
Abstract
Presenilin 1 gene (PSEN1) mutations are the most common cause of familial Alzheimer’s disease (FAD). One of the most abundant FAD mutations, PSEN1 A431E, has been reported to be associated with spastic paraparesis in about half of its carriers, but the determining mechanisms of this phenotype are still unknown. In our study we characterized three A431E mutation carriers, one symptomatic and two asymptomatic, from a Mexican family with a history of spastic paraparesis in all of its affected members. At cognitive assessment and MRI, the symptomatic subject showed an atypical non-amnestic mild cognitive impairment with visuospatial deficits, olfactory dysfunction and significant parieto-occipital brain atrophy. Furthermore, we found several periventricular white matter hyperintensities whose progression pattern and localization correlated with their motor impairment, cognitive profile, and non-motor symptoms. Together, our data suggests that in this family the A431E mutation leads to a divergent neurological disorder in which cognitive deterioration was clinically exceeded by motor impairment and that it involves early glial and vascular pathological changes.
Keywords
INTRODUCTION
Familial Alzheimer’s disease (FAD) is the autosomal dominant form of Alzheimer’s disease (AD) and accounts for less than 1% of the total of cases reported worldwide [1]. It is caused by mutations in the Amyloid Precursor Protein (APP), Presenilin 1 (PSEN1), or Presenilin 2 (PSEN2) genes, or APP duplications, with mutations in PSEN1 representing the most frequent cause of the FAD [2, 3]. Despite its low incidence, FAD provides a model for the study of the pathogenic mechanisms behind AD-linked neurodegeneration, mainly because the majority of the pathogenic mutations show complete penetrance and segregate with the presentation of dementia. Furthermore, FAD pathology closely resembles the pathology seen in patients with the sporadic form of AD, so that both types of AD predominantly cause an amnestic dementia syndrome and manifest the pathognomonic brain changes of amyloid plaques and neurofibrillary tangles [4, 5].
Nevertheless, some FAD mutation carriers manifest various concomitant motor features such as ataxia, parkinsonism, spastic paraparesis (SP), and myoclonus, among others [6–8]. Intriguingly, SP is exclusively seen in individuals affected by PSEN1 mutations [9] and in some of the mutation carriers a mild non-amnestic dementia is preceded and clinically surpassed by SP [10–12]. Besides their clinical uniqueness, these patients also show particular neuroimaging and histopathologic features including white matter hyperintensities (WMH) on magnetic resonance imaging (MRI), amyloid deposition in vessel walls as cerebral amyloid angiopathy (CAA), and diffuse and cotton-wool amyloid plaques [7, 13]. The underlying causes of these phenomena and their relationship with other aspects of AD pathology are poorly understood. However, motor impairment, WMH, and CAA have all been observed more frequently in association with PSEN1 mutations located beyond codon 200 [8, 15].
SP has been reported to be caused by the PSEN1 A431E mutation, which is arguably the second most prevalent FAD mutation in the world. While approximately 45% of the PSEN1 A431E carriers develop SP, all patients manifest some degree of muscular rigidity in the advanced stages of the disease [16]. Brain imaging studies of A431E mutation carriers have revealed white matter (WM) and conduction defects in areas of the motor cortex and relative preservation of cortical grey matter, with no significant differences between SP affected and non-affected individuals [17, 18]. These findings raise the possibility that this mutation has a particular detrimental effect on WM integrity and functions. Here we present the clinical features and MRI data of three subjects from a Mexican family carrying the PSEN1 Ala431Glu mutation who shared a common clinical presentation of spastic paraparesis followed by atypical non-amnestic dementia.
MATERIALS AND METHODS
Subjects and samples
A Mexican-mestizo family with a three-generation known history of progressive paresis of the lower limbs consulted our research group for genetic assessment. A total of 13 subjects were asked to sign an informed consent form for the compilation of their clinical history and biological samples, complying to the statutes of the Bioethics Committee on Human Research (COBISH, folio # 038/2016) of the Center for Research and Advanced Studies, CINVESTAV.
