
Abstract
Background:
Dysbiotic microbiota in the gastrointestinal tract promotes and aggravates neurodegenerative disorders. Alzheimer’s disease (AD) has been shown to correlate to dysbiotic bacteria and the immune, metabolic, and endocrine abnormalities associated with abnormal gut-brain-axis signaling. Recent reports also indicate that brain dysbacteriosis may play a role in AD pathogenesis.
Objective:
To evaluate the presence and differences of brain-region dependent microbiomes in control and AD subjects and the contribution of study bias.
Methods:
Two independent cohorts of postmortem AD brain samples were collected from separate locations, processed with different extraction protocols and investigated for the presence of bacterial DNA indicative of a brain microbiome with V4 16S next generation sequencing.
Results:
In both cohorts, few differences between the control and AD groups were observed in terms of alpha and beta diversities, phyla and genera proportions. Independent of study in both AD and control subjects the most abundant phyla were Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. Variations in beta diversity between hippocampal and cerebellum samples were observed indicating an impact of brain region on the presence of microbial DNA. Importantly, differences in alpha and beta diversities between the two independent cohorts were found indicating a significant cohort- and processing-dependent effect on the microbiome. Finally, there were cohort-specific correlations between the gut microbiome and subject demographics indicate that postmortem interval may have a significant impact on brain microbiome determination.
Conclusions:
Regardless of the study bias, this study concludes that bacterial DNA can be isolated from the human brain suggesting that a brain microbiome may exist; however, more studies are required to understand the variation in AD.
INTRODUCTION
Gastrointestinal (GI) dysbiosis in the microbiome has broad-acting effects on the health of both the gut and the brain with important implications in neurodegenerative diseases such as Alzheimer’s disease (AD) [1, 2]. One of the mechanisms that drives dysbiosis-induced neurodegeneration is a compromised GI and blood-brain barriers (BBB) facilitating the infiltration of immune cells, neuroactive products, viruses and even bacterial cells from the GI lumen into the brain. This also supports the inflammatory hypothesis of AD where a dysbiotic microbiome creates an aberrant innate-immune crosstalk between the GI tract and the brain, promoting the neuroinflammatory milieu: a recognized risk factor of AD and other neurological disorders. One group has even postulated a “cooperative pathogenesis” where less virulent organisms, such as the dysbiotic microbiota, can enable infection of typically non-virulent organisms in the brain by facilitating their incursion into the brain [3]. These mechanisms challenge the previously assumed sterility of the brain. Several studies have demonstrated that there indeed exists bacterial and fungal species in the brain, and that these species could drive neurodegenerative pathogenesis. Indeed, some studies have detected, albeit in low quantities, bacteria in the brain of healthy individuals [4–6] insinuating the possibility of a brain microbiome.
It is highly possible that the opening of the BBB and consequent infiltration of pathogenic bacteria into the brain is a driving force for neurodegenerative pathologies [7]. 16S rRNA sequencing of frozen multiple sclerosis (MS) patients’ brain sections revealed that Proteobacteria, associated with increased inflammatory gene expression, may be the dominant phylum with restricted diversity in cerebral white matter compared to non-MS patients. Both clinical groups displayed 1,200–1,400 bacterial genomes/cm3 [4]. Several groups have also demonstrated the possibility of polymicrobial infections in AD patients. In both formalin fixed and frozen sections, postmortem AD tissue samples were identified to have greater incursion of bacterial species compared to controls including Firmicutes, Staphylococcus, and the family Propionibacteriaceae [5]. Another group identified immunolabelled prokaryotic structures in the postmortem brain specimens from AD patients [8], which were later confirmed using anti-peptidoglycan, anti-Clamidophyla, and anti-Borrelia antibodies [6]. This group went further with NGS to identify that the phylum Proteobacteria was the most prominent in both AD patients and controls, followed by Firmicutes, Actinobacteria, and Bacteroidetes whereas at the family level, Burkholeriaceae and Staphylococcaceae were found in greater abundance in the AD brains compared to control brains. Based on these results, this group also investigated the brain-region specificity of bacterial infiltration in amyotrophic lateral