
Abstract
Background:
Epidemiological studies suggest a relationship between posttraumatic stress disorder (PTSD) and dementia.
Objective:
This study assessed whether Alzheimer’s disease (AD) imaging biomarkers were elevated in Vietnam veterans with PTSD.
Methods:
The study compared cognition, amyloid-β, tau, regional brain metabolism and volumes, and the effect of APOE in 83 veterans with and without PTSD defined by the Clinician-Administered PTSD Scale.
Results:
The PTSD group had significantly lower education, predicted premorbid IQ, total intracranial volume, and Montreal Cognitive Assessment score compared with the controls. There was no difference between the two groups in the imaging or genetic biomarkers for AD.
Conclusion:
Our findings do not support an association between AD pathology and PTSD of up to 50 years duration. Measures to assess cognitive reserve, a factor that may delay the onset of dementia, were lower in the PTSD group compared with the controls and this may account for the previously observed higher incidence of dementia with PTSD.
Keywords
INTRODUCTION
Posttraumatic stress disorder (PTSD) is a chronic and disabling condition with a lifetime prevalence of 1–9% in the general population and an increased prevalence in military veterans [1, 2]. Previous studies that provided compelling evidence for cognitive impairment in combat-related PTSD sparked a series of investigations that examined the association between PTSD and dementia in Vietnam veterans [3, 4]. Several epidemiological studies have recently demonstrated an increased incidence of dementia including Alzheimer’s disease (AD) on clinical criteria in older veterans with PTSD compared with those without PTSD [5–9]. Furthermore, structural and functional magnetic resonance imaging (MRI) studies have shown reduced volumes of structures involved in cognition such as hippocampi, amygdala, and anterior cingulate cortex along with reduced activity in the anterior cingulate cortex in PTSD [10, 11]. Metabolic imaging studies with 18F-fluorodeoxyglucose positron emission tomography (FDG PET) have produced inconsistent findings in PTSD with a tendency to hypometabolism in anterior cingulate, limbic, and temporal regions [12]. These studies had small, middle aged cohorts. In contrast, more specific patterns of brain hypometabolism occur in most dementia syndromes [13]. Brain hypometabolism reflects synaptic dysfunction and loss and is regarded as a measure of neurodegeneration that may precede the onset of symptoms by several years [14, 15]. The hypometabolic findings characteristic of AD relate to posterior brain regions, in particular the posterior cingulate gyrus and parietotemporal cortex.
The previous studies did not examine the specific pathological biomarkers of AD, amyloid-β (Aβ) and tau, in PTSD. Deposition of extracellular neuritic plaques and intracellular neurofibrillary tangles is the pathological hallmark of AD [16]. Enabling in vivo and early detection of Aβ and tau using specific radioactive ligands, amyloid and tau PET represents a breakthrough in AD research. The uptake of these radioactive tracers is a proxy measure of Aβ and tau although not the same as the autopsy, which is the gold standard for AD diagnosis. Aβ plaques can be detected on PET up to 20 years before the onset of dementia due to AD [17, 18]. Compared with Aβ plaques, the progression of neurofibrillary tangles is believed to occur closer to the development of symptoms and consequently has shown a better correlation with neuronal damage and cognitive deterioration in patients with AD [19]. Current knowledge of the role of Aβ and tau in the pathogenesis of AD, although not fully understood, is expanding and the relationship may be best described as synergistic [20]. Animal studies have observed elevated Aβ in PTSD [21]. Exposure to psychological trauma and corticotrophin-releasing factor have been reported to enhance both Aβ plaque formation and tau phosphorylation [22].
A recent study by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) group in Vietnam veterans (ADNI-DOD study) did not find an increased risk of AD with PTSD according to the global Aβ burden estimated from 18F-florbetapir PET [23]. Whether tau accumulation or other types of neurodegeneration explain the elevated incidence of dementia in PTSD remains unknown. The aim of this study was to use 18F-florbetaben and 18F-AV-1451 to quantify Aβ and tau burden, respectively, 18F-FDG to estimate regional brain metabolism, and MRI to measure regional brain volume in Vietnam veterans with and without PTSD. Our hypothesis is that veterans with PTSD have increased AD pathology as detected by neuroimaging biomarkers for AD, and neurodegeneration prior to a diagnosis of mild cognitive impairment or dementia.