DNA extraction and sequencing
Leukocyte genomic DNA was extracted using Blood DNA Preparation Kit (Jena Bioscience). Amplification of the PSEN1, PSEN2, and APP genes was performed using custom made oligonucleotide primers. The PCR amplified fragments were then sequenced by a standard Sanger sequencing method using Big Dye terminator v3.1 Cycle Sequencing kit on an ABI Prism 310 Sequencer (Applied Biosystems). Sequencing data was analyzed using CLC Main Workbench version 7.7.1 software (Qiagen).
Cognitive and neurological assessment
Cognitive performance was evaluated using the Spanish-translated versions of Mini-Mental State Examination (MMSE), Montreal Cognitive Assessment (MoCA), and Clinical Dementia Rating Scale (CDR). Olfactory function was determined with an adapted version of the University of Pennsylvania Smell Identification Test (UPSIT) for the Mexican population, in which unfamiliar scents (from the original version) were exchanged with more common and recognizable odors. As a control, the same test was performed on twenty age-matched healthy individuals.
Brain imaging
Magnetic resonance images were acquired on a 3T GE Discovery MR750 scanner (General Electric). 3D-FLAIR image processing, analysis and rendering was performed using Horos version 3.3.4 image viewer software (The Horos Project) and ITK-SNAP version 3.6.0 software (University of Pennsylvania). Volumetric analysis of brain structures and WMH was carried from T1-weighted images out using VolBrain version 1.0 and from FLAIR weighted images using LesionBrain version 1.0 pipelines through their web interface (http://volbrain.upv.es/).
RESULTS
Case 1 (Proband)
The proband (Fig. 1A, subject III-2) was a 54-year-old male with a 6-year history of progressive lower limb weakness, diagnosed elsewhere with an idiopathic upper motor neuron disease. Although no formal cognitive testing was carried out at the time of his assessment, he could establish a coherent conversation and was oriented in time and space. Through genetic analysis of FAD associated genes, a heterozygous C to A change at position c.1292 of the PSEN1 cDNA sequence was found, that results in the substitution of Alanine 431 into a Glutamic acid (A431E) (Fig. 1B, III-2). Shortly after his assessment, at the age of 55, the patient died from a cardiac arrest during a hospital admission for respiratory distress, which was attributed to a degenerative motor disease. According to his family, his mother (Fig. 1A, subject II-4) died at the same age after five years of illness. Besides the lower limb spasticity and paresis, she also developed prosopagnosia (i.e., the inability to recognize familiar faces) in the last stage of her disease. He was survived by four siblings, two of whom were found to also be carriers of the A431E mutation in PSEN1.

A) Family pedigree of the study subjects. Males are illustrated as squares and females as circles. Crossed figures depict deceased individuals. Symptomatic and asymptomatic A431E mutation carriers are colored in black while symptomatic subjects with no genetic confirmation are in grey. Subjects with a question mark were not probed for the mutation. B) Sequence electropherogram of the mutation carriers. The A to C heterozygous substitution is seen as two different peaks arising from the same position.
Case 2 (Symptomatic subject (SS))
This subject, a sibling of the proband, was a 54-year-old male with a 7-year history of a motor disorder (Fig. 1A, subject III-4). His illness began with bilateral lower limb fatigue that progressed over the course of three years into an incapacitating condition that required physical assistance. He also complained of consecutive worsening of speech fluency which he referred to as “stuttering”. At the age of 51, he started having shortness of breath that prevented him from eating or drinking normally and increasing visual deterioration that did not improve with subsequent changes to his glasses prescription. However, the patient denied having symptoms related to his memory, attention, orientation, or mood. At 53 years old, he scored 18/30 on the MMSE and 16/30 points on the MoCA test, failing in the questions that assess visuospatial abilities, attention, delayed memory, and orientation to time (Table 1). In spite of this, he lived alone and could perform all of his main activities of daily life (ADL) independently, except for transferring. He scored 0.5 on the CDR scale and was therefore classified as having a mild cognitive impairment. On neurological examination, the patient exhibited typical signs of upper motor neuron disease with lower limb spasticity, generalized brisk reflexes and positive Babinski, Chaddok, Bing, and Gonda/Stranski signs. He also exhibited a re-emergence of palmar grasp, glabellar, and snout primitive signs, indicative of frontal lobe pathology. Furthermore, he performed very poorly on the smell identification test, obtaining a score of 16/40, which qualifies as anosmia on the UPSIT scale grading.
