
Abstract
Background:
Advanced glycation end products (AGEs) are an important risk factor for the development of cognitive decline in aging and late-onset neurodegenerative diseases including Alzheimer’s disease. However, whether and how dietary AGEs exacerbate cognitive impairment and brain mitochondrial dysfunction in the aging process remains largely unknown.
Objective:
We investigated the direct effects of dietary AGEs on AGE adducts accumulation, mitochondrial function, and cognitive performance in mice.
Methods:
Mice were fed the AGE+ diet or AGE– diet. We examined levels of AGE adducts in serum and cerebral cortexes by immunodetection and immunohistochemistry, determined levels of reactive oxygen species by biochemical analysis, detected enzyme activity associated with mitochondrial respiratory chain complexes I & IV and ATP levels, and assessed learning and memory ability by Morris Water Maze and nesting behavior.
Results:
Levels of AGE adducts (MG-H1 and CEL) were robustly increased in the serum and brain of AGE+ diet fed mice compared to the AGE– group. Furthermore, greatly elevated levels of reactive oxygen species, decreased activities of mitochondrial respiratory chain complexes I & IV, reduced ATP levels, and impaired learning and memory were evident in AGE+ diet fed mice compared to the AGE– group.
Conclusion:
These results indicate that dietary AGEs are important sources of AGE accumulation in vivo, resulting in mitochondrial dysfunction, impairment of energy metabolism, and subsequent cognitive impairment. Thus, reducing AGEs intake to lower accumulation of AGEs could hold therapeutic potential for the prevention and treatment of AGEs-induced mitochondrial dysfunction linked to cognitive decline.
INTRODUCTION
Advanced glycation end products (AGEs) are the irreversible products of nonenzymatic glycation of proteins by reducing sugars and/or dicarbonyls [1 –4]. The formation of AGEs occurs both endogenously and exogenously. Endogenous AGEs form slowly and continuously under physiological conditions throughout life [5]. Ingested (exogenous) AGEs from foods are thought to be a vital contributor to AGEs within body tissues where they become structurally and functionally indistinguishable from endogenous AGEs [6, 7]. Although AGEs accumulation is part of the normal aging process [8], when significantly accelerated, AGEs accumulation detrimentally affects nearly every type of cell and molecule in the body, having roles in the development and progression/aggravation of many degenerative diseases, including diabetes, atherosclerosis, cardiovascular disease, and Alzheimer’s disease (AD) [9 –13]. In AD, AGEs can induce crosslinking of long-lived proteins such as amyloid-β peptide (Aβ) and hyperphosphorylated tau, which are hallmarks of AD pathology [11 –14]. Additionally, excessive AGEs accumulation disturbs synaptic transmission and reduces long-term potentiation [2], contributing to a decline in cognitive ability during the aging process and an acceleration in progression from mild cognitive impairment to AD [15, 16].
Despite the evidence of mitochondrial dysfunction in AD-affected brains and cognitive impairment in AD patients and AD transgenic mouse models, there are major gaps in our knowledge of the causal relationships between environmental factors such as excessive dietary AGEs and physiological effects such as mitochondrial dysfunction and cognitive impairment. Here, we first characterize a methylglyoxal (MG)-derived AGEs diet additive by evaluating the extent of protein structure modification through a variety of physicochemical and biochemical techniques. Second, we investigate the direct effects of dietary AGEs on AGE adducts formation and accumulation, mitochondrial function, and cognitive performance in mice. These studies address the following key questions: 1) How do dietary AGEs affect accumulation of circulatory and cerebral AGEs? 2) Do dietary AGEs elevate levels of reactive oxygen species (ROS), thereby promoting oxidative stress? 3) Do dietary AGEs cause deterioration in mitochondrial function, contributing to impairment in learning and memory? Our results indicate that dietary AGEs accelerate accumulation of AGE adducts in the brain, resulting in mitochondrial dysfunction and cognitive impairment.
MATERIALS AND METHODS
Animal study
Both male and female mice (Charles River C57BL/6 strain) were used in this study. Mice were divided into two groups and placed on either AGE– or AGE+ diets. Mice were housed 5 per cage in a temperature-controlled room with a 12 h light–dark cycle and provided food and water ad libitum. Food and water intake were recorded during the treatment period. For biochemical analysis, mice were kept on their respective diets for 6 months starting at 3 months of age before serum was collected and brain cortexes (including hippocampi) were dissected, snap frozen in liquid nitrogen, and stored at –80°C. A second set of mice were fed AGE– or AGE+ diet for 17 months starting at 4–5 months of age and then had their learning and memory function assessed by Morris Water Maze (MWM) and nesting behavior tasks. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Kansas.
In vitro glycation of BSA
Glycation of BSA was conducted in accordance with our previous method with modifications [1, 17]. Briefly, native BSA (75 mg) was dissolved in 1× phosphate buffered saline (PBS), containing 1 mM EDTA (pH 7.4), followed by incubation with 100 mM MG at 37°C for 12 days. Aliquots were taken at different time intervals and frozen for later analysis. The glycated BSA (MG-BSA) on day 12 was extensively dialyzed at 4°C with PBS to remove any free MG and then passed through a 0.22 μm filter. As a control, native BSA was incubated under the same conditions in the absence of MG.
Spectroscopic analysis
The absorption spectra of native and glycated BSA between 200 and 800 nm were recorded using an Ultrospect 3100 Pro UV/Visible spectrophotometer (Amersham Biosciences, Piscataway, NJ) [18]. Increase in absorbance (hyperchromicity) at 280 nm was calculated by the following equation:
Fluorescence spectra were recorded using a Hitachi F-7000 Fluorescence Spectrophotometer (Japan) at 25±0.1°C. Native and glycated BSA samples were excited at 350 nm and emission intensities were recorded between 350–500 nm. Presence of fluorogenic AGEs in the glycated sample was verified with AGE-specific fluorescence emission at 450 nm [17]. Increase in fluorescence intensity (FI) was calculated from the following equation:
Far-UV circular dichroism (CD) measurements of native and glycated BSA were recorded on a Chirascan™ qCD spectrometer (Applied Photophysics, UK) between 190 and 250 nm. All scans were recorded at wavelength intervals of 1 nm. Baselining and analysis were done using Chirascan V100 control software. Protein samples were prepared at 0.5 mg/ml for both the far and near UV CD measurements [19]. All CD spectra were collected in cells of 1 or 10 mm path length respectively, for the far-and near-UV CD measurements. The scan speed was 100 nm/min and a response time of 1 s was used for all measurements as reported earlier [20].
