
Abstract
Background:
Neuroinflammation has been recognized as an important factor in the pathogenesis of Alzheimer’s disease (AD). One of the most recognized pathways in mediating neuroinflammation is the prostaglandin E2-EP1 receptor pathway.
Objective:
Here, we examined the efficacy of the selective EP1 antagonist ONO-8713 in limiting amyloid-β (Aβ), lesion volumes, and behavioral indexes in AD mouse models after ischemic stroke.
Methods:
Transgenic APP/PS1, 3xTgAD, and wildtype (WT) mice were subjected to permanent distal middle cerebral artery occlusion (pdMCAO) and sham surgeries. Functional outcomes, memory, anatomical outcomes, and Aβ concentrations were assessed 14 days after surgery.
Results:
pdMCAO resulted in significant deterioration in functional and anatomical outcomes in the transgenic mice compared with the WT mice. No relevant differences were observed in the behavioral tests when comparing the ONO-8713 and vehicle-treated groups. Significantly lower cavitation (p = 0.0373) and percent tissue loss (p = 0.0247) were observed in APP/PS1 + ONO-8713 mice compared with the WT + ONO-8713 mice. However, the percent tissue injury was significantly higher in APP/PS1 + ONO-8713 mice compared with the WT + ONO-8713 group (p = 0.0373). Percent tissue loss was also significantly lower in the 3xTgAD + ONO-8713 mice than in the WT + ONO-8713 mice (p = 0.0185). ONO-8713 treatment also attenuated cortical microgliosis in APP/PS1 mice as compared with the vehicle (p = 0.0079); however, no differences were observed in astrogliosis across the groups. Finally, APP/PS1 mice presented with characteristic Aβ load in the cortex while 3xTgAD mice exhibited very low Aβ levels.
Conclusion:
In conclusion, under the experimental conditions, EP1 receptor antagonist ONO-8713 showed modest benefits in anatomical outcomes after stroke, mainly in APP/PS1 mice.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the most common memory disorder, and its prevalence is expected to triple by the year 2050 [1]. The hallmark of AD is extracellular aggregation and deposition of amyloid-β (Aβ) and intracellular hyperphosphorylated tau protein in various regions of the brain [1]. The pathogenesis of AD has been the subject of numerous studies. One of the widely recognized processes implicated in the pathogenesis and progression of AD is neuroinflammation [2]. In fact, it has been proposed that neuroinflammation has early and substantial involvement in the pathogenesis of AD, and recent studies have shown genetic overlap between AD and immune-mediated diseases [3]. Therefore, events that cause inflammation, such as a cerebrovascular event, can contribute to the pathogenesis of AD. Indeed, cerebrovascular diseases are a recognized risk factor for AD and stroke. This is supported by studies that have shown a significantly higher prevalence of cerebrovascular diseases in patients with AD and in autopsy series [4]. In this context, neuroinflammation triggered by the cerebrovascular event is considered a key factor in linking ischemic stroke to AD.
One of the main pathways involved in mediating neuroinflammation is the prostaglandin E2 (PGE2) pathway [5]. This pathway is considered one of the underlying mechanisms in exacerbating the effect of ischemic stroke on AD. This is supported by findings such as elevated concentrations of the proinflammatory PGE2 in patients with probable AD [6], as well as increased cerebrospinal fluid levels of PGE2 in early-stage AD [7]. In addition, epidemiological studies have suggested that the use of nonsteroidal anti-inflammatory drugs (NSAIDs), which inhibit prostaglandin production, can prevent the development of AD [8]. On the other hand, controlled clinical trials with cyclooxygenase-2 selective and nonselective NSAIDs in patients with AD have not supported such epidemiological evidence; it was thereafter postulated that the chronic use of NSAIDs may be beneficial only in the normal brain or early stages of AD by inhibiting the production of Aβ42 [9–11]. Based on these premises, early intervention through inhibition/blockade of the prostaglandin pathway in high-risk patients may be an effective strategy in AD management. However, long-term NSAID use is associated with multiple side effects that render them inefficacious for long-term use [12, 13]. Selecting and targeting receptor(s) downstream of PGE2 based on the physiological characteristics of these receptors may provide a better alternative to NSAIDs as an effective strategy for the management of early-phase AD.
PGE2 mainly activates four distinct G-protein-coupled receptors: EP1–EP4. The activation or inhibition of these receptor subtypes results in different effects, but in general, EP2 and EP4 are involved in neuroprotective pathways while EP1 and EP3 seem to exacerbate neurodegeneration in AD, ischemic stroke, and other neurological conditions [5]. EP1 is a PGE2 receptor expressed in the brain under basal conditions in the cerebral cortex, hippocampus, and cerebral Purkinje cells [14]. The neurotoxic effects of this receptor have been shown in previous studies [15–20]. The genetic deletion and the pharmacological inhibition of this receptor with selective antagonists resulted in significantly reduced cerebral injury in the post-ischemic stroke setting in different experimental models [16–19]. In addition, genetic deletion of the EP1 receptor in transgenic AD mice significantly reduced the neuronal damage and decreased basal and post-ischemic levels of Aβ compared to AD transgenic mice without EP1 deletion [21]. Such characteristics make this receptor a plausible therapeutic target for delaying the progression of AD, particularly in the setting of an acute brain insult such as stroke, by mitigating the triggered neuroinflammation.
In this study, we examined the efficacy of a selective EP1 antagonist, ONO-8713, on the soluble Aβ load along with lesion volume and changes in behavioral indexes in the setting of unilateral permanent distal middle cerebral artery occlusion (pdMCAO). More specifically, we were interested in the efficiency of the antagonist in the period before insoluble Aβ accumulation and deposition in various regions of the brain. We hypothesized that stroke is followed by activation of the EP1 receptor, which contributes to neurodegenerative processes and exacerbates AD pathology and outcomes and that the pharmacological blockade with a selective antagonist would improve outcomes after acute stroke in genetic animal models of AD. The study was conducted in two cohorts of AD transgenic mice at pre-symptomatic ages that we believe can simulate an early AD stage [22]. Ischemic stroke was introduced 14 days before termination of animals, and behavioral studies were conducted during this period. Lesion volumes, microgliosis, and astrogliosis were measured and Aβ40 and Aβ42 were quantified.
MATERIAL AND METHODS
Mice
All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Florida and were conducted in accordance with guidelines established by the National Institutes of Health. We used 264 male mice, with 72 mice for each transgenic strain and 60 for their respective age-matched wildtype (WT) controls. The following mice were purchased from Jackson Laboratories (Bar Harbor, ME): first cohort - B6C3-Tg(APPSwe,PSEN1dE9)85Dbo (stock# 004462/HEMI, 4.5 months old); second cohort - B6;129-Psen1 Tg(APPSwe,tauP301L)1Lfa (stock# 034830, 3 months old), referred to herein as APP/PS1 and 3xTgAD, respectively. The WT mice had the following genetic background: C57BL/6 x C3H non-carrier (first cohort; stock#004462/NCAR) and C57BL/6 x 129S1/SvlmJ (second cohort; stock# 101045). These are the closest recommended controls for APP/PS1 and 3xTgAD, respectively. The animals were maintained in our animal facilities in a temperature-controlled environment (23±2°C) on a 12-h reverse dark-light cycle so that behavioral testing could be performed during the awake phase. Mice were allowed ad libitum access to food and water before and after surgical procedures.