METHODS
Participant recruitment
The institutional review board of Austin Health, one of the major metropolitan hospitals in Melbourne, provided ethics approval and written informed consent was obtained from all participants.
Participants were Australian male Vietnam veterans recruited from the community via advertisement. Interested veterans went through preliminary screening. We applied the following exclusion criteria: substance abuse in the past six months, traumatic brain injury, psychosis, bipolar affective disorder, dementia, existing diagnosis of mild cognitive impairment (MCI), and any unstable medical condition that could have made participation difficult or have a significant impact on cognitive assessment. MCI and dementia were excluded to reduce recruitment bias and avoid confounding of the diagnosis of PTSD. Veterans who passed the initial screening had a face-to-face evaluation with a psychiatrist.
Brain imaging
All participants underwent 20-min PET scans acquired 90 min after a slow IV bolus administration of 250 MBq (±10%) of 18F-florbetaben and 70 min after the injection of 370 MBq of 18F-AV-1451. Scans were acquired on a Siemens PET/CT mCT128 and CT attenuation correction was applied. Image reconstruction used the Ordered Subset Expectation Maximization algorithm. There was no correction for partial volume effect. We analyzed the PET scans with the Computational Analysis of PET from AIBL and calculated standardized uptake value ratio (SUVR) of 18F-florbetaben using cerebellar grey matter uptake as the reference to quantify global Aβ burden [24]. Amyloid burden was also calculated in Centiloid units using the standard Centiloid method cortical region of interest normalized to whole cerebellum as previously described [25]. 18Florbetaben scan was read visually by three readers and the classification into negative or positive scan was based on majority results. The visual inspection was based on brain amyloid plaque load, which was derived from the regional cortical tracer uptake in the four regions: lateral temporal cortex, frontal cortex, posterior cingulate cortex/precuneus, and parietal cortex. Typical transverse PET slices were judged as negative if the tracer uptake in the grey matter was lower than that of the white matter and positive if the uptake in the grey matter was equal to or more than that in the white matter. The SUVR of 18F-AV-1451, using cerebellar grey matter uptake as the reference region, estimated global and regional tau deposition. We measured tau in the following regions: mesial temporal; temporoparietal; and rest of the neocortex.
A part of the data analyzed in the preparation of this article were obtained from the ADNI database (http://adni.Ioni.usc.edu). The primary goal of ADNI has been to test whether serial MRI, PET, other biological markers and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. For up-to-date information, see http://www.adni.info.org.
For the acquisition of 18F-FDG PET scan, participants fasted for four hours and then had an injection of 200 MBq of 18F-FDG. As per standard practice, they remained in a quiet, darkened room for 30 min with eyes open in order to keep the occipital metabolism consistent. Acquisition was commenced 30 min after the injection on a Philips Allegro PET camera. A post-injection transmission scan for attenuation correction was performed. Acquisition time was 20 min. Reconstruction was performed with a RAMLA filter. SUVR was calculated for frontal, mesial temporal, and rest of the neocortex. Participants underwent 3-Tesla Siemens Trio brain MRI for the measurement of hippocampal volume and the total intracranial volume (TICV). A three-dimensional (3D) T1 magnetization-prepared rapid gradient echo (MPRAGE) was acquired with the following parameters: FoV = 260×256, Matrix = 240×256, 160 slices, 1.0×1.0×1.2 mm voxels, TR = 2300 ms, TE = 2.98 ms, flip angle = 9°. The T1 weighted images were rigidly registered to the Montreal Neurological Institute (MNI) average brain and segmented into grey and white matter and cerebrospinal fluid space with Expectation Maximisation Segmentation algorithm. Partial tissue classification and cortical thickness were then estimated using a software Computational Quantification program [24]. The Harmonized Hippocampus protocol was used to define the hippocampus region of interest. Volumetry was adjusted for the TICV by dividing the regional volume by TICV.