Thermal denaturation of native and glycated BSA and their controls were evaluated by temperature scan from 30 to 95°C at an increment of 1.5°C/min on a Genesys 20 spectrophotometer (Cole-Parmer) coupled with a temperature programmer and controller assembly [17]. The change in absorbance at 282 nm was recorded along with the melting temperature (Tm) and the percent denaturation was computed using the following equation:
AT is the absorbance at a given temperature T (in °C), Amax is the final maximum absorbance upon completion of denaturation at 95°C, and A30 is the initial absorbance at 30°C [17].
Dietary formulas
AGE– diet is normal diet without the addition of MG-BSA and was used as control. AGE+ Diet is modified LabDiet® Advanced Protocol® PicoLab® Verified-75 IF, 5V75, containing 1000 ppm MG-BSA (1.33%). The diets were produced using the highest quality ingredients to ensure minimal inherent biological variation in long-term studies. Irradiation and special 4-ply packaging were used to provide a virtually bacteria-free diet. This diet is especially suited to animals maintained in barrier facilities. Both dietary formulas were pelleted by the manufacturer and kept at 4°C.
Determination of protein-bound carbonyl groups, HMF contents, and ketoamine moieties
Protein bound carbonyl content was determined using 2,4-dinitrophenylhydrazine. Absorbance was measured at 360 nm and carbonyl content was estimated using an extinction coefficient of 22,000 M–1 cm–1 [21].
Hydroxymethylfurfural (HMF) content was estimated by thiobarbituric acid assay as described earlier [21].
The ketoamine moieties in serum were determined by nitroblue tetrazolium (NBT) reduction assay as described [21]. The absorbance of protein samples was recorded at 525 nm and the content (nmol mL–1) was estimated using an extinction coefficient of 12,640 M–1 cm–1.
Measurement of superoxide anion, hydroxyl radical, and hydrogen peroxide (H2O2)
To detect the presence of O2– anion in mouse serum samples, cytochrome c reduction assay was performed as previously described [1 , 13]. The reduction rate was determined by measuring increases in absorbance at 550 nm for 10 min at room temperature. Absorbance was recorded at 1 min intervals.
To detect generation of hydroxyl radicals in mouse serum samples, thiobarbituric acid reactive substances (TBARS) assay was conducted [1 , 21]. The conversion of 2-deoxy-D-ribose into TBARS was measured by adding l mL of 2.8% (w/v) trichloroacetic acid and 1 mL of 1% (w/v) thiobarbituric acid (TBA) followed by heating at 100°C for 10 min. After cooling, the absorbance was read at 532 nm against an appropriate blank.
H2O2 levels in AGE– and AGE+ mouse serum samples were determined using the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Cat#A22188; Invitrogen) according to the manufacturer’s instructions. Absorbance of reaction product at 560 nm was measured using the standards from 0–5 μM with no H2O2 as a negative control.
Electron paramagnetic resonance (EPR) measurements
Evaluation of intracellular ROS levels was accessed by EPR spectroscopy [22]. Brain tissues were incubated with CMH (cyclic hydroxylamine 1-hydroxy-3-methoxycarbonyl-2, 2, 5, 5-tetramethyl-pyrrolidine, 100 μM) for 30 min, then washed three times with cold PBS. The brain tissues were collected and homogenized with 100 μl of PBS for EPR measurement. The EPR spectra were collected, stored, and analyzed with a Bruker EleXsys 540×-band EPR spectrometer (Billerica, MA, USA) using the Bruker software Xepr (Billerica, MA, USA).
Immunodetection of AGE-adducts
Glycation has major influences on the folding, function, and stability of many different proteins. Analysis of non-enzymatic AGE modification is challenging using western blot because of the complex band pattern as a result of non-specific pairing. We therefore use immunodot blot to represent the distribution of groups of modified protein as points on a simple scale, allowing us to confirm the total amount of glycation adducts in each. For immunoblotting, equal amounts of protein (2 μg) from BSA and MG-BSA samples were resolved by 10% SDS-PAGE and transferred onto a nitrocellulose membrane (Catalog# 162-0094, Bio-Rad Laboratories, Hercules, CA, USA). For immunodot blot, equal amounts of total protein extracts (1 μg) from AGE– and AGE+ diet samples, serum, and homogenates of cerebral cortex (including hippocampus) from mice were spotted onto nitrocellulose membranes (Bio-Rad). Ponceau staining of protein spots was used as the protein loading control for dot blot. Primary antibodies against MG-H1 (HM5017, Hycult Biotech), or CEL (HPA008023, Sigma-Aldrich) at 1:3000 and the corresponding secondary antibody HRP-conjugated anti-mouse or anti-rabbit IgG at 1 : 10000, and Chemiluminescent Substrate (Catalog #34580, ThermoFisher Scientific) were used to detect AGE-adducts of proteins. Protein signals were visualized using a FluorChem HD2 imaging system. The intensity of signal was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). For ELISA, total AGEs in mouse serum and brain cortex homogenates were determined using mouse AGEs ELISA kit (Catalog #MBS704846, MyBioSource), according to the manufacturer’s protocol.