Experimental groups and drug treatment
ONO-8713, a selective EP1 receptor antagonist, was obtained from ONO Pharmaceuticals (Osaka, Japan). Stock solutions were prepared in dimethyl sulfoxide and subsequently aliquoted and stored at –20°C. Solutions for injections were freshly diluted daily from stock aliquots in sterile saline such that the final concentration of dimethyl sulfoxide was <0.01%, and mice were appropriately rehydrated. The control group received an equivalent volume of saline injection. Injections of the drug (10 μg/kg) or vehicle were performed intraperitoneally immediately after the surgical procedure and then daily after the behavioral tests for 14 days. The stock solution of drug and vehicle were prepared by an independent investigator and identified as A and B. The drug/vehicle coding was kept confidential during all the treatment and postmortem studies to ensure a blind analysis throughout this work. Each cohort of transgenic mice and their respective WT controls were randomly assigned to different experimental groups (n = 12–24) and identified by ear tags before the beginning of the procedures: pdMCAO without drug (pdMCAO + Veh), pdMCAO with drug (pdMCAO + ONO-8713), sham without drug (Sham + Veh), and sham with drug (Sham + ONO-8713).
Permanent distal middle cerebral artery occlusion (pdMCAO)
Ischemic stroke was induced by pdMCAO, as previously described [23, 24]. Surgical procedures were performed under anesthesia using isoflurane for induction and delivered through a nose cone for maintenance throughout the procedure (1.5/3.0% oxygen/air balance). An incision of approximately 1 cm was made between the right eye and ear, and the temporal muscles were dissected and retracted outward to visualize the temporal bone. A craniotomy was performed by carefully removing a piece of bone approximately 2 mm2 to expose the distal middle cerebral artery (MCA). The distal MCA branches were occluded permanently using a bipolar electrocoagulation forceps (Bovie Medical Corp., Clearwater, FL). Complete occlusion was confirmed by severing the vessel. Body temperature was maintained at 37°C with a heating pad during the surgery, and animals were allowed to recover in a temperature- and humidity-controlled chamber for approximately 2 h before returning to their cages. The sham-operated control mice were subjected to the same surgical procedure, except that the distal MCA was not occluded. The body weight and animal health status were monitored and all efforts were made to minimize animal suffering.
Open-field locomotor activity
Locomotor activity was measured using an automated open field activity monitor and video tracking interface system (MED Associates Inc., St. Albans, VT) before the surgery (baseline) and 1, 3, and 7 days after pdMCAO. Briefly, mice were placed individually in four transparent acrylic cages, and their locomotion and distance traveled were recorded over a 30-min test period. Data are represented as distance traveled omitting the first 5 min to exclude for initial habituation/anxiety responses [25].
Cylinder test
Mice were evaluated in this test on the same days as the open field test: before the surgery (baseline) and 1, 3, and 7 days after pdMCAO. Briefly, each mouse was placed in a transparent acrylic cylinder mounted above a solid surface and recorded for 8 min. A 45° -inclined mirror was used to videotape animal movements in all directions. The videos were watched in slow motion to assess forelimb use as identified by the number of contacts of each or both forelimbs with the cylinder wall during full rear [23, 24]. Data are presented as the percentage of left forelimb use.
Passive avoidance test
This test was carried out on days 13 and 14 after the surgery. The step-through passive avoidance task was performed in a shuttle box apparatus (Gemini Avoidance System, San Diego Instruments, San Diego, CA) that consists of light and dark compartments of equal size separated by a connecting gate. The test was performed as previously described with some modifications [21]. On the first day, each mouse was placed in the light compartment, and after 10 s, a potent light was turned on in the start chamber at the same time the gate was opened. The time the mouse took to cross to the dark compartment (acquisition time) was recorded. Once the mouse entered the dark chamber, the door automatically closed and an electrical shock of 1 mA lasting 1 s was delivered to the animal’s paw. After 10 s the mouse was removed from the cage and placed in its home cage. Approximately 24 h later, the animal was again placed in the light compartment and the time it took to cross to the dark chamber (retention time) was recorded until the maximum of 300 s. If the mouse did not enter the dark chamber during the testing, it was assigned the maximum latency, which represented successful retention of the task. Results are presented as latency time to cross the gate in each phase.
Brain harvesting
The animals were sacrificed 14 days after surgery. For histopathological analysis, mice were deeply anesthetized and transcardially perfused with phosphate-buffered saline (PBS), pH 7.4, followed by 4% paraformaldehyde. Brains were collected and kept in 4% paraformaldehyde for at least 24 h before cryopreservation in a 30% sucrose/PBS solution. For Aβ analyses, the mice were deeply anesthetized and transcardially perfused with PBS. The cortex was immediately dissected, flash-frozen, and stored at –80°C.
Aβ quantification by enzyme-linked immunosorbent assay (ELISA)
Aβ was extracted from cortical brain samples with radioimmunoprecipitation buffer containing protease inhibitor cocktail (Roche, Indianapolis, IN) and used for sandwich ELISAs as described previously with some modifications to improve the lower detection limit [26]. Briefly, Aβ40 was captured with 20 μg/mL of monoclonal antibodies (mAb) 13.1.1 (human Aβ35–40 specific; T.E. Golde, University of Florida, Gainesville, FL) and Aβ42 was captured with 20 μg/mL of mAb 2.1.3 (human Aβ35–42 specific; T.E. Golde). Both Aβ40 and Aβ42 were detected using horseradish peroxidase-conjugated mAb 33.1.1 (T.E. Golde). ELISA results were analyzed using SoftMax Pro software (Molecular Devices, Sunnyvale, CA).
Histology and quantification
To reduce any potential bias, an investigator blinded to experimental groups performed the staining and quantification. Furthermore, to eliminate inter-individual variability in quantification, a single individual quantified all slides for a particular stain. Last, a single individual reviewed all stains and quantifications to confirm the consistency throughout the project. Ten sets of 16 sections equally distributed throughout the anteroposterior brain regions were processed on a CM 1850 cryostat (Leica Biosystems, Buffalo Grove, IL) at 30 μm and stored at –80°C. Before staining, slides were thawed at room temperature for 48 h to allow firm tissue adherence to the glass. Once stained, all slides were scanned using a Scanscope XT and analyzed using ImageScope software (Aperio Technologies, Vista, CA). Cresyl violet staining was used to assess lesion volume, tissue injury, and cavitation. Immunohistochemical staining was performed to identify microgliosis and astrogliosis using the following primary antibodies: ionized calcium-binding adaptor protein 1 (Iba1; 1:1500; Wako, Richmond, VA) and glial fibrillary acidic protein (GFAP; 1:1500; Dako, Carpinteria, CA).
For quantification procedures in which total brain pathology was analyzed (lesion volume, cavitation, tissue injury), all sections that had lesions were quantified for each animal. To assess astrogliosis and microgliosis, four sections for each animal representing the maximal lesion area were analyzed.
Lesion volume
Injured brain areas were outlined and these areas were then abstracted from the ImageScope Software. Using known distances between sections and section thickness, these areas were used to calculate total brain lesion volume.