Clinical and cognitive assessment
We used Clinician-Administered PTSD Scale (CAPS) - DSM-IV version to assess PTSD [26]. A score of 40 or more and a history of combat exposure constituted the inclusion criteria for the PTSD group. This score was based on the existing diagnostic utility data for a range of selected cut-off scores which indicated that a CAPS score of 40 had 93% sensitivity and 80% specificity for dichotomous classification of PTSD [27]. A CAPS score of 30 or less and military experience formed the inclusion criteria for the control group. We have chosen these scores to ensure a clear separation between the diagnostic groups and controls. Subjects who scored between 30 and 40 were excluded from the analysis based on the presumption that they had fluctuating and sub-threshold symptoms. The Geriatric Depression Scale (GDS) measured depressive symptoms [28]. Participants underwent APOE ɛ4 genotyping. A vascular risk factor score was calculated by giving one point to each of the following: hypertension, ischemic heart disease, previous history of stroke, atrial fibrillation, current smoking, diabetes mellitus, body mass index over 30, and hypercholesterolemia. Sum of all points gave cumulative vascular risk score. Veterans with mild, moderate, or severe traumatic brain injury according to the criteria set by the U.S Department of Defense (DoD) were excluded.
A standardized neuropsychological test battery assessed cognitive functions [29–35]. The following tests measured memory component: the delayed paragraph recall from the Logical Memory Test II, Rey Auditory Verbal Learning Test 30-min delay, and the Rey-Osterrieth Complex Figure Test (ROCFT) 30-min delay. We calculated a composite memory score from these three tests using standard deviation and mean scores from the control population. We used Trail Making Test part A to assess the visual orientation and processing speed, part B to measure executive function, Wechsler Adult Intelligence Scale to measure attention, categorical fluency test to assess semantic fluency, and ROCFT to assess visuospatial orientation and constructional skills. The Mini-Mental State Examination and the Montreal Cognitive Assessment (MoCA) measured global cognitive function [36, 37]. Along with the measurement of attention, delayed recall, and visuospatial abilities, MOCA assesses executive function using an alternation task adapted from the Trail Making Test Part B, phonemic fluency task and two-item verbal abstraction task. Wechsler’s Test of Adult Reading (WTAR) estimated premorbid Intelligent Quotient (IQ) [35]. WTAR was adjusted for age and then predicted IQ was calculated using published criteria.
Statistical analysis
The PTSD and the control groups were compared for the following outcome variables: 18F-florbetaben SUVR and Centiloid units, 18F-AV-1451 and 18F-FDG SUVRs, MRI regional volumes, and cognitive test scores. Age, APOE ɛ4, and vascular risk factors were analyzed as covariates in a multivariable regression analysis because of their known association with amyloid retention. For this study, current PTSD was the explanatory variable for the primary analysis given a priori that it is the persistence of symptoms that is related to the risk of AD in late life. Life-time history of PTSD was also examined. The two-tailed results were corrected for multiple comparisons using the Benjamini-Hochberg procedure. Chi-square (χ2) was used to analyze categorical variables, such as positive or negative 18F-florbetaben scan, and APOE ɛ4 status. Pearson correlation test was performed for the whole sample to test correlation between the tracer SUVRs and CAPS scores. The analyses were performed on SPSS version 21. Using florbetapir Aβ PET results from normal controls in the ADNI study, we calculated that to detect a group difference with an effect size of 0.75, with 80% power at α= 0.05, required 29 subjects in each group.
RESULTS
From 2014 March to May 2017, 169 male Vietnam veterans expressed interest in the study. 20 veterans later withdrew from the study for reasons of inconvenience and perceived distress. After excluding 66 veterans (including three veterans who had CAPS score between 30 and 40) for the reasons shown in Fig. 1, we collected the neuropsychological and PET data from the remaining 83 veterans. A diagnosis of lifetime PTSD was present in 53 veterans, and 30 veterans were controls. Among veterans with lifetime PTSD, a diagnosis of current PTSD was present in 30. All veterans experienced PTSD symptoms either during or soon after military service.