Cognitive tests scores from the AS, PSS, and SS
AS, asymptomatic subject; PSS, pre-symptomatic subject; SS, symptomatic subject; MoCA, Montreal Cognitive Assessment; MMSE, Mini-Mental State Examination; CDR, Clinical Dementia Rating Scale.
Case 3 (Pre-symptomatic subject (PSS))
The patient, another sibling of the proband, was a 46-year-old female (Fig. 1A, subject III-8) who was referred as asymptomatic but on direct questioning admitted experiencing mild fatigue in both legs, with left predominance. At 45 years of age, she scored 27/30 on the MMSE and 26/30 on MoCA test, failing on the questions that evaluated visuospatial cognition, attention, and episodic memory (Table 1). Nevertheless, her scores fall within the normal range of cognitive function. On physical examination, she exhibited generalized hyperreflexia, perhaps to a lesser extent than the SS, and an incomplete but pathological response to the stimulation of Babinski, Chaddok, and Bing reflexes. She scored 30/40 in the smell identification test, which corresponds to mild hyposmia on the UPSIT scale.
Case 4 (Asymptomatic subject (AS))
The subject, a cousin of the proband, was an asymptomatic 38-year-old man (Fig. 1, subject III-10). At age 35 he scored 26/30 on the MMSE and 28/40 on the smell identification test corresponding to normal cognition and olfaction, but lower to the mean score of his age-matched control group (Table 1). His medical record included an MRI scanning of the brain performed while 36 years of age that reported periventricular WHM as the only finding.
PSEN1 A431E mutation carriers showed early evidence of periventricular white matter lesions, white matter volume loss, and enlarged Virchow-Robin Spaces
MRI assessment revealed an atrophy pattern restricted to parieto-occipital areas with a relative predominance in the right hemisphere of the PSS and no asymmetry in the SS (Supplementary Figure 1A). In the SS, this atrophy pattern along with the referred visual dysfunction, visuospatial abnormalities, and upper motor neuron disease are suggestive of a diagnosis of posterior cortical atrophy associated to AD (PCA-AD) [19]. Moreover, hippocampal size appeared normal by visual estimation (MTA scale: 0 and 1, in the PSS and SS, respectively) and was later confirmed to be within the volume range of healthy age-matched controls (Table 2 and Supplementary Figure 1B). However, computational analysis of brain volume-loss failed to localize atrophy to the cortex but to cerebral WM where we also found numerous < 1 cm hypointense signals that appear to be enlarged Virchow-Robin spaces (VRS). These enlarged VRS were also found in the PSS and the AS, but in reduced quantities. Besides, we detected several periventricular WMH that were classified at level 1 in the Fazekas scale for the AS and PSS, and level 2 in the SS (Supplementary Figure 1C). While these WM lesions were readily seen in a normal FLAIR-weighted MRI, we decided to make a 3D rendering of these images to more precisely localize them anatomically (Fig. 2 and Supplementary Videos 1 and 2). Thus, in the SS we were able to identify several affected WM structures within the frontal, temporal, parietal, and occipital lobes. Notably, the most prominent lesions were found within the cingulate gyrus (CG) and the optic radiation (OR) with no apparent involvement of the corpus callosum, except for its inferior border or roof of the lateral ventricles (LV). Nevertheless, there were numerous small WMH dispersed in areas more eccentric to the LV, that involved structures like the internal (IC) and external capsules (EC). On the other hand, the PSS largest WMHs were found on the CG above the genu of the corpus callosum and near the occipital horns of the LV. Moreover, we also identified a particular linear pattern that seemed to follow the shape of the fornix (FX) on both patients and a strong signal on the inferior border of the frontal lobes that most likely correspond to the olfactory nerves (OLN) (Fig. 2, red arrows).