Cavitation
The area between injured tissue and the hemispheric border was outlined and abstracted from the ImageScope Software.
Penumbral tissue injury
Tissue injury was assessed by subtracting the cavitation from the total lesion volume. For immunohistochemical stains, an ImageScope Positive Pixel Count algorithm was used for quantification after the appropriate brain regions were outlined (see below). Each algorithm was tuned for each stain such that the appropriate signal and strength of the signal were evaluated [27]. Thresholds were set intermediately between the signals seen in the experimental groups on a representative slide in each group such that the algorithm allowed for optimal detection in either direction (i.e., more intense versus less intense). After running an algorithm, all slides were checked for specificity and accuracy and to ensure minimal interference from artifact before the data was abstracted from ImageScope Software.
Microgliosis and astrogliosis
This analysis was performed by a blinded investigator, as previously explained in lesion quantification. Cortical gliosis was analyzed by outlining the cortex in both the contralateral and ipsilateral hemispheres. Hippocampal gliosis was analyzed by circling the hippocampus in both hemispheres. Then positive pixel counts were obtained using the appropriated algorithm from the ImageScope Sofware following the protocol we described earlier [28].
Statistical analysis
SAS-JMP (SAS Institute, Cary, NC) was used for statistical analyses. Data are shown as the mean±SEM (except as otherwise indicated), and the significance level was set at p < 0.05. One-way analysis of variance followed by a Tukey post-test or two-way analysis of variance followed by a Bonferroni post-test was used in case of parametric distribution while Kruskall-Wallis followed by Mann-Whitney was used to compare nonparametric data.
RESULTS
Physiologic data
At the baseline evaluation, the average body weight of APP/PS1 mice (27±3 g) was similar to the WT controls (26±2 g) and remained similar throughout experiments. However, the 3xTgAD mice weighed less at baseline (25±3 g) compared with the WT controls (29±3 g) and this difference was maintained throughout the study. In both cases, surgery or treatment did not influence weight.
Lesion volume
To evaluate the influence of EP1 antagonism in the setting of ischemic stroke and AD pathology, lesion volumes were measured 14 days after pdMCAO (Figs. 1 and 2). No significant differences in total lesion volume (cavitation + tissue injury) were seen between transgenic and WT controls with or without EP1 antagonism (Figs. 1B and 2B). In the APP/PS1 cohort, when compared with WT + ONO-8713, APP/PS1 + ONO-8713 mice demonstrated significantly less cavitation (p = 0.0373; Fig. 1 C) and percent cavitation relative to total lesion volume (p = 0.0247; Fig. 1D). APP/PS1 + ONO-8713 mice had significantly greater tissue injury (p = 0.0185; Fig. 1E) when compared with the APP/PS1 + Veh group and greater percent tissue injury relative to total lesion volume (p = 0.0373; Fig. 1F) when compared with the WT + ONO-8713 group.

Effect of EP1 selective antagonism using ONO-8713 treatment on anatomical outcomes of APP/PS1 and wildtype (WT) control mice 14 days after permanent middle cerebral artery occlusion (pdMCAO). A) Representative Cresyl violet stained images are shown for transgenic and WT pdMCAO and sham-operated mice that received ONO-8713 or vehicle control daily post-surgery until sacrifice. The area of tissue injury is outlined, and high-magnification inserts are provided to show the penumbral tissue injury in detail. Square selections on low-magnification images denote the location of magnified regions. B) Quantification reveals that treatment with ONO-8713 has no significant effect on total lesion volume. C, D) The APP/PS1 + ONO-8713 mice have significantly less cavitation and percent tissue loss relative to total lesion volume compared with the WT + ONO-8713 group. E) Compared with the APP/PS1 + Veh group, the APP/PS1 + ONO-8713 mice have significantly greater tissue injury. F) Treatment with ONO-8713 leads to a significantly greater percent penumbral tissue injury relative to total lesion volume in APP/PS1 mice compared with their WT controls. Scale bars in hemispheric images, lesion inserts, and high magnification tissue injury images represent 1 mm, 300 μm, and 50 μm, respectively. Comparisons include 9–11 mice per group. *p < 0.05.
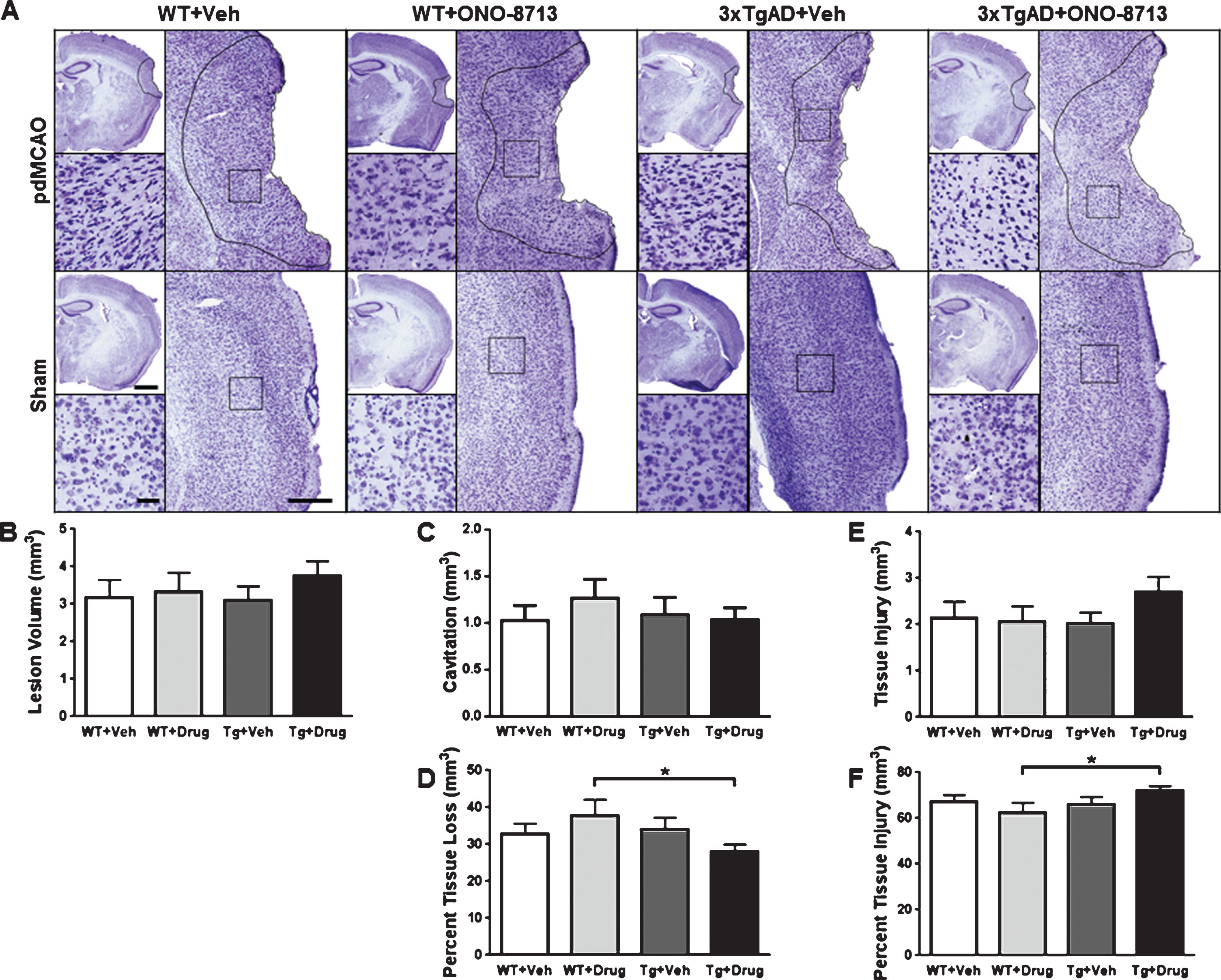
Effect of EP1 selective antagonism using ONO-8713 treatment on anatomical outcomes of 3xTgAD and wildtype (WT) control mice 14 days after permanent middle cerebral artery occlusion (pdMCAO). A) Representative Cresyl violet stained images are shown for transgenic and WT pdMCAO and sham-operated mice that received ONO-8713 or vehicle control daily post-surgery until sacrifice. The area of tissue injury is outlined, and high-magnification inserts are provided to show the penumbral tissue injury in detail. Square selections on low-magnification images denote the location of magnified regions. B, C, E) Quantification reveals that treatment with ONO-8713 has no significant effect on lesion volume, cavitation, or penumbral tissue injury. D, F) Treatment with ONO-8713 leads to significantly greater percent penumbral tissue injury and less percent cavitation relative to total lesion volume in 3xTgAD mice compared with WT controls. Scale bars in hemispheric images, lesion inserts, and high-magnification tissue injury images represent 1 mm, 300 μm, and 50 μm, respectively. Comparisons include 10–13 mice per group. *p < 0.05.