Immunohistochemistry
Mice under deep anesthesia with ketamine/xylazine were perfused intracardially with 0.9% saline and then 4% paraformaldehyde in PBS (PFA). Brains were removed from skulls and additionally fixed in PFA for 14–18 h (4°C). 40 μm sections were prepared with a vibratome (Leica VT1000S) and stored in cryoprotectant solution at –20°C. For double immunostaining free floating sections were incubated with anti-AGE antibody (generated in our laboratory, guinea pig polyclonal, 1 : 150) [1 , 13] and anti-NeuN (pan neuronal marker, mouse monoclonal, 1 : 150, Millipore, # MAB377) in 0.2% Triton X-100 in PBS (4°C, overnight) followed by goat anti-guinea pig Alexa Fluor 594 (1 : 300, Thermo Fisher Scientific, Eugene, OR) or goat anti-mouse (Alexa Fluor 633, 1 : 300). Sections were mounted on slides using Vectashield (Vector Laboratories, Burlingame, CA). As controls for the specificity of immunostaining, primary antibodies were omitted and substituted with appropriate normal serum. Slides were viewed using a confocal microscope (Nikon Ti Eclipse). Levels of AGE immunofluorescence were evaluated in the images (merged from stacks of 5 adjacent images with 1024×1024 pixel resolution, observed area 295×295 μm, captured with confocal microscope at a distance of 0.5 μm from each other) obtained from brain sections stained for AGE. Quantification of AGE immunofluorescent intensity was performed using MetaMorph® Image Analysis Software.
Measurement of enzyme activities associated with mitochondrial respiratory chain complexes
Activity of key enzymes associated with the respiratory chain was measured using homogenates of cerebral cortex including hippocampus as previously described [22 –24]. Briefly, sections of cerebral cortex were homogenized in Cell Lysis buffer (Cell Signaling Technology). Homogenates containing 50 μg of protein were used for complex I (NADH-ubiquinone oxidoreductase) and IV (cytochrome c oxidase) assays. Complex I activity was determined in 25 mM potassium buffer containing 3 M KCl, 1 M Tris-HCl, and 0.5 M EDTA (pH 7.4). The change in absorbance was monitored at 340 nm wavelength every 20 s for 6 min using an Amersham Biosciences Ultrospect 3100 pro spectrophotometer. Complex IV activity was determined by cytochrome c reduction rate. Briefly, the protein samples were added to a cuvette containing 0.475 ml of assay buffer (10 mM Tris-HCL, pH 7.0, and 120 mM KCl), and the reaction volume was brought to 0.525 ml with enzyme dilution buffer (10 mM Tris-HCl, pH 7.0 and 250 mM sucrose). The reaction was initiated by the addition of ferrocytochrome c substrate solution (0.22 mM) into the cuvette. The rate of change in absorbance at 550 nm was recorded using a kinetic program with 5 s delay and 10 s intervals for 6 readings on Amersham Biosciences Ultrospect 3100 pro spectrophotometer.
Measurement of ATP levels
Adenosine 5’-triphosphate (ATP) levels were assessed using an ATP Bioluminescence assay Kit (Sigma/Roche) following the manufacturer’s instructions. Briefly, mouse cortical tissues were homogenized in the lysis buffer provided, incubated on ice for 30 min, and centrifuged at 12,000×g for 10 min at 4°C. ATP levels were then measured in the subsequent supernatants using a Luminescence plate reader (Molecular Devices). A 1.6 s delay time after substrate injection and 10 s integration time were used.
Morris water maze
Mice were subjected to the MWM hidden platform test as described in our previous studies [22, 23]. Briefly, mice were trained for five consecutive days with four trials per mouse per day. On the last day, a probe trial was performed to assess the spatial memory of the mice. Traces of mice were recorded and data were analyzed using HVS Image 2014. All animals were individually coded and investigators were blinded to the mouse genotypes for the duration of behavioral testing.
Nesting behavior study
Each cage is supplied with a ‘Nestlet’, a 5×5 cm square of pressed cotton batting (Ancare, Cat# NES3600), for three days prior to measuring nesting capability. For assessment, mice were housed individually with normal bedding and given an intact, pre-weighed cotton nestlet (3.0 g; Ancare). After 24 h, images were taken for each nest. Nest quality was scored on a scale of 1 to 5, according to previously published criteria [25, 26], with 1 being largely untorn nestlets and 5 being near perfect nests.
Statistical analysis
One-way ANOVA was used for repeated measure analysis, followed by Fisher’s protected least significant difference for post hoc comparisons. p < 0.05 was considered significant. StatView statistics software was used. All data were expressed as the mean±SEM.
RESULTS
Preparation and characterization of MG-AGEs (glycated BSA) in vitro
To establish the reaction conditions for preparation of MG-derived AGEs in vitro, the structural modifications in BSA were evaluated by spectroscopic analysis following incubation of BSA with MG for 1–12 days. The UV visible absorbance spectra from 200–800 nm of nonglycated BSA (BSA) and MG-glycated BSA (MG-BSA) at different time points during 1–12 days of incubation were assessed. Hyperchromicity, at 282 nm, of MG-BSA increased in a time-dependent manner, reaching a higher level at day 12 (Fig. 1A, B). Incubation of BSA in the absence of MG under the same conditions did not result in increased absorbance at 282 nm. Thus, the change in hyperchromicity of the MG-BSA samples may be attributed to AGE formation and crosslinking by MG.