sclerosis (ALS) and AD patients. Using a nested polymerase chain reaction (PCR) approach to specifically amplify bacterial species instead of human, the bacterium Cutibacterium acnes (formerly known as Propionibacterium acnes) was found in abundance in all regions investigated in the ALS patients. Other species detected included Corynebacterium sp., Fusobacterium nucleatum, Lawsonella clevelandesis, and Streptococcus thermophilus. To get a comprehensive view of the bacterial species in a region-specific manner, NGS was performed in the motor cortex, medulla, and spinal cord of 11 ALS patients. The phyla Actinobacteria and Proteobacteria were found in all three regions of the 11 ALS patients, while the order Sphyngomonodales, the family Methilobacteriaceae, and the genus Cupriavidus seemed to be the most prevalent bacterial groups in ALS patients. The phyla Firmicutes and Bacteroidetes were also prevalent, whereas other phyla were less represented. Multivariate principal coordinates analysis (PCoA) showed no significant clustering among the three central nervous system (CNS) regions. Interestingly, region-dependent bacterial enumeration of ALS patients were significantly different compared to the enthorrinal region from AD patients as previously reported [6]. For instance, the bacterial genera Acrobacter, Thermomonas, Hemophylus, Propionibacterium, and Corynebacterium were more represented in motor cortex, medulla, and spinal cord regions in ALS patients compared to the AD patients. Many of these variations were also confirmed using immunohistochemical techniques confirming that there is the infiltration of GI bacteria into the brain, with possible enrichment in AD patients [9].
There are several caveats to investigating bacterial populations in human brain sections. One is the possibility of contamination of brain sections with the skin microbiome during the autopsy, unless proper sterilization techniques are observed. Storage, processing, and timing (postmortem interval; PMI) are also significant issues and difficult to control when utilizing postmortem human tissue specimens. As such, the thanatomicrobiome has recently been coined to describe the colonization of microbes in internal organs and orifices after death, a study normally used in forensics to identify time of death [10]. One study concluded that there are time-, organ-, and sex-dependent alterations in the thanatomicrobiome, including samples taken from the brain [11]. Finally, extraction, amplification, and sequencing protocols also induce bias into NGS results compromising comparative and meta-analyses within and between studies [12, 13].
To address these caveats and gather more information on the potential microbial load in the brain of AD patients, we conducted NGS sequencing of two independent study groups from samples collected from three independent sites in two separate brain regions: hippocampus and cerebellum. Samples from two independent cohorts (Study 1 and Study 2) were extracted with different methods yet all samples were processed with the same NGS protocol. We also conducted correlative analyses between subject demographics collected from the AD patients with the variations in brain microbiome samples to understand if specific bacteria are associated with AD pathophysiology. Despite there being few differences in the gut microbiome between control and AD cohorts, we did identify significant study biases that need to be addressed as studies move forward in investigating the possibility of a brain microbiome.
METHODS
Tissue specimens
Two tissue cohorts are included in this study. In the first study (Study 1, Supplementary Table 1), frozen postmortem hippocampal formation specimens from 10 neurological control (7 females and 3 males) and 10 AD cases (7 females and 3 males) were provided by The Mount Sinai/JJ Peters VA Medical Center NIH Brain and Tissue Repository. The second cohort (Study 2, Supplementary Tables 2 and 3) was received from the Brain Endowment Bank from the University of Miami Health System containing 22 AD specimens (16 females and 7 males) and 19 neurological controls (13 females and 6 males) from the hippocampus. Included in Study 2 were 12 control (8 females and 4 males) and 20 AD cerebellum samples (15 female and 5 males) that were obtained from The Mount Sinai/JJ Peters VA Medical Center NIH Brain and Tissue Repository. Sample details and demographic information can be found in Supplementary Tables 1–3. Information regarding the sterilization and autopsy technique were not provided.
DNA extraction
In study 1, samples were extracted with the Qiagen Stool Kit (catalogue # 19590) following company instructions. In study 2, specimens were placed into a MoBio PowerMag Soil DNA Isolation Bead Plate. DNA was extracted following MoBio’s instructions on a KingFisher robot.
PCR, sequencing, and sequence processing
DNA from extracted samples was amplified using Invitrogen’s AccuPrime High Fidelity kit (Cat# 12346094). The primers used for amplification contain adapters for MiSeq sequencing and single-end barcodes allowing pooling and direct sequencing of PCR products. As all samples were amplified using the same method, there should not be any significant hindrance in relation to sequencing due to extraction method. Each PCR reaction is prepared by combining 16μl of the master mix (13.85μl water, 2μl 10X reaction buffer, and 0.15μl Taq DNA polymerase), 2μL template DNA, 4μL forward (515F 5′ AATGATACGGCGACCACCGAGATCTACACTATGGTAATTGTGTGCCAGCMGCCGCGGTAA- 3′ and reverse (806R 5′-CAAGCAGAAGACGGCATACGAGATTCCCTTGTCTCCAGTCAGTCAGCCGGACTACHVGGGTWTCTAAT-3′) primers. The DNA samples are amplified using the following thermocycler program: 95°C for 2 min followed by 30 amplification cycles of 20 s at 95°C, 45 s at 50°C and, 90 s at 72°C followed by final extension at 72°C for 10 min. The PCR product was quantified using Invitrogen’s Quant-iT Picogreen dsDNA assay kit (Cat # P7589). Amplicons are pooled and purified using the QIAquick PCR purification kit (Cat# 28104).
16S Sequencing
The 16S rRNA gene sequencing was conducted by Diversigen (Houston, TX) [14]. The 16S rRNA V4 gene region was amplified from the extracted community DNA by PCR and sequenced in the MiSeq platform (Illumina) using the 2×250 bp paired-end protocol. This generates paired-end reads that overlap almost completely. The primers used for amplification contain adapters for MiSeq sequencing and single-end barcodes allowing pooling and direct sequencing of PCR products [15]. Sequences that passed chimera slaying post sequencing were merged and blast against the 16S specific curated Silva database (v132). Of the kept sequences, only 35.56% were successfully mapped due to the exclusion of host 16S mitochondrial contamination. Samples were subsequently rarefied to 356 to effectively eliminate noise but retain as many samples as possible. With this cut-off, 18 samples from Study 2 were excluded from the analysis including 21, 23, 24, 25, 26, 29, 33, 34, 37, 39, 42, 44, 46, 49, 64, 70, 81, and 84.
Statistical analysis
Alpha diversity was calculated for the Shannon diversity index and observed operational taxonomic unit (OTU) abundances. As microbiome data are usually non-parametric in nature, a Mann-Whitney U test was used for comparisons between two groups. Beta diversity was estimated both qualitatively with unweighted UniFrac and quantitatively with weighted UniFrac analysis distance matrices. Variation in community structure was assessed with permutational multivariate analyses of variance (PERMANOVA) between groups with visualization of the data using PCoA. Between group statistics for control and AD subjects of the phyla and genera were conducted with the Mann-Whitney U test with false discovery rate (FDR) corrections using the Benjamini-Hochberg method. To preform correlation data with patient metadata, we used the Spearman’s Rho test and incorporated FDR for multiple testing corrections. All metrics and statistical analyses were conducted in R (Version 3.5.1 with the exception of the Spearman’s Rho which was conducted in GraphPad Prism (version 8.0)).
RESULTS
Microbiome abundances in AD versus control subjects in Study 1
In samples derived from The Mount Sinai/JJ Peters VA Medical Center NIH Brain and Tissue Repository (Study 1) consisting only of hippocampal specimens, there was no difference in the alpha diversity shown with observed OTUs or Shannon index estimation between the AD and control subjects (Fig. 1a). In addition, beta diversity estimated with both unweighted (not shown) and weighted (Fig. 1b) UniFrac representation showed no clustering between the AD and control subjects. The most abundant phyla in both the AD and control subjects were the Proteobacteria followed by Firmicutes, Actinobacteria, and Bacteriodetes with lower abundances of Cyanobacteria, Deinococcus-Thermus, Acidobacteria, Verrucomicrobia, and Gemmatimonadetes. Regardless, there were no differences between AD and control subjects for any of the observed phyla (Fig. 1c, upper) and the variable distribution between individuals in Study 1 can be observed in Fig. 1d (left). The most abundant genera included Acinetobacter, Pseudomonas, Escherichia-Shigella, Staphylococcus, Chryseobacterium, Stenotrophomonas, Massilia, Bacillus, Corynebacteriaceae, and Lawsonella, with again, no differences between the AD and control subjects (Fig. 1c, lower). Again, the significant variation between individuals in Study 1 for the enumeration of genera can be observed in Fig. 1d (right). Although not shown, the potential gender differences within the Study 1 cohort were also examined, but no differences or