In the 3xTgAD cohort, there were no significant differences in lesion volume, cavitation, or tissue injury (Fig. 2B, C, E). However, the 3xTgAD + ONO-8713 mice had significantly lower percent cavitation relative to total lesion volume (p = 0.0185; Fig. 2D) and greater percent tissue injury relative to total lesion volume (p = 0.0176; Fig. 2F) compared with the WT + ONO-8713 group.
Open-field test
Open-field locomotor activity, defined by the total distance traveled, was used as a measure of functional outcomes. For the first cohort, there was no difference among groups, except in the baseline evaluation where mice from the WT + ONO-8713 group had lower locomotor activity compared with the APP/PS1 + ONO-8713 group (p = 0.0089; Fig. 3A), although at this time point the treatment had not started yet. On the other hand, many differences were seen for the second cohort, though most differences were related to genotype (Fig. 3B and Supplementary Figure 1B). The 3xTgAD + Veh mice had lower locomotor activity compared with the WT + Veh group at baseline (p = 0.0036), and 1 (p = 0.0001), 3 (p = 0.0005), and 7 (p = 0.0011) days post-pdMCAO surgery (Fig. 3B). Similarly, the 3xTgAD + ONO-8713 mice had lower locomotor activity compared with the WT + ONO-8713 group at baseline (p = 0.0001), and 1 (p = 0.0004), 3 (p = 0.0003), and 7 (p = 0.0001) days post-pdMCAO surgery (Fig. 3B). Furthermore, the WT + Veh mice had lower locomotor activity compared with the WT + ONO-8713 group on days 3 (p = 0.0449) and 7 (p = 0.0219), but these differences (drug effect) were not observed in transgenic mice (Fig. 3B).
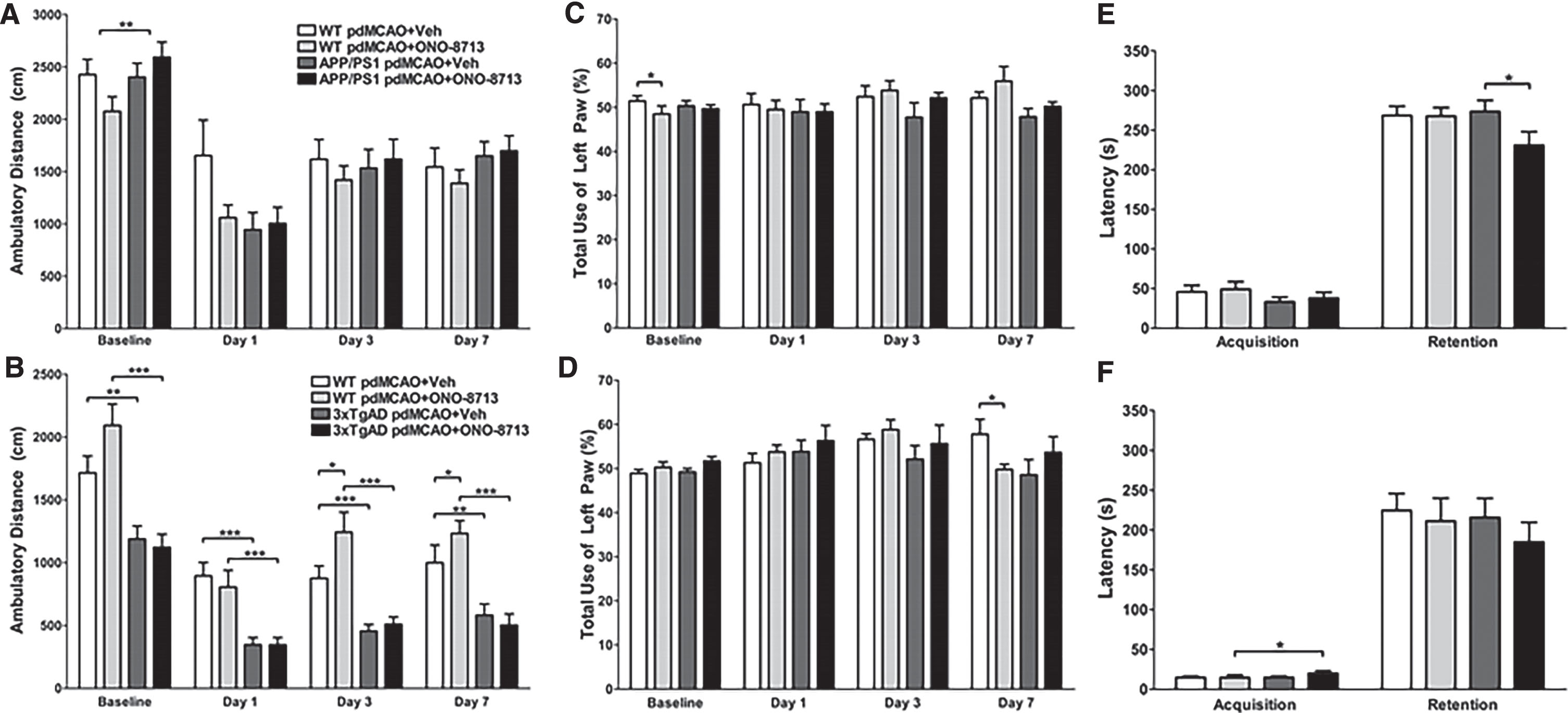
Effect of EP1 selective antagonism using ONO-8713 treatment on functional outcomes of transgenic and wildtype (WT) control mice after permanent middle cerebral artery occlusion (pdMCAO). A, B) Open-field locomotor activity was measured at baseline, 1, 3, and 7 days post-pdMCAO for cohorts 1 and 2. A) The WT + ONO-8713 group has less baseline locomotor activity than the APP/PS1 + ONO-8713 treated mice. B) For all time points, 3xTgAD + Veh mice have significantly less locomotor activity compared to WT + Veh control mice. Similarly, the 3xTgAD + ONO-8713 mice have significantly less locomotor activity compared with WT + ONO-8713 mice at all time points. At days 3 and 7, WT + ONO-8713 mice ambulate greater distances compared with WT + Veh control mice. C, D) Total use of the contralateral left forelimb in the cylinder test was examined at baseline, 1, 3, and 7 days post-pdMCAO for cohorts 1 and 2. C) The WT + ONO-8713 mice use their left paw comparatively less than the WT + Veh control group at baseline. D) Similarly, on day 7, WT + ONO-8713 mice use their left paw comparatively less than the WT + Veh control group. E, F) In the passive avoidance test, latency to cross from the light to dark compartments on the 13th day (acquisition, training phase) and 14th day after pdMCAO surgery (retention, test phase) were assessed for cohorts 1 and 2. E) There are no differences in acquisition, but the APP/PS1 + ONO-8713 group has a significantly lower latency of retention than the APP/PS1 + Veh group. F) The 3xTgAD + ONO + 8713 mice have a higher latency of acquisition compared to the WT + ONO-8713 group. All comparisons include 16–25 mice per group. *p < 0.05, **p < 0.01, ***p < 0.001.