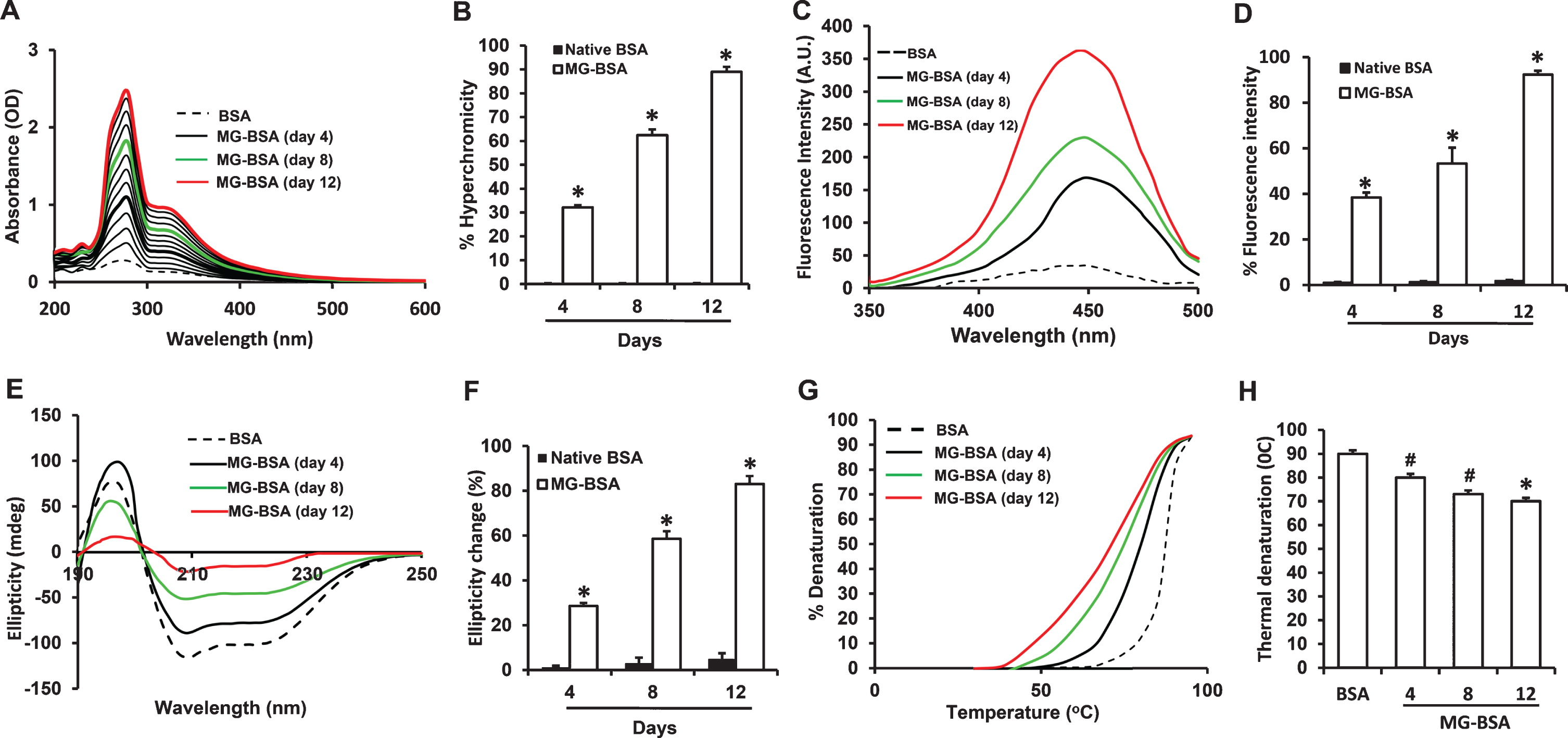
Characterization of MG-glycated BSA. A) Ultraviolet absorption spectra of native BSA (BSA) and BSA modified with 100 mM MG (MG-BSA) during 1–12 days of incubation. B) Increased % hyperchromicity of MG-BSA compared to day 4 BSA. C) Fluorescence emission spectra of BSA and MG-BSA. Excitation wavelength was 350 nm. D) % change in fluorescence intensity of MG-BSA compared to BSA. E) Far UV CD spectra of BSA and MG-BSA. F) % change in ellipticity (structural loss) of MG-BSA (*p < 0.01 versus day 4 MG-BSA). G) Thermal melting profile of BSA and MG-BSA. H) % decrease in melting temperature (°C) of MG-BSA versus BSA. Data presented as mean±SEM, # p < 0.05, *p < 0.01 versus day 4 glycated sample or BSA group. All spectra/profile shown are the averages of three determinations.
To confirm the formation of fluorogenic AGEs in glycated BSA, fluorescence spectroscopy was performed by exciting MG-BSA at 350 nm and recording the emission maxima at 450 nm. BSA incubated without MG did not show any fluorescence. The maximum fluorescence intensities for MG-BSA increased with incubation time by 37.43%, 54.19%, and 90.5% after 4, 8, and 12 days of incubation, respectively, as compared to BSA alone (Fig. 1C, D).
To further determine the effect of MG-induced glycation on the conformational characteristics of BSA, we recorded far-UV CD (circular dichroism) spectra between 190–250 nm (Fig. 1E). The CD spectra of BSA indicated that it is a predominantly α-helical protein as shown by negative minima at 208 nm (Fig. 1E). The changes in ellipticity by day 4, 8, and 12 were 23.64%, 55.69%, and 81.88%, respectively (Fig. 1F). Such changes in ellipticity may be associated with decrease in α-helicity and increase in β-sheet structure, providing an environment for the intermolecular interactions required for aggregation and AGE formation. These data are indicative of time-dependent alterations in the structure of BSA as a result of glycation by MG.
The effect of MG-induced protein structure alteration on stability was evaluated by monitoring thermal denaturation of MG-BSA and BSA alone between 30 and 95°C (Fig. 1G). Glycation by MG shifted the thermal denaturation curves to lower temperatures in a time-dependent manner. By day 12 of incubation, the melting temperature (Tm) of MG-BSA had decreased by 17°C compared to BSA, suggesting decreased resistance of glycated BSA to thermal unfolding and lower thermal stability (Fig. 1H).
Table 1 summarizes the physicochemical parameters of MG-BSA.