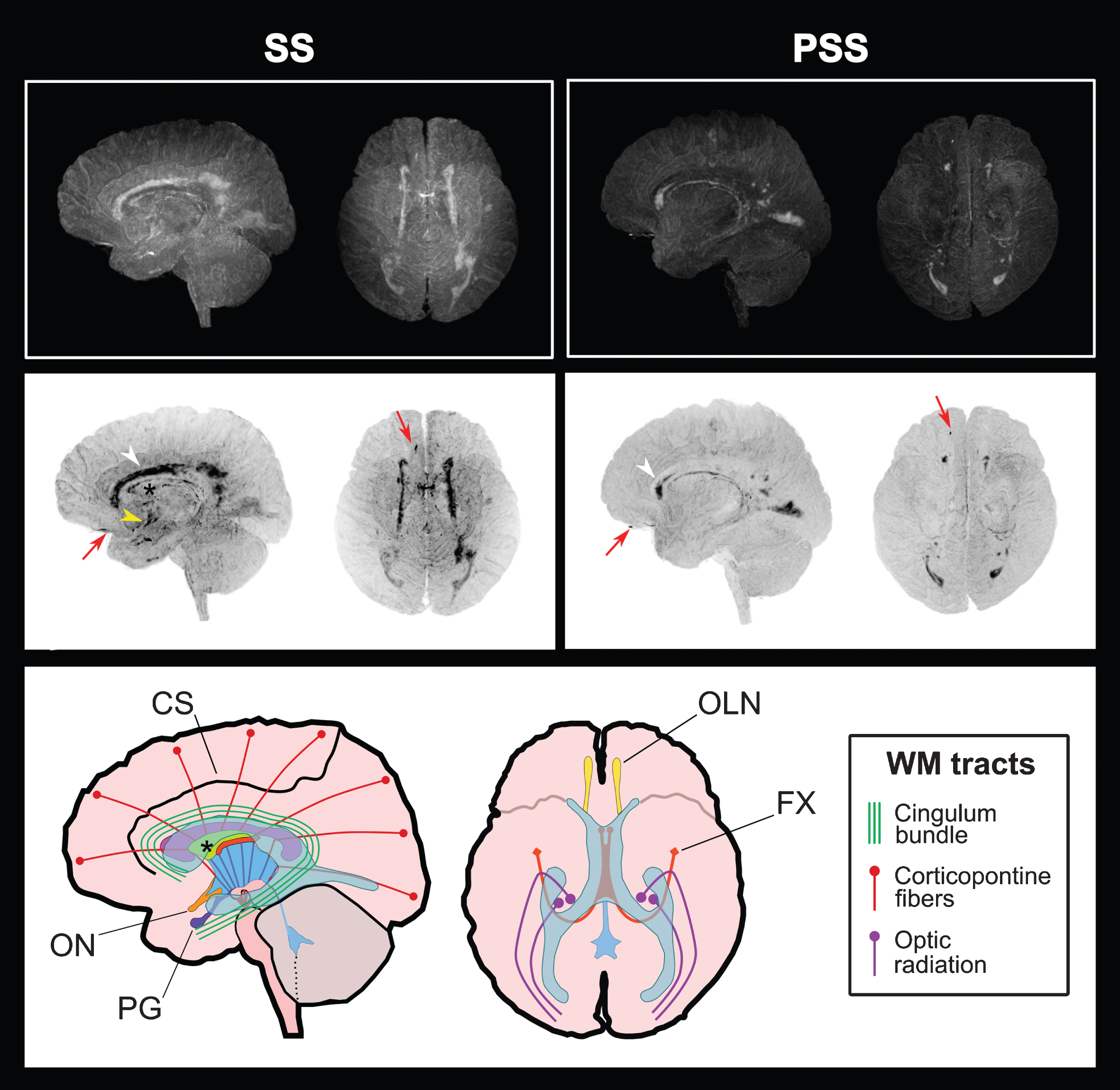
Periventricular WMH localization through 3D FLAIR-weighted MRI. (Top) Unmodified images of the SS and PSS brains in left and superior views. (Middle) Inverse projection (negative) of the images above for improved visualization of WMH. (Below) Schematic representation of the brain and relevant structures. The ventricular system is depicted in blue and the corpus callosum in fuchsia. WMH were classified according to their distribution pattern in three types: the first type represented by thick spots involving areas like the cingulum bundle and the optic radiation (white arrowhead), the second one seen as a thin line delimiting structures like the septum pellucidum (black asterisk) and the fornix, and the third dotted pattern involving the internal and external capsules. Also, an intense signal was perceived in the location of the olfactory nerve (red arrow) of both subjects and in the supraoptic recess near the optic chiasm in the SS (yellow arrowhead). CS, cingulate sulcus; FX, fornix; OLN, olfactory nerve; ON, optic nerve; PG, pituitary gland; PSS, pre-symptomatic subject; SS, symptomatic subject.
Calculated volumes of intracranial structures and white matter lesions. Volumes are presented in absolute values measured in cm3 and in relative values measured in relation to the intracranial cavity. Values between brackets show expected limits (95%) of normalized volume in function of sex and age for reference purpose. Numbers in bold indicate that the volume is under the expected volume limits
DISCUSSION