Cylinder test
Only minimal differences were seen in the use of the contralateral left forelimb between groups. For cohort 1, we found some casual differences in the baseline evaluation not related to the surgery or treatment (Fig. 3C and Supplementary Figure 1C). On day 1, sham-operated WT + ONO-8713 mice had significantly more left forelimb use compared with the WT + Veh (p = 0.0374) and APP/PS1 + ONO-8713 (p = 0.0333; Supplementary Figure 1C) mice. For cohort 2, on day 7, the WT + Veh mice used their left forelimb more than the mice in the WT + ONO-8713 (p = 0.0288; Fig. 3D) group did. In the case of sham-operated animals, the WT + ONO-8713 mice used their left forelimb comparatively less than the 3xTgAD + ONO8713 group (p = 0.0288; Supplementary Figure 1D) on day 1.
Passive avoidance
After pdMCAO, the APP/PS1 + ONO-8713 had significantly less latency to cross from the light to the dark chamber in the retention phase compared with the APP/PS1 + Veh (p = 0.0369; Fig. 3E) group. For sham-operated, the WT + ONO-8713 mice had significantly less latency in the retention phase compared with the APP/PS1 + ONO-8713 (p = 0.0255; Supplementary Figure 1E) mice. No significant differences in retention were seen between groups for cohort 2, although 3xTgAD + ONO-8713 mice had significantly greater latency in the acquisition phase compared with the WT + ONO-8713 (p = 0.0360; Fig. 3F) group. For sham-operated, 3xTgAD + ONO-8713 mice had significantly greater latency in the acquisition phase compared with the 3xTgAD + Veh (p = 0.0069; Supplementary Figure 1F) group.
Microgliosis
To evaluate microgliosis, Iba1 immunohistochemistry was performed on brain sections from pdMCAO- (Fig. 4) and sham-operated (Supplementary Figure 2) mice for cohort 1. We measured microgliosis in the ipsilateral and contralateral cortex and hippocampus and analyzed the ratio ipsilateral/contralateral positive count. Following pdMCAO, APP/PS1 + ONO-8713 mice had significantly less unbalance between ipsilateral and contralateral cortical microgliosis compared with the APP/PS1 + Veh (p = 0.0079; Fig. 4B) group. No differences in hippocampal microgliosis were seen between groups in pdMCAO-operated mice. No differences in cortical or hippocampal microgliosis were seen between groups in sham-operated mice (Supplementary Figure 2).
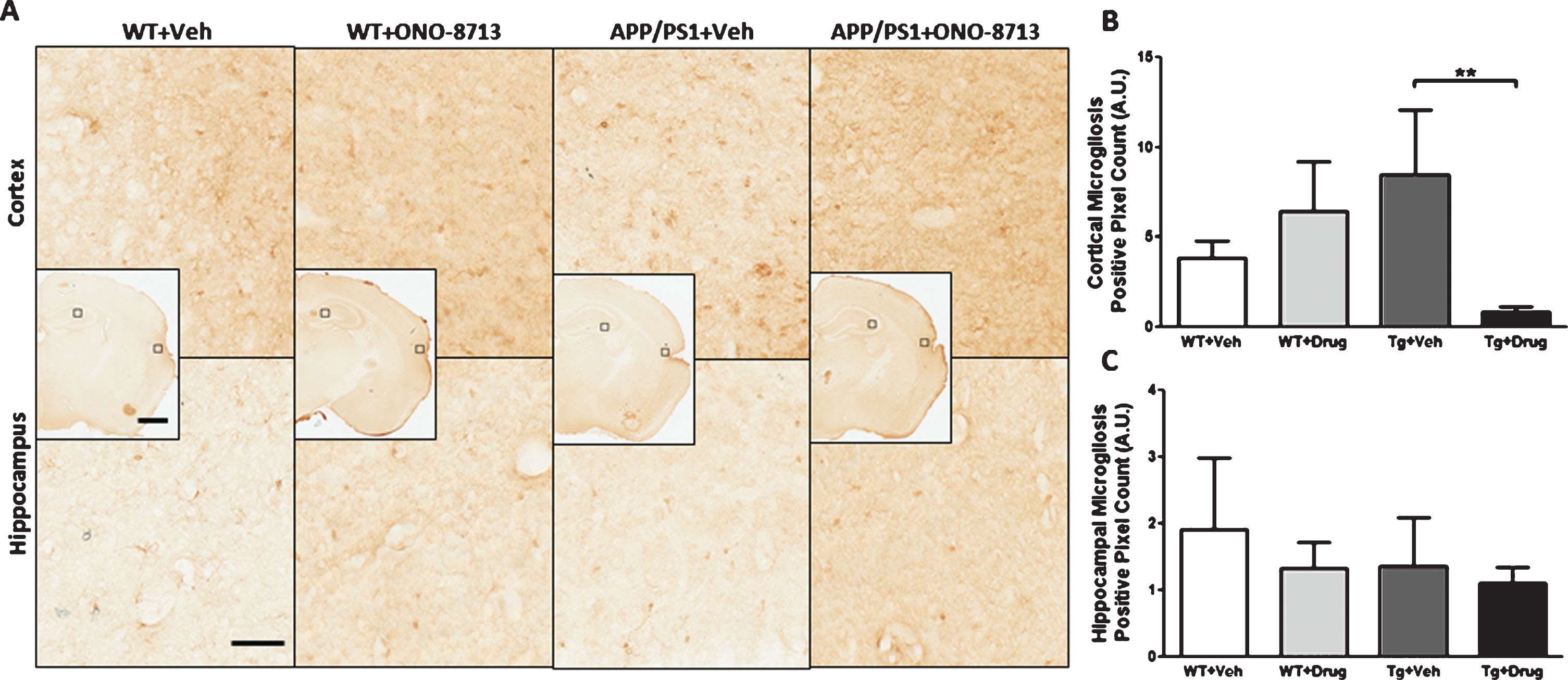
Effect of EP1 antagonism using ONO-8713 treatment daily post-permanent middle cerebral artery occlusion (pdMCAO) on microgliosis in APP/PS1 and wildtype (WT) control mice 14 days after pdMCAO. The 3xTgAD mouse brains were not evaluated. A) Representative high-magnification images of coronal brain sections depicting microgliosis in the ipsilateral cortex and hippocampus are shown for transgenic and WT control mice. Square selections in the inserts denote magnified regions. B) Ipsilateral/contralateral ratio for positive Iba1 count in cortex. APP/PS1 mice treated with ONO-8713 have significantly less cortical microgliosis compared to APP/PS1 + Veh group. C) Ipsilateral/contralateral ratio for positive Iba1 count in the hippocampus. No differences in hippocampal microgliosis are seen between any groups. Scale bars in hemispheric and magnified inserts represent 1 mm and 50 μm, respectively. All comparisons include 3–7 mice per group. **p < 0.01.