Characterization of native BSA and MG-BSA samples
# p < 0.05; * p < 0.01.
Determination of MG-derived AGEs in glycated BSA and AGE+ diet
Given that the MG-AGEs methylglyoxal-hydroimidazolone (MG-H1) and N ɛ –(1-carboxyethyl)-L-lysine (CEL) are markers of MG-induced modification of arginine and lysine residues, respectively [27], we verified whether these adducts were present in the MG-BSA preparation by immunnodetection. Analysis of BSA and MG-BSA samples by SDS-PAGE and Coomassie blue staining showed bands at approximately 66 kDa and 70 kDa, respectively (Fig. 2A, Lane 1 & 2). The increase in molecular weight of glycated BSA is due to MG modification, leading to intramolecular crosslinking. Immunoblot analysis using specific antibodies revealed the presence of MG-H1 and CEL in MG-BSA (Fig. 2A, lanes 4 & 6) but not in nonglycated BSA (Fig. 2A, Lanes 3 & 5); thus, verifying the formation of MG-AGEs. These results indicate that MG-AGEs, including MG-H1 and CEL, are major components of the MG-BSA preparation used in the formulation of our customized mouse diet for subsequent in vivo experiments.
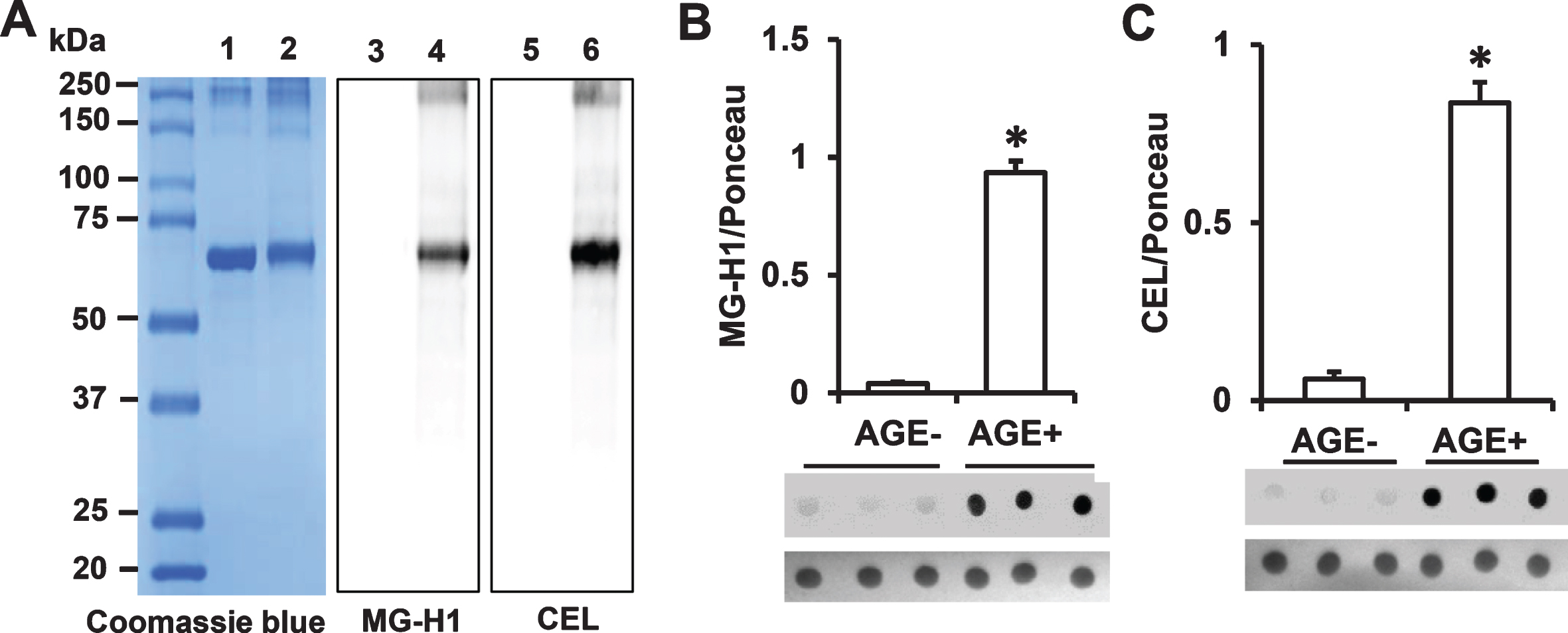
Determination of AGE adducts in the AGE additive and AGE diet. A) Protein (2 μg) from BSA or MG-BSA samples following 12 days of incubation were subjected to SDS-PAGE. Protein bands were stained by Coomassie Blue (A, Lane 1 & 2). Glycation was detected by immunoblotting with antibody to MG-H1 (A, Lane 3 & 4) or CEL (A, Lane 5 & 6). Lane 1, 3, 5, BSA; lane 2, 4, 6, MG-BSA. Quantification of immunodot intensity of MG-H1 (B), and CEL (C) normalized to Ponceau staining intensity (as a protein loading control) in protein extracted from the samples of AGE– and AGE+ diet. Data are presented as mean±SEM (n = 3), *p < 0.01 versus AGE– diet.
Next, we further determined the MG- derived AGEs (MG-H1 and CEL) content of our customized diet containing the MG-BSA additive (AGE+) versus that of the normal diet used as control (AGE–) by immunodetection. Immunodot blot analysis of proteins extracted from AGE+ and AGE– diet revealed the presence of MG-H1 and CEL in AGE+ diet but negligible levels of these MG-derived AGEs in AGE– diet (Fig. 2B, C). Collectively, these results indicate the presence of high levels of AGEs in the AGE+ diet, making it appropriate for in vivo determination of the effect of dietary intake of AGEs on mitochondrial and cognitive function.