The PSEN1 A431E mutation was first described more than a decade ago in eleven families originating from the Jalisco state of Mexico and thirteen families from the United States with Mexican ancestry [16, 20]. Their phenotype was described as an early-onset amnestic dementia (mean age at onset of 40) with a variable presentation of SP among mutation carriers. Here, we report a family with five confirmed A431E mutation carriers, two of which were symptomatic and manifested lower limb weakness as their initial symptom. Furthermore, as told by other family members, all of the affected subjects from the first two generations of their pedigree had initial motor symptoms and were cognitively unimpaired or affected until a later stage of the disease. To our knowledge, this is the only A431E carrier family that has been reported to demonstrate such consistency in the appearance of initial motor features among its members. The early appearance of pyramidal symptoms preceding dementia has been seen in carriers of different SP-linked PSEN1 mutations, such as Y154 N, P88 L, and R278K [10–12]; however, it is not common.
Irrespective of the order in which cognitive and motor features manifest, many FAD patients affected by SP suffer atypical dementia syndromes characterized by visuospatial abnormalities and slower cognitive deterioration compared to most FAD mutation carriers [21–23]. In our study, we identified a very mild non-amnestic cognitive impairment in the SS that allowed him to perform most of his daily life activities independently. As expected, he scored 0.5 on the CDR, which is the lowest score reported for carriers of this mutation. Considering that the SS was 54 years old when evaluated, which is a year younger than the mean age of death (AOD) calculated for his family (i.e., 55 years) and six years older than the AOD previously reported for other individuals carrying the A431E mutation (i.e., 48 years) [16, 20], it appears that his main condition was motor dysfunction.
