice, compared with the normal mitochondrial morphology (Fig. 5B, D, arrow) in A8-treated group. The number of abnormally enlarged mitochondria decreased in A8-treated mice compared with APP/PS1 control mice and the IgG-treated control group (p < 0.01, Fig. 5E).
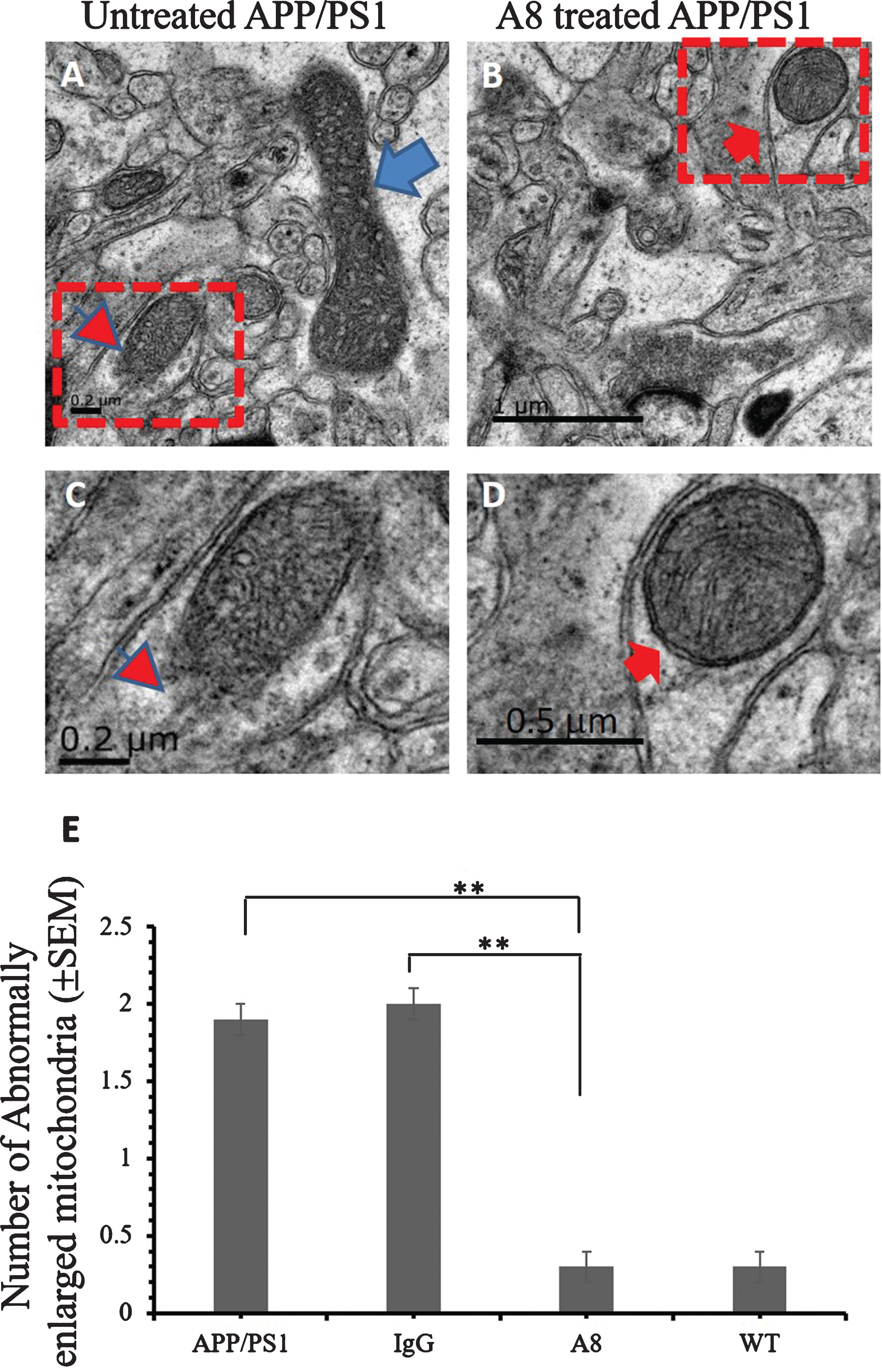
Ultrastructural improvement of mitochondria in the brains of A8-treated APP/PS1 mice. The ultrathin sections were prepared and observed via TEM. TEM images from the hippocampus of the APP/PS1 mouse (A, and C) brain and of the A8-treated APP/PS1 mouse (B, and D) brain were showed here. A) Abnormally enlarged mitochondria (arrow), and damaged mitochondria (arrowhead) (scale bar = 0.2 μm). B) Relatively normal mitochondria (arrow) (scale bar = 1 μm). C) The enlargement image of dotted line frame in (A) (scale bar = 0.2 μm). D) The higher magnified image of dotted line frame in (B) (scale bar = 0.5 μm). E) Quantitative graph of the numbers of abnormally enlarged mitochondria in CA3. The mitochondria with an average inner diameter greater than 1 μm were identified as enlarged mitochondria. The numbers of enlarged mitochondria were counted in three fields of view on each ultrathin section, and three ultrathin sections from different mice were analyzed in each group (One-way ANOVA and Student’s t-test, n = 3 per group, error bar represent SEM, **p < 0.01).