Administration of AGE+ diet increases AGEs in the serum and cerebral cortex
Immunodot blot showed robustly increased MG-H1 and CEL in the serum of AGE+ fed mice (Fig. 3A, B). Accordingly, elevation of total AGEs was found in the serum samples from AGE+ fed mice by quantitative ELISA (Fig. 3C). Thus AGE+ fed mice had higher levels of circulating AGEs in the serum than AGE– fed mice. We further determined the accumulation of AGEs in the brain by measuring levels of glycated adducts in AD-affected regions (cerebral cortex containing hippocampus) of AGE+ and AGE– fed mice. Immunodot blots showed that levels of MG-H1 and CEL in the cortex of AGE+ fed mice were increased by 4.5- and 3-fold, respectively (Fig. 4A. B). Similarly, the AGE+ mouse cortex revealed robust elevation of total AGEs by quantitative ELISA (Fig. 4C). We performed immunostaining with an AGE specific antibody to validate the distribution of AGEs in the brain. AGE+ fed mice revealed significantly higher levels of AGEs in neocortical neurons, as compared with AGE– fed mice (Fig. 4D, E). Thus, exogenous AGEs promote accumulation of AGEs in the serum and brain.

Measurement of levels of AGEs in serum from AGE diet fed mice. Quantification of immunodot intensity of MG-H1 (A) and CEL (B) normalized to Ponceau staining intensity in serum of AGE– and AGE+ diet fed mice. Ponceau staining of membranes of corresponding dot blots was used as a protein loading control (A & B, lower panel). ELISA for the measurement of AGE (C) levels in serum samples of AGE– and AGE+ fed mice. Data are presented as mean±SEM (n = 5), *p < 0.01 versus AGE– fed mice as a control group.
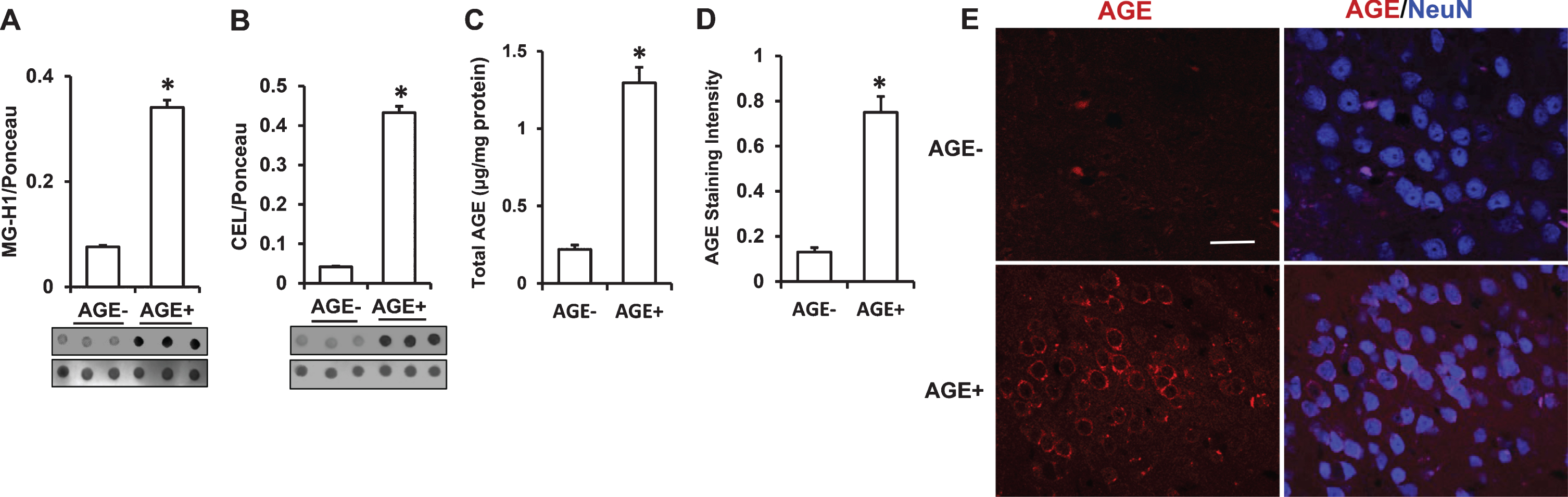
Measurement of levels of AGEs in cerebral cortex from AGE diet fed mice. A–C) Quantification of immunodot intensity of MG-H1 (A), and CEL (B) normalized to Ponceau staining intensity in cerebral cortex of AGE– and AGE+ fed mice (A & B, lower panel). ELISA for measurement of AGE (C) levels in cerebral cortex of AGE– and AGE+ fed mice. D) Quantification of AGE immunofluorescence intensity in the cortex of the indicated group of mice using MetaMorph® Image Analysis Software. E) Representative AGEs staining images for AGEs (red) and nucleus (NeuN, neuronal marker, blue) under confocal microscopy. Scale bar = 25 μm. Data are presented as mean±SEM (n = 3), *p < 0.01 verses AGE– fed mice.
AGE+ diet increases ROS and oxidative stress
AGE-modified proteins, including glycated tau and Aβ, generate ROS and induce neuronal oxidative stress [1 , 11–13]. We therefore determined whether increased intake of exogenous AGEs would elevate ROS and oxidative stress. Indeed, levels of ROS, including superoxide anion, hydrogen peroxide, and hydroxyl radicals (Fig. 5A–C and Table 2) were elevated by ∼16-, 1.4-, and 12-fold, respectively, in serum of AGE+ fed mice compared to that of AGE– fed mice. We further examined whether AGE-diet-stimulated ROS production elevated the reactive intermediates facilitating oxidative stress. Ketoamine Amadori products, glycation intermediates, were elevated by ∼5-fold in AGE+ serum versus AGE– serum. This paralleled increases in other reactive intermediates in AGE+ serum such as carbonyl species and hydroxymethylfurfural (HMF) which were elevated by 14- and 3.75-fold, respectively (Table 2). Consistent with these observations, we quantitatively measured intracellular ROS levels in brain tissues by highly specific EPR spectroscopy. The intracellular ROS levels indicated by EPR peaks were significantly elevated in AGE+ fed mice compared to AGE– fed mice (Fig. 5D, E). These data indicate that dietary AGEs accumulate in the brain resulting in elevated ROS and oxidative stress.