Astrogliosis
To evaluate astrogliosis, GFAP immunohistochemistry was performed on brain sections from pdMCAO- (Fig. 5) and sham-operated (Supplementary Figure 3) mice for cohort 1 in the ipsilateral and contralateral cortex and hippocampus. No significant differences in cortical or hippocampal astrogliosis were seen between groups in pdMCAO-operated mice after analyses of the ipsilateral/contralateral ratio. Sham-operated APP/PS1 + ONO-8713 mice had significantly less unbalance between ipsilateral and contralateral hippocampal astrogliosis compared with the APP/PS1 + Veh (p = 0.0423; Supplementary Figure 3C) group, although no differences were seen between groups for cortical astrogliosis.
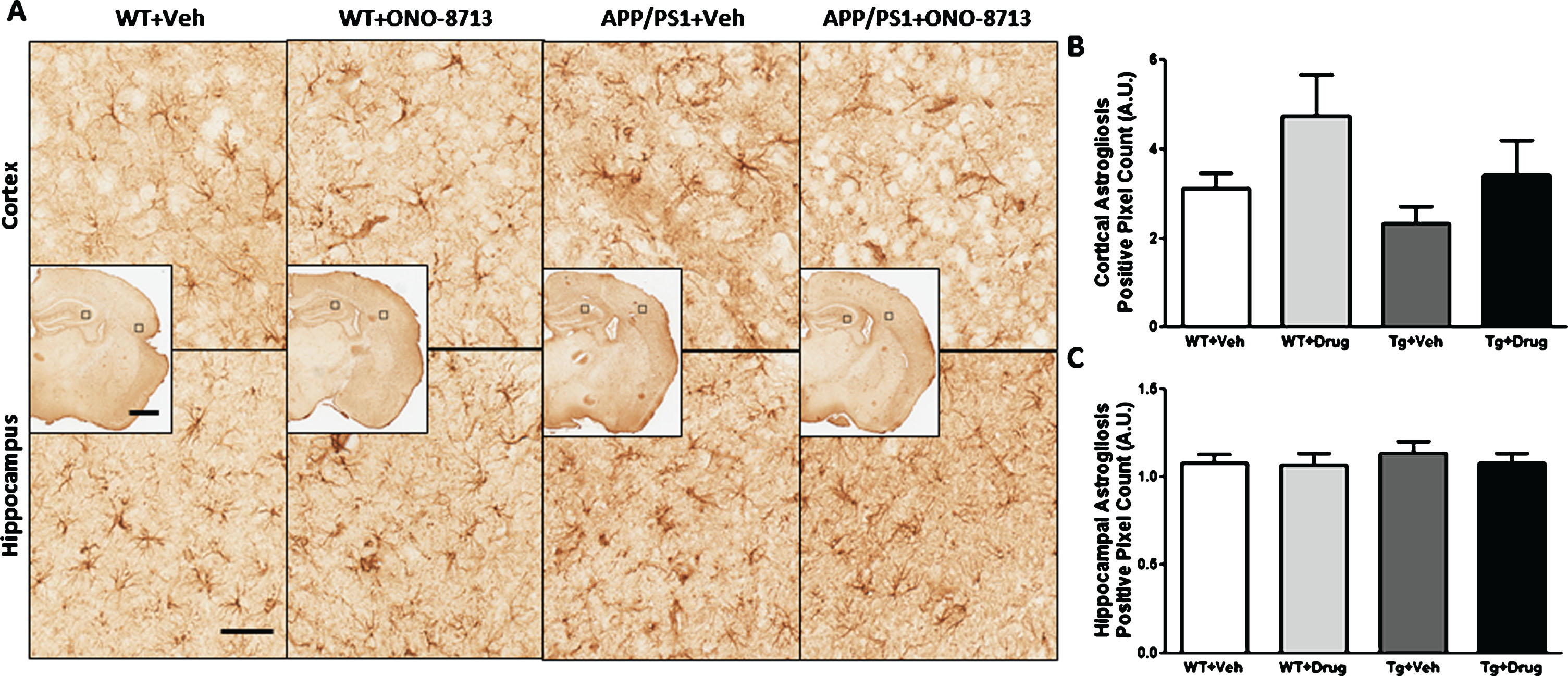
Effect of EP1 antagonism using ONO-8713 treatment daily post-permanent middle cerebral artery occlusion (pdMCAO) on astrogliosis in APP/PS1 and wildtype (WT) control mice 14 days after pdMCAO. The 3xTgAD mouse brains were not evaluated. A) Representative high-magnification images of coronal brain sections depicting astrogliosis in the ipsilateral cortex and hippocampus are shown for transgenic and WT control mice. Square selections in the inserts denote magnified regions. B) Ipsilateral/contralateral ratio for positive GFAP count in cortex. No differences are seen in cortical astrogliosis between any groups. C) Ipsilateral/contralateral ratio for positive GFAP count in the hippocampus. No differences are seen in hippocampal astrogliosis between any groups. Scale bars in hemispheric and magnified inserts represent 1 mm and 50 μm, respectively. All comparisons include 8–10 mice per group.
Aβ quantification
To determine whether selective EP1 antagonism had an effect on Aβ levels after ischemic stroke, we measured ipsilateral cortical Aβ40 and Aβ42 concentrations using end-specific ELISAs. No detectable differences in the Aβ40, Aβ42, or Aβ40/Aβ42 ratio were found for APP/PS1 mice (Table 1). In the 3xTgAD cohort, the levels of Aβ40 and Aβ42 were very low and near the limit of quantification (data not shown).