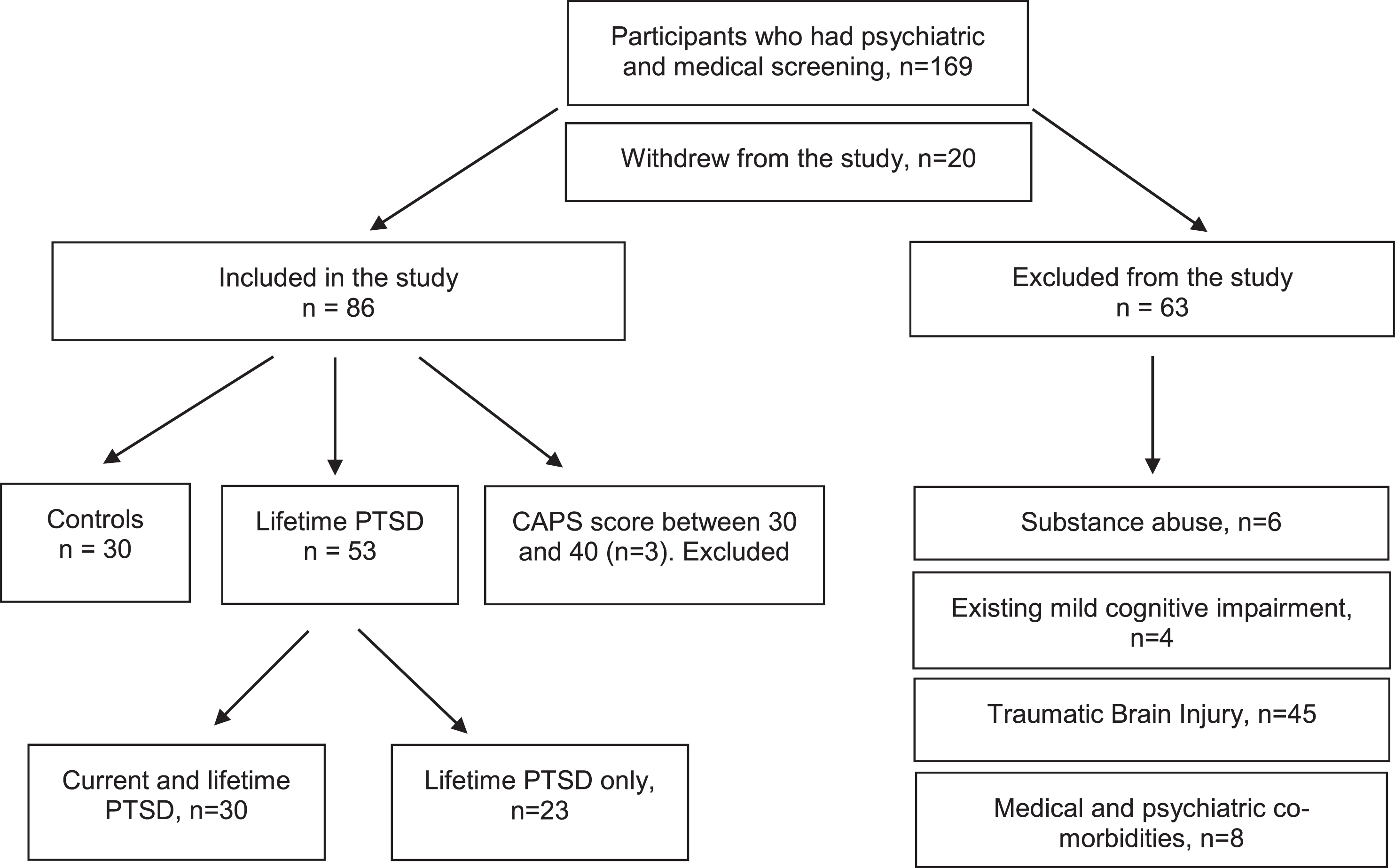
Participants recruitment.
The participants’ characteristics are shown in Table 1. The median current CAPS score for the PTSD group was 52.50 compared with 4 for the control group (Mann-Whitney U = 0.000, p < 0.001). Veterans with current PTSD were slightly younger than those without PTSD (PTSD group mean age: 67.80±2.48 versus the control group mean age: 70.23±5.46; p = 0.043; CI = 0.220–4.64; Cohen’s d = 0.57). Median predicted premorbid IQ (104 versus 114; U = 201.00; p < .001) and years of education (11 versus 12; U = 305.00; p = 0.043) were significantly lower whereas the median GDS score (5.50 versus 1, U = 130.00, p < .001) was significantly higher in the PTSD group than in the controls. The TICV was significantly lower in the PTSD group than in the control group (1565.173±1114.31 cm3 versus 1674.12±1474.64 cm3; p = 0.010; CI = 33.59–184.307; Cohen’s d = 0.40). The median vascular risk factor score did not differ significantly between the groups (1 versus 2, U = 384.50, p = 0.318). APOE ɛ4 carrier status was available for 55 (92%) participants, and at least one allele was present in 7 (24%) veterans with PTSD and 2 (9%) controls (χ2 = 2.70, p = 0.10).