DISCUSSION
In our previous work [16], the specific monoclonal antibody A8, which targets Aβ42 oligomers, was used in passive immunotherapy in the SAMP8 mouse model of AD. We determined that A8 disturbed Aβ fibrillation, suppressed Aβ oligomer cytotoxicity and improved learning and memory in SAMP8 mice. In the present study, A8 mAb was administered to the double-transgenic amyloid precursor protein/presenilin 1 (APP/PS1) mouse model of AD, and similar effects on cognitive behavior and Aβ clearance were detected. Furthermore, we determined for the first time that Aβ immunotherapy by mAb A8 increased the density of synapses and ameliorated mitochondrial lesions and synaptic pathology in the hippocampi of APP/PS1 mice.
Our findings have shown mAb A8 to be effective immunotherapy in two AD mouse models: SAMP8 and APP/PS1. The underlying rationales on which we administered mAb to these two AD mouse models are as follows: (1) approximately 1% of AD represents familial cases, while more than 95% of AD cases are sporadic [24]; (2) the APP/PS1 mouse line used for this study expressed the Mo/Hu APP695swe construct in conjunction with the exon-9-deleted variant of human presenilin 1 (PS1-dE9) [25]; (3) age dependent increases in APP expression levels, Aβ plaque formation, and tau protein phosphorylation in SAMP8 mice have also been demonstrated [26]; (4) the non-transgenic SAMP8 mice have the advantage of late-onset/age-related AD, which models sporadic AD.
Aβ42 and p-tau have been recognized as early biomarkers for AD diagnosis, although Aβ40 and unphosphorylated tau are also involved in the pathogenesis of AD. The western blot results in the present study showed that Aβ42 but not Aβ40 levels in the brains of APP/PS1 mice decreased (p < 0.05) after eight-week treatment with mAb A8 compared with the levels in the IgG-treated group (Fig. 2A, B). The results were consistent with the specificity of A8 mAb (Supplementary Figure 1). Therefore, Aβ42 but not Aβ40 in the brain would be a precise indicator for monitoring the effect of AD immunotherapy. Our results showed that p-tau231 decreased in APP/PS1 mice after mAb A8 immunotherapy, and p-tau404 decreased in SAMP8 mice after A8 administration. Further research should be conducted to elucidate the underlying mechanism.
The tau pathology includes three types: NFTs (tau fibers are in the cell bodies), neuropil threads (tau in process fibers), and neuritic plaques (tau in plaque) here in the cortex (Fig. 3C). However, the staining of the neurons in the hippocampus is not strong enough to be called NFTs (data not shown). We focused on the study of the early effects of A8 immunotherapy; therefore the mice was only six month when detected by IHC. When the mouse get older, maybe the NFTs will show up.
Cognitive deficits are one of the pathological features of AD, and pathological ultrastructural changes in the number and morphology of synapses seem to be widespread alterations in the APP/PS1 AD model. In our study, the number of synapses per unit area declined less in the A8 immunotherapy group than in the untreated APP/PS1 mouse model. Meanwhile, we also found an improvement of pathological synaptic injury in A8-treated mice. Therefore, synapse density should be used in the future to evaluate synaptic effects in AD antibody and drug screening.
In the present study, very large mitochondria could be detected under TEM observation in APP/PS1 mice (Fig. 5A), but not in the mAb A8 treatment group (Fig. 5B). These data suggest that A8 may be useful for improving mitochondria dynamics in the brain by inhibiting the toxicity of Aβ oligomers via immunotherapy. Mitochondrial dynamics are regulated by a delicate balance of two opposing processes: mitochondrial fusion and mitochondrial fission, which are critical for mitochondrial integrity in eukaryotic cells. Therefore, mitochondrial signal cascades could be suitable targets for AD therapeutic strategies in the future.
Taken together, our data indicated that mAb immunotherapy has the potential to reduce cognitive deficits and synaptic lesions in AD. The future research direction is to elucidate the regulatory mechanism of A8 mAb in improving synaptic pathology and energy metabolism in the brain. Therefore, identifying antibody variants that retain amyloid clearance and improve synaptic function remains a major focus of translational research in the field of AD immunotherapy.
Footnotes
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81100809 and 81271417), the Beijing Natural Science Foundation (7152090) and the Fundamental Research Funds for the Central University of China (2015JBM096) to YZ. We thank Drs. William L. Klein and Mary P. Lambert (Northwestern University) for their advice on antibody preparation.