Aβ concentrations in ipsilateral cortical brain lysates of APP/PS1 mice
DISCUSSION
Stroke and AD are major neurodegenerative diseases that may worsen the prognosis of each other, but cofactors shared by these two diseases remain unclear [29, 30]. It has been reported that PGE2 levels are elevated in the brains of both AD and stroke patients [31]. Epidemiologic studies have documented a putative beneficial effect of anti-inflammatory drugs in AD; however, data from experimental studies suggest that NSAIDs are beneficial only in the early stages of AD process [11, 33]. Furthermore, the clinical use of NSAIDs or selective cyclooxygenase-inhibitors are associated with significant side effects [12, 13], which makes the search for new drugs of vital importance, with PGE2 being an important target for selective drugs [31, 34]. A large body of evidence suggests that pharmacological treatment with selective antagonists of PGE2 subtypes EP1 are neuroprotective in in vitro and animal models of stroke and AD. Our preliminary studies demonstrated that a brief ischemic insult caused more severe neuronal damage and neurobehavioral deficits in the APP/PS1 mice than in wildtype controls, and genetic deletion of the EP1 receptor significantly attenuated neuronal damage and memory loss [21]. In line with these results, the objective of this study was to examine the efficacy of ONO-8713, a selective EP1 antagonist, at a given dose regimen in attenuating Aβ accumulation and behavioral indexes, as well as lesion volume, in the setting of ischemic stroke in young AD transgenic and WT mice.
The study was conducted in two separate cohorts with animals of 3 or 4.5 months of age in order to evaluate whether the ischemic injury could anticipate the cognitive deficits in a pre-symptomatic stage of AD. The APP/PS1 is a double transgenic strain that expresses the human mutant amyloid precursor protein and the mutant presenilin 1, which results in the acceleration of amyloid pathology and precocious manifestation of AD [35]. The 3xTgAD mice possess equivalent mutations of APP/PS1 plus the insertion of a tau protein mutation [36]. These mice exhibit different AD anatomical and vascular pathologies at different ages, including amyloid deposition, tau pathology, cerebral amyloid angiopathy, and other cerebrovascular pathologies [22, 36]. Therefore, we chose these complementary AD mouse models to test whether the stroke would augment AD outcomes or vice versa and whether the EP1 receptor antagonist could limit inflammation in these models.
The pdMCAO model induced only subtle differences in the behavioral tests. This model was selected because the aim here was to test whether a minor infarct would aggravate AD pathology after a long period of survival. This model is also a better choice to test long-term outcomes and has lower mortality rates. In the open field test, no relevant differences were observed for APP/PS1 mice treated with vehicle or ONO-8713, and the only difference occurred between WT and transgenic mice at basal evaluation. When we evaluated the second cohort of mice, we observed less ambulatory activity in 3xTgAD mice compared with the WT, both in the sham as well as the pdMCAO groups. This difference occurred on basal and post-ischemic evaluations but was not modified by the treatment. Similarly, no relevant alterations were observed in APP/PS1 mice for the cylinder test. In the second cohort, we found a higher imbalance of left/right paws for WT + Veh mice subjected to pdMCAO compared with the WT + ONO-8713 group on day 7. However, considering that the use of the left paw varied for the different groups over time, it is likely that this alteration was causal and cannot be attributed to the treatment. Sensorimotor dysfunction is the earliest and most prominent symptom of pdMCAO-induced ischemia in mice. The asymmetry is better observed in the first two days after the surgery and perhaps the sensorimotor deficit could have been detected using different parameters [24].
We also evaluated the memory of the animals on the passive avoidance test 14 days after the surgery (pdMCAO lesion or sham group). As expected, the WT animals showed a low latency of acquisition (first phase) and a high latency of retention (second phase). However, the same performance was observed in the sham and pdMCAO groups both in WT and transgenic mice, with only minor alterations. We observed a significant decrease in retention latency in the APP/PS1 mice submitted to pdMCAO and treated with ONO-8713 compared with the vehicle group, meaning that the treatment worsened the performance of the animals. No differences were observed in the retention phase for 3xTgAD mice. In our previous study, 12-month-old APP/PS1 mice showed shorter retention latency than WT mice did in the passive avoidance task [21]. In the same study, global cerebral ischemia induced a cognitive deficit in APP/PS1 mice compared with sham animals, and the deletion of EP1 receptor (APP/PS1xEP1-/-) tended to induce greater memory.
The results from our current study were not consistent with this previous study [21], but some aspects can be discussed to justify this discrepancy. First, we used younger mice in our current study because our objective was to evaluate the effect of EP1 antagonism at the given dose regimen after a brief ischemic insult. Second, we employed a different model of stroke, pdMCAO, instead of global ischemia, which exclusively affects the hippocampus and thus cognition. But the most plausible explanation for not observing a cognitive deficit is the level of Aβ production at 3 and 4.5 months of age (pre-symptomatic and pre-pathology stages of AD). This is particularly relevant to the 3xTgAD cohort (3 months old) in which the Aβ level was very low. Because this strain of AD transgenic mouse presents a triple mutation with faster Aβ onset, we intended to test whether the pdMCAO surgery could aggravate the cognitive deficits already demonstrated for older animals in other studies. The Aβ deposition in the cortex of AD transgenic mice is dependent on the strain and age [22, 36–39]. For instance, the repetitive mild traumatic brain injury in AD Tg2576 mice induced the acceleration of amyloid deposition and the impairment of cognitive function compared with WT mice at 9 months [40].
In the case of APP/PS1 mice, the presenilin mutation results in the acceleration of amyloid pathology, but cognitive deficits are observed only at 6 months, and mice do not show extensive vascular amyloid pathology [35]. Literature describes how cerebral amyloid angiopathy for APP/PS1 mice starts around 6 months of age and is more severe in female than male mice [38]. Soluble Aβ40 was observed in 4-month-old mice while soluble and insoluble Aβ40 and Aβ42 were detected at 6 months, with an overall increase between 4 and 12 months [41]. Similarly, senile plaques were detected as early as 4 months of age, and there was an overall increase in number and plaque burden with age [41]. In fact, we observed Aβ load in the 4.5-month-old APP/PS1 mice, but there was no difference between pdMCAO and sham groups or vehicle and treated groups.
According to a recent review, 3xTgAD mice exhibit amyloid plaques at 3 months of age, with neurofibrillary tangles appearing much later, at 12 months of age in the hippocampus and cortex [22]. The authors discuss how Aβ deposition occurs early in this transgenic strain, prior to any significant cognitive impairment, which is reported to start around 6 months of age [22]. In general, it is accepted that the age and disease stage at which cognitive deficits are observed depends in part on the task being used to measure it [22, 36]. However, the levels of Aβ in 3-month-old 3xTgAD mice in our study were inadequate for quantification and meaningful analysis. It is likely that the Aβ production in this particular strain of transgenic mice at the age of 3 months does not reach a level where any type of mitigating intervention can appropriately be assessed.
It is known that the activation of COX-2 and production of PGE2 (among other eicosanoids) seems to modulate amyloid-β protein precursor (AβPP) processing and promotes amyloidosis in the brain. APP/PS1/COX-2 mice expressing human COX-2 potentiated the formation of amyloid plaques and elevated the concentration of Aβ1-42 and Aβ1-40 without changing the total AβPP expression, suggesting that COX-2 can promote Aβ generation and amyloid burden by influencing AβPP processing [32]. Several other pieces of evidence also support the idea that elevated levels of COX-2 and prostaglandins are involved in the pathogenesis of AD, influencing the abnormal cleavage of AβPP, the formation of Aβ plaques, and the inclusion of phosphorylated tau in neurofibrillary tangles (for a recent review, see [33]).