Participant characteristics
PET imaging
There was no significant difference between the PTSD group and the controls in the mean SUVR of the Aβ tracer, 18F-florbetaben (p = 0.927, Cohen’s d = 0.02). According to the visual inspection of 18F-florbetaben scans, 7 (21.8%) veterans with current PTSD and 4 (10.5%) veterans without PTSD had a positive scan. This difference was not significant (χ2 = 0.57, p = 0.48). In the multivariable regression analysis with 18F-florbetaben SUVR as the dependent variable and current PTSD, age, APOE ɛ4, and vascular risk factors as the independent variables, current PTSD did not predict 18F-florbetaben SUVR (standardized coefficient β= –0.093, p = 0.513), whereas APOE ɛ4 did (R2 = 0.126, standardized coefficient β= 0.185, p = 0.043). The regional and global uptake of the tau tracer, 18F-AV-1451 did not differ between the PTSD and control groups (Table 2). 18F-AV1451 SUVR did not correlate with any cognitive score. There was no significant correlation between the severity of PTSD as measured by CAPS score and global or regional 18F-AV-1451 SUVRs. There were no differences in the 18F-FDG SUVRs between the groups (Table 2). There was no correlation between the severity of PTSD as measured by CAPS and 18F-FDG SUVR in any region. We repeated the analysis for subjects with lifetime PTSD with and without current PTSD (n = 53) against the same control group (n = 30). There was no significant difference in the uptake of any tracer globally or regionally between the two groups.
PET Tracer binding expressed in SUVR
Adding the U.S. ADNI-DOD study amyloid PET data expanded the sample size to a total of 97 Vietnam veterans with PTSD and 85 controls. Centiloid units were calculated to allow the merging of scans obtained with the different Aβ PET tracers, florbetapir and florbetaben. The combined data did not reveal a significant difference between the two groups in the Centiloid values (PTSD Mean: 9.01±20.73; versus Control Mean 14.37±26.12 Cohen’s d = 0.22; Median rank: 89.85 versus 93.39, p = 0.651; Mann-Whitney U = 3962.00). More veterans in the control group than in the PTSD group had a positive amyloid scan based on Centiloid score of 25 or more (13 versus 7, χ2 = 7.47, p = 0.024, uncorrected). With the additional ADNI data APOE ɛ4 was present in 23 veterans with PTSD and 17 veterans without PTSD (χ2 = 0.567, p = 0.451, uncorrected).
MRI results
Forty of the lifetime PTSD group including 30 with ongoing or current PTSD, and 25 controls underwent MRI. Others did not have MRI for reasons of inconvenience and metal safety. The TICV was slightly but significantly lower in the current PTSD group than in the control group (1565.173±1114.31 cm3 versus 1674.12±1474.64 cm3; p = 0.01; CI = 33.59–184.307; Cohen’s d = 0.40). There was no significant difference between veterans with and without current PTSD in the adjusted volumes of the hippocampus, amygdala, anterior cingulate cortex, middle frontal, or orbitofrontal cortex on either side (Table 3). The analysis was repeated with subjects who had a lifetime diagnosis of PTSD. The TICV was significantly lower in the lifetime PTSD group compared with the control group, but volumetric grey matter measures did not significantly differ between the groups. There was no significant correlation between volumetric measure and severity of current PTSD as quantified by the CAPS score.