Effect of AGE diet on ROS generation and mitochondrial function. Quantification of superoxide anion (A), hydrogen peroxide (H2O2) (B), hydroxyl radicals (C) in serum of AGE– and AGE+ fed mice. Representative in vitro EPR spectra measured in cortical brain homogenates (D, E) are shown. The peak height in the spectrum indicates levels of ROS. Quantification of EPR spectra in the indicated AGE– & AGE+ fed mice (D). *p < 0.01 compared to other groups of mice. Data are expressed as fold-increase relative to AGE– fed mice. N = 5 mice per group. Activities of complexes I (F) and complex IV (G) and ATP levels (H) were determined in the cortex of AGE– and AGE+ mice. Data presented as mean±SEM (n = 5), *p < 0.01 versus AGE– control group.
Quantification of glycation adducts or intermediates in serum
# p < 0.05; * p < 0.01.
AGEs impair mitochondrial function
Next, we evaluated the effect of AGEs on mitochondrial respiratory function and energy metabolism by assessing activities of mitochondrial respiratory chain complexes I (NADH: ubiquinone oxidoreductase) and IV (cytochrome c oxidase) and ATP levels. Cortical mitochondria of AGE+ fed mice showed significant reduction in both complex I and IV activities and ATP levels compared to AGE– fed mice (Fig. 5F–H), suggesting that dietary AGEs induce defects in mitochondrial respiratory function.
AGEs impair cognition and daily task performance
Given the vital role of mitochondria in generating the energy necessary for cellular function and modulating calcium homeostasis and neurotransmitter release [22 , 28–31], we next assessed whether AGEs-mediated mitochondrial dysfunction was reflected in altered neuronal function as evidenced by impairment in learning and memory and changes in daily task performance. To assess spatial learning and memory, mice were subjected to the MWM test. MWM is a test of spatial learning and working memory for assessment of a long-lasting form of memory. It has proven to be a robust and reliable test that is strongly correlated with hippocampal synaptic plasticity [32]. It has been broadly used in behavioral assessment in the AD mouse model and used in our published studies in AD mouse models [22 , 34]. Compared to AGE– fed mice, AGE+ fed mice displayed significantly longer escape latencies during the 5-day training session (Fig. 6A) and reduced number of crossings of the target area and time spent in the target quadrant during the probe trial (Fig. 6B, C, E). There were no significant differences in swimming speed as measured by the visual swimming speed test (Fig. 6D), indicating that the observed difference in spatial learning and memory is due to cognitive decline rather than differential motility or motivation. These data indicate that intake of an AGEs-enriched diet induces deficits in learning and memory.

Effect of AGE diet on learning and memory. Mice fed AGE– or AGE+ diet for 17 months starting at the age of 5 months were subjected to the Morris water maze test. A) Escape latency during MWM hidden platform task in AGE– and AGE+ fed mice. B) Time spent in the quadrant with the hidden platform. C) Mean number of crossings of the target during the probe test. (D) Average swim speed of mice in MWM. (NS, no significance between AGE– and AGE+ group). E) Representative traces during the probe test. Data are shown as mean±SEM. *p < 0.01 versus AGE– fed mice (n = 10 mice per group). F, G) Evaluation of nest building performance. F) Representative nest photos showing the nest building behavior in the indicated mice. G) Nest score on the basis of nest building behavior. Nesting deficits indicated by incomplete nests and low nest score. Nesting ability indicated by complete nests, high nest score. *p < 0.01 compared with AGE– fed mouse nesting ability.
Given that deterioration in the ability to perform activities of daily living is an early sign of AD and cognitive decline, as well as hippocampal lesions, daily task performance was measured by assessing mouse nest-making ability [25]. AGE+ fed mice scored significantly lower on the nestlet task (mean score = 2.9), compared to AGE– fed mice (mean score = 4.8) (Fig. 6F, G), suggesting that consumption of a diet with high AGEs content could lead to deficits in daily task performance.
DISCUSSION
Growing evidence points to the importance of foods as an exogenous source of AGEs [6 , 35]. Human exposure to AGEs is widespread, especially because most Western foods are processed at high temperature and are therefore rich in AGEs. AGEs accumulate in the human brain with increasing age [8], being found in neurofibrillary tangles and senile plaques in patients with AD [12 –14]. AGEs are associated with neuronal dysfunction, synaptic damage, and oxidative stress. However, the underlying mechanisms are poorly understood. Because mitochondria are vital for controlling cell survival and death as well as synaptic function, it is essential to investigate the effect of AGEs on mitochondrial function, which may provide a mechanistic link to cognitive decline. In this study, we determined the direct effects of dietary AGEs on mitochondrial and cognitive function after long-term AGEs feeding in wild-type mice. Our results demonstrated that a diet high in AGEs perturbs mitochondrial and cognitive function, which advances our understanding of the etiology of neurodegeneration and cognitive decline in aging and age-related neurodegenerative disease. Greater understanding of AGEs-mediated mitochondrial pathology may assist in the development of new therapeutic avenues for the prevention and treatment of age-related dementia and AD.
MG reacts with lysine and arginine residues on proteins to generate AGEs [27, 36]. The present studies generated MG-derived AGEs in vitro by incubation of MG with BSA at 37°C for 12 days. Indeed, the shorter incubation time necessary for the glycation of BSA by MG compared to the previous 6–8 week incubation for the glycation of BSA by glucose or glucose 6-phosphate [1, 2] aligns with the significantly stronger glycation ability of MG compared to glucose or glucose 6-phosphate [3, 4].
Lysine and arginine modified by MG have been reported to give electron paramagnetic resonance signals [37] indicative of the formation of free radicals (·OH and O2–). Furthermore, accumulation of AGE-modified Aβ and tau triggers production of reactive oxygen intermediates and free radicals resulting in neuronal oxidative stress [1 , 11–13]. In line with these observations, we demonstrated that dietary MG-glycated AGEs elevated circulating AGEs levels, leading to increased formation of ROS, including superoxide anion, hydroxyl radicals, and hydrogen peroxide, in the serum and brain of AGE+ fed mice compared to AGE– fed mice. Consumption of AGE+ diet also elevated serum levels of glycation intermediates such as ketoamines and HMF. HMF has been shown to induce mitochondria-associated cell death and ROS generation [38]. Such increases in ROS and oxidative stress may lead to deterioration in neuronal and mitochondrial function, accelerating the progression of AD [13 , 39].