We also identified several periventricular WMH in the PSS and SS, as well as in the AS. This observation is consistent with the high prevalence of WMH found in patients carrying SP-linked mutations that lie within the C-terminal portion of Presenilin 1 [15]. Even though early occipital WMH have been identified in pre-symptomatic FAD patients even 22 years before their estimated age of onset [24], their clinical correlation and causation are still a matter of discussion. Nonetheless, we consider that the WMH observed in both our patients may be related with their clinical stage and cortical atrophy pattern. For example, in the SS the extensive damage seen in the CG, IC, EC, and OR may be associated with the loss of brain volume found in both parietal and occipital lobes since these highly myelinated structures contain several axons from neurons of the motor and visual cortexes. Meanwhile, in the PSS such areas were almost spared from WMH except for the anterior portion of the cingulate bundle and a small area near the OR, which could potentially explain her mild attention and visuospatial deficits shown during the cognitive assessment. Nevertheless, we also detected WMHs delimitating the perimeter of the FX in both subjects. This structure has been linked to age-related hippocampal grey matter damage and episodic memory decline [25]. One could speculate that this may explain why the PSS, but mostly the SS, failed to answer the questions that evaluated delayed memory while maintaining normal hippocampal volumes. Besides the distinguishable signal found in the OLN of both subjects, we also found WMH in the vicinity of the anterior commissure, which is a WM tract that contains several nerve fibers from both temporal lobes including the olfactory tract. Olfactory dysfunction is a frequent finding among patients with mild cognitive impairment and AD dementia and has been mainly associated with tau pathology (i.e., neurofibrillary tangles) and neurodegeneration at different levels of the olfactory pathway [26, 27]. Nonetheless, this condition is also found in other several demyelinating diseases such as neuromyelitis optica and multiple sclerosis (MS) [28].
Since not all myelinated areas of the brain were affected by WMH but only those closely related to the lateral ventricles, we speculate that these WM lesions were caused by a primary pathogenic mechanism that is not intrinsic to myelin. One possibility is that they reflect dysfunction of the blood-brain barrier. For instance, in neuromyelitis optica and MS, early blood-brain barrier disruption causes a cascade of events that induces demyelination and results in neurodegeneration [29–31]. Moreover, enlarged VRS in MS affected subjects have been proposed as a marker for progression of brain atrophy [32]. In our study, we found numerous enlarged VRS in the brain parenchyma of our patients that constitute a sign of impaired drainage of perivascular interstitial fluid and is strongly associated with CAA [33, 34]. In CAA, amyloid-β peptides accumulate in the basement membranes in the walls of cerebral vessels [35, 36] and have been shown to cause capillary constriction, endothelial autophagy, and delayed vascular regeneration [37, 38]. Moreover, CAA has been reported in A431E mutation carriers [39, 40] and also in several other SP-linked FAD mutations. Thus, the relationships between these findings and the motor and cognitive alterations caused by the A431E mutation should be further explored through experimental assays and detailed clinic-pathological assessments.
In conclusion, the cognitive features displayed by the PSEN1 A431E mutation carriers of this family and the possible relationship of WMH with their clinical stage and cognitive, visual, and motor dysfunctions are evidence of an atypical disease course that involves important glial and vascular changes that resemble to those seen in demyelinating diseases like MS. This should be considered for the study of the pathogenic mechanisms associated with alternative phenotypes caused by SP-linked FAD mutations.
Footnotes
ACKNOWLEDGMENTS
We would like to thank Dr. Rocío Gómez Ortega from the Laboratory of Population Genomics for kindly providing reagents used in sequencing analysis and to MSc. Pilar Figueroa Corona for her technical support. Also, to Dr. Luis Concha Loyola and Dr. Erick Pasaye Alcaraz from the National Laboratory for Magnetic Resonance Imaging for their invaluable help on the planning and performance of the MRI study. Finally, to Alicia García Rubí and Estefanía Gutiérrez Castañeda for their considerate assistance during the conduction of clinical tests.
This work was supported by grants from the National Council for Science and Technology of México (CONACYT), Problemas Nacionales 2017-01-5359. RASM is supported by a PhD fellowship from CONACYT (fellowship No. 425721). MAMR is a member of the National System of Researchers (SNI) of CONACYT. NSR is supported by a University of London Chadburn Lectureship in Medicine.