MRI volumes in PTSD and controls, adjusted for total intracranial volume
Cognitive functions
The performance of both groups was within the normal range of age-adjusted published norms of the neuropsychological tests except on the MoCA where the mean score for the PTSD group (25.27) was fractionally below the conventional cut-off score of 26. In comparison with the controls, veterans with current PTSD scored lower on global cognitive function as measured by the MoCA (Median: 26 versus 28; U = 250.00; p = 0.027). We performed a multivariable regression analysis after log-transformation of the total MoCA score. The predicted premorbid IQ and the GDS score significantly correlated with the total MoCA score and also differed between the groups. Therefore, these variables were included in the model along with current PTSD as explanatory (independent) variables with the MoCA as the dependent variable. The regression analysis met all assumptions. In the regression analysis (adjusted R2 = 0.249), the difference in the performance on the MoCA (β= –0.252, p = 0.081) did not retain significance when predicted premorbid IQ (β= –0.341, p = 0.009) and depression score (β= 0.006; p = 0.611) were adjusted. The cognitive scores are summarized in Table 4. There was no significant difference between the PTSD groups and the controls in the other cognitive measures. The correlation between predicted premorbid IQ and the TICV was significant (r = 0.351, p = 0.013).
Cognitive functions in PTSD and controls
RAVLT, Rey Auditory Verbal Learning Test; MMSE, Mini-Mental State Examination; MoCA, Montreal Cognitive Assessment.
DISCUSSION
There is abundant literature on cognitive function, structural brain imaging, and incidence of dementia in Vietnam veterans with PTSD, but little is known about the AD pathological biomarkers in PTSD. This study measured Aβ and tau in vivo in Vietnam veterans with PTSD against veterans without PTSD. This ensured a homogenous population with military experience in both groups and an age range in which preclinical AD pathology is developing and detectable in the general population. The present study did not show a significant association between PTSD and increased amyloid deposition in the brain as measured by 18F-florbetaben. Global amyloid burden or regional amyloid as reflected in the visual inspection was not significantly different between the PTSD and the control groups. Amyloid deposition is the earliest detectable marker of AD and amyloid PET scans become abnormal up to 20 years before the clinical diagnosis of AD dementia [17, 18]. Our finding is consistent with the results from the US-based ADNI Veterans study that also did not find an association between PTSD and increased Aβ deposition [23]. Like our findings, the PTSD cohort in the ADNI-DOD veterans study had worse global cognition than controls. The ADNI-DOD data revealed a significantly lower level of education in the PTSD group, and our study showed the same.
To the best of our knowledge, this is the first study to report tau deposition in PTSD along with Aβ, regional brain metabolism and brain volumetry along with neuropsychological data. This is perhaps the most comprehensive assessment of biomarkers of AD in PTSD. We found no increase in binding of the tau tracer 18F-AV-1451 in PTSD. Similarly, there was no difference between the PTSD group and the controls in regional brain metabolism or regional brain volumetry. If PTSD causes neurodegeneration associated with various dementia syndromes, then it would be likely that after 40 or more years of symptoms in the age group under study, some change would be present. 18F-FDG PET scan has been reported as showing abnormality several years before the onset of AD dementia [14, 15]. Similarly, alterations in hippocampal and regional cortical volumes in PTSD could be temporary, or the mild volume reductions as previously described may be masked by the atrophy that occurs with normal aging. Our participants were in their 60 s and 70 s, and the effect of aging may have had more impact on volumetric changes than PTSD itself, in cortical as well as hippocampal regions. A longitudinal study is required to address these possibilities.
Commensurate with the finding of no difference between PTSD and the control status regarding Aβ, tau, and regional brain metabolism and volumes, there was no evidence of an independent association between PTSD and cognitive impairment. Although previous studies, mostly done in younger veterans, have shown impaired cognitive function in PTSD, our findings suggest that cognitive functions are mostly intact in older veterans with PTSD and the mild impairment we found in the global cognitive performance was reflective of predicted premorbid IQ and the affective state. Our finding is in line with previous data that suggested lower premorbid IQ as a vulnerability factor for the development of PTSD upon trauma exposure and associated cognitive impairment [38, 39]. A co-twin-control study of veterans found that premorbid cognitive ability, as measured by Armed Forces Qualification Test, predicted the future risk of PTSD in a dose dependent manner indicating the role premorbid intellectual function in the adaptive coping ability after trauma [39]. It should be noted that patients with PTSD do not form a low IQ group because the premorbid IQ in the PTSD group was consistent with the expected general population average, but the veteran controls were above average.