Neurons require a large number of mitochondria to meet their high energy demands, particularly with respect to synaptic function. Mitochondria synthesize ATP, the body’s energy carrier, through the electron transport chain and ATP synthase. Complexes I, III, and IV are proton pumps that generate the transmembrane proton gradient necessary to drive ATP generation by ATP synthase. Thus, perturbations of the electron transport chain produce excessive ROS and disrupt downstream mitochondrial and cellular processes. AGEs accumulate over a person’s lifetime as a part of the normal aging process [8], but significantly elevated accumulation is seen in patients with diseases such as diabetes and AD [9, 10]. Significant declines in ATP levels and mitochondrial respiratory chain complexes were observed in the cerebral cortex of AGE+ fed mice, suggesting that dietary AGEs-driven accumulation of AGEs in the brain causes cumulative oxidative damage and subsequently impacts mitochondrial function, thereby disrupting brain energy metabolism.
Altered mitochondrial function and oxidative stress are well-established causes of cognitive decline [22 , 34]. Increased levels of AGEs have been reported in cortical neurons of older adults and are positively correlated with severity of cognitive impairment [15]. However, the impact of dietary AGEs on mitochondrial function and cognition is under explored. Therefore, we investigated whether AGE-mediated mitochondrial dysfunction links to altered cognitive performance related to neuron and brain activity. AGEs treatment of hippocampal slices alters synaptic structure and function as shown by impaired long-term potentiation and loss of synapses [2]. Given that mitochondria are vital for maintaining synaptic transmission and plasticity and that intact synaptic structure and function are essential for the synaptic encoding that forms stable memories, we hypothesized that AGE-mediated deficits in synaptic plasticity linked to mitochondrial function might contribute to the learning and memory deficits seen in age-related cognitive decline. Extending our previous findings of AGEs–induced synaptic and mitochondrial dysfunction in vitro, our present in vivo studies showed that long-term consumption of a diet high in AGEs led to significant impairment in learning memory and daily task performance. These impaired learning behaviors provide evidence that exogenous MG-derived AGEs induced memory loss through mitochondrial dysfunction, suggesting that accumulation of exogenous AGEs is an important risk factor in age-related dementia and the development and progression of AD.
AGEs not only induce changes in structure and function of cellular proteins, but are also recognized by a receptor for AGE (RAGE). AGEs act as a signaling ligand by interaction with RAGE in response to metabolic changes that occur during aging and age-related neurodegenerative disease [1 , 13]. Expression of RAGE is elevated in AD patients and AD transgenic mice [40 –42]. RAGE also functions as a signal transduction receptor for Aβ to induce Aβ accumulation, oxidative stress, and inflammation [1 , 43–45]. Overexpression of RAGE in neurons or microglia of AD mice accelerates Aβ accumulation and aggravates spatial learning/memory impairment and neuropathological and biochemical changes [43, 44]. Interaction of RAGE with AGEs enhances oxidative stress and exacerbates synaptic dysfunction by reducing long term potentiation [1 , 13]. Blockade or genetic deletion of RAGE or RAGE signaling in neurons or microglia of AD mice attenuates Aβ induced deterioration, preserves spatial learning and memory, and protects against synaptic dysfunction [2 , 43–45]. Therefore, blockade of AGE-RAGE interactions may prevent mitochondrial dysfunction and cognitive impairment resulting from AGE accumulation and RAGE-dependent signaling.
In this study, we compared results generated from a diet containing high levels of AGEs with those from a diet containing small amounts of naturally produced AGEs without additional AGEs-modified BSA. Since the AGEs were orally administered, it is possible for free AGEs to enter and affect the entire brain in addition to the cortex and hippocampus. Thus, we could not exclude the effects of AGEs on other brain areas. AGE content in the cerebral cortex of AGEs fed mice was about 3–4.5-fold higher than that of the normal diet fed mice based on AGE determination assay. Under these conditions, we provide concrete evidence of the detrimental effects of a high AGE diet compared to a low AGE diet on mitochondrial and cognitive function, which has significant implications for the treatment and prevention of aging-related dementia and cognitive decline relevant to neurodegenerative diseases including AD. Given that many commonly cooked foods, especially those that have been exposed to high heat, for example through barbecuing, grilling, roasting, frying, and toasting, may have AGE content 10–100 times that of uncooked foods, diets high in those types of food may result in potentially harmful levels of AGE intake [46, 47]. Limiting dietary AGEs may lower the risk of many chronic diseases and premature aging [48 –50].
In conclusion, excessive AGEs intake is an important contributor to the accumulation of AGEs in the serum and brain, causing significantly decreased mitochondrial respiratory chain complex I & IV enzyme activity, reduced ATP levels, and deficits in learning and memory and daily task performance. Increase in dietary AGEs intake may be a risk factor for accelerating age-related dementia and cognitive dysfunction. Reducing AGEs uptake and blockade of AGEs toxicity may hold therapeutic potential for the prevention and treatment of AGEs-involved mitochondria dysfunction linked to cognitive decline.
Footnotes
ACKNOWLEDGMENTS
This study was supported in part by the National Institute of Aging. We thank Drs. Russ Middaugh and Jian Xiong from the University of Kansas School of Pharmacy-Pharmaceutical Chemistry for assistance in CD structure analysis, and Justin T. Douglas from the Nuclear Magnetic Resonance Laboratory at University of Kansas for assistance using the EPR instrument.
This study was supported by NIH/NIA (R37AG037319, R01AG044793, R01AG053041, RF1AG054320, R56AG06434, and R01AG061324).