In regard to the histopathologic analysis, we found some differences related to phenotype and treatment. The APP/PS1 + ONO-8713 mice submitted to pdMCAO had less cavitation and lower percent tissue loss than the WT + Veh group, but the effect on percent tissue injury was the opposite, i.e., the transgenic mice presented greater tissue injury compared with the respective WT. In addition, tissue injury was found to be higher in treated transgenic mice compared with their respective vehicle treatment. We also observed less cortical microgliosis in transgenic APP/PS1 mice submitted to pdMCAO compared with their counterparts treated with vehicle, but no differences were seen in hippocampal microgliosis or in either cortical and hippocampal astrogliosis. These data suggest, at least in part, some benefit of the ONO-8713, at the dose that was selected based on previous efficacy in preclinical studies, but the results are very subtle and are not enough to prove the efficacy of the treatment under the analyzed parameters.
We did not observe alterations in lesion volume, cavitation, or tissue injury in 3xTgAD mice submitted to pdMCAO treated with ONO-8713 or vehicle and its respective WT. The only differences observed were in percent tissue loss (lower for transgenic treated mice compared with the WT treated group) and percent tissue injury (higher for 3xTgAD treated mice compared with the WT treated group). Because these differences were moderate and opposite, it is likely that they did not reflect a robust drug effect, but perhaps some difference in the lesion profile in WT and transgenic mice. Based on our anatomical findings (negative results in most lesion parameters analyzed), we elected not to evaluate gliosis in the 3xTgAD mice. Another study that employed the pdMCAO model in 3xTgAD mice observed opposite and age-dependent changes. The transgenic background was associated with a slightly decreased neurobehavioral deficit score for 3- and 12-month-old mice, but the proportion of mice with a lesion that was visible simultaneously in the neocortex, striatum, and hippocampus decreased significantly with age in both WT and transgenic mice [42]. On the other hand, qualitative and quantitative data showed more severe degeneration of endothelial cells and astrocyte endfeet in 3xTgAD mice [42].
Using a relatively brief treatment protocol, we did not detect strong differences in lesion volumes and Aβ load between different treatment groups in either cohort. This is in contrast to our previous study in which loss of function of EP1 resulted in a remarkable attenuation of Aβ load as well as lesion volumes in APP/PS1 mice [21], as this could be explained by complete loss of activity as compared to the drug-treated mice. Another explanation for the difference is the smaller lesion size produced by pdMCAO and consequently a limited involvement of areas of the brain with EP1 receptors in this study compared with the aforementioned studies. In previous studies, other lesion models were used and greater lesion size encompassed areas with a higher EP1 receptor presence, consequently rendering the antagonist more efficacious. The model of transient cerebral occlusion, which was employed in previous studies, may have rendered the ischemic area more accessible to the drug by virtue of subsequent reperfusion, whereas the permanent occlusion model, used in our study, might have prevented the drug from reaching the affected regions and rendered the antagonist inefficacious.
EP1 is recognized as a proinflammatory PGE2 receptor in the brain. EP1 activation contributes to excitotoxicity and impairs the Na+-Ca2+ exchange necessary for Ca2+ homeostasis [15]. Several studies have suggested a neuroprotective effect for the EP1 selective antagonist SC-51089. The drug increased the viability of a human neuroblastoma cell line and murine primary cerebral cortical neurons incubated with Aβ1-42 [43] and increased the survival rate of cerebral cortical cells after in vitro hypoxia/reoxygenation [44]. An in vivo study showed that SC-51089 ameliorated motor coordination, balance dysfunction, and long-term memory deficit in a mouse model of Huntington’s disease and reduced the number of huntingtin nuclear inclusions in the striatum and hippocampus of 18-week-old animals [45]. The drug also reduced neuronal damage in models of hypoxic-ischemic injury in neonatal rats [46] and ischemia/reperfusion by bilateral common carotid artery occlusion [47]. However, recent studies have suggested that the effect of EP1 receptor modulation on neuroinflammation and neurotoxicity is complex. Although several studies showed a neuroprotective effect of EP1 antagonists or genetic deletion of EP1 [15, 48], these two strategies failed to protect mice from traumatic brain injury [49]. Moreover, EP1 deletion and the EP1 antagonist SC-51089 exacerbated the brain injury and neurobehavioral impairment induced by intracerebral hemorrhage [25, 50]. These results suggest that the efficacy of the EP1 antagonist may depend on the kind of injury and the regions involved as well as the adequacy of the reperfusion in the ischemic area. In our study, the lesion produced by pdMCAO was very focused after 14 days and was not severely aggravated in AD transgenic mice. As mentioned, the low production of Aβ in 3- or 4.5-month-old mice may not have been enough to contribute to the lesion severity.
It is important to discuss some potential limitations of the study: 1) We evaluated only the most characteristic anatomical findings in this study by sacrificing the mice at 14 days after the stroke. It is likely that the evaluation of other biomarkers of inflammation and AD pathology along with more comprehensive time points of assessments might have provided more insight into the role of the EP1 receptor in AD pathology after stroke. Moreover, future studies should include the ischemia/reperfusion model of stroke in aged transgenic AD mice to further investigate our hypothesis. 2) It is also important to note that we used relatively young AD transgenic mice with the assumption that stroke onset would aggravate AD pathology and we would be able to observe that during the time frame of our investigation. However, based on the data, AD transgenic mice of different ages need to be used to test the effect of stroke on AD pathology. 3) Other points of consideration are the drug dosage and the mode and duration of treatment. The dose used in this study was based on previous studies that pointed out that ONO-8713 has a wide therapeutic window [18], as also observed for another EP1 antagonist, SC51089 [19]. The neuroprotective effect of ONO-8713 was achieved through intracerebroventricular and intraperitoneal routes in different cerebrovascular models using acute or subacute treatment [16, 18]. Our results were recorded at an effective dose that has been reported previously [51], while others have also used a higher dose for this EP1 receptor antagonist [52, 53]. However, it is possible that the dose selected did not reach the therapeutic level in the current model, and a dose-response study is needed to achieve the best therapeutic dose in studies where stroke pathology is investigated in AD mouse models.
Overall, the treatment with ONO-8713 produced limited beneficial effects in transgenic and WT mice submitted to a model of focal ischemic injury. Other studies using different stroke models, as well as the use of older transgenic mice or a different strain with earlier Aβ accumulation onset, may provide more conclusive results with regard to the efficiency of the antagonist in attenuating Aβ production.
Footnotes
ACKNOWLEDGMENTS
We thank other Doré Lab members, notably, Morgan Carson, Andrew Lampert, James Catlin, Todd Sahagian, Alexander V. Glushakov, and Ana Paula F. Mendonça for helping with some experiments and analyses. Special thanks to Dr. David R. Borchelt’s and Dr. Todd A. Golde’s laboratories, notably Thomas Lad, and also Dr. Terrie Vasilopoulos for all their insightful comments. We also acknowledge the contribution of Dr. Takayuki Maruyama for his constructive suggestions. This work was supported by the NIH grant R01NS046400 (SD) and by the Florida Department of Health. FRM had a postdoctoral fellowship from São Paulo Research Foundation (#2014/18702-4).