The findings of the present study and those of the ADNI-DOD veterans study do not lend support to a direct link between PTSD and AD pathology. Therefore, it is worth exploring the potential explanatory factors behind the previously reported association between PTSD and dementia including AD [5–9]. The present study suggests a low cognitive reserve (CR) in PTSD. Cognitive reserve is a potential mechanism to buffer the impact of the pathological process associated with AD [40]. With high CR more pathological load is required to produce the same degree of cognitive impairment than with low CR possibly because of recruitment of alternate functional circuits particularly in the dorsolateral prefrontal cortex [41]. High CR affords protection against the onset of dementia, not the neuropathological markers of AD suggesting the role of CR as neurocompensation rather than neuroprotection [42].
According to our findings, predicted premorbid intelligence and education, the proxy measures of CR and the TICV which is a measure of brain reserve (BR) were significantly lower in the PTSD group than in the controls. A recent study demonstrated that increased intracranial volume mitigated adverse effects of dementia pathology on cognitive function, particularly attention and executive function [43]. Likewise, premorbid cognitive performance predicted the onset of dementia independent of the potent genetic risk factor for AD, APOE ɛ4 [44]. Furthermore, physical and cognitive engagement and leisure activities contribute to CR, but evidence suggests that avoidant behavior and hyperarousal symptoms of PTSD may preclude such activities [45, 46]. We postulate that the relatively low CR and other dementia risk factors associated with PTSD may account for previously observed higher incidence of dementia with PTSD. It may be noted, however, that it is not the abnormally low CR or BR that may explain the association of PTSD and dementia. It is a relative concept in comparison with the controls that is worth considering and the previously reported increased risk of dementia with PTSD was also in comparison with the controls. This may suggest that while subjects with PTSD may have the same risk as that of general population, those without PTSD may have additional protection against the onset of dementia from increased CR or BR and the previous epidemiological studies may be reflective of this difference. The clinical implication is that treatments that aim to restore physical and cognitive activities in patients with PTSD may increase CR and lead to dementia risk reduction. In this context, reverse causation hypothesis is also valid: i.e., people with higher reserve may more frequently choose to engage in these types of activities.
This study has several limitations. We could not access direct measurements of pre-military IQ; premorbid IQ in our study was an indirect measure based on the WTAR. Nonetheless, WTAR was validated and used in the cognitive assessment of traumatic brain injury studies [47]. Secondly, given the negative findings, type II error is an important consideration. Although there were small numbers of veterans with a positive amyloid scan and the samples were underpowered for Chi-square test, the analysis for the global burden of amyloid by SUVR had 80% power to detect a difference with effect size of 0.75 or more. For the amyloid imaging, adding the ADNI data did give improved power sufficient to detect an effect size of 0.43. Another limitation of the study was that large numbers of veterans met the exclusion criteria. Among the excluded veterans some declined participation because of perceived stress. Whether they had more severe PTSD or would have altered the results is unknown. The ADNI-DOD study also reported a large number of excluded veterans. Head injury, substance abuse, and medical illnesses are comorbid with PTSD and such factors may lead to restricted inclusion of veterans. Limited inclusion could impact the generalizability of findings. The literature of CR and BR is complex and still evolving, and an explanation of the relationship between PTSD and dementia, being far from conclusive, needs more research.
In conclusion, our findings indicate that PTSD is not associated with an increased prevalence of the biomarkers for the specific pathology of AD or other forms of progressive neurodegeneration. Compared with the controls, veterans with PTSD had a relatively low cognitive reserve. Given that high cognitive reserve may delay the onset of dementia, low cognitive reserve in PTSD may explain its previous association with dementia.
Footnotes
ACKNOWLEDGMENTS
The authors thank the U.S. Department of Defense and Piramal Pharmaceuticals for supporting this study. The study utilized the infrastructure and resources of Australian Imaging Biomarkers and Lifestyle (AIBL) Study of ageing.
Part of the data analyzed in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: ![]()
ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.;Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (![]() ). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
This study was modelled on the U.S. Alzheimer’s Disease Neuroimaging Initiative - Department of Defense veterans study. Authors presented this work in the Annual Meeting of American Psychiatric Association, May 2016 in Atlanta and also in the Alzheimer’s Association International Conference, July 2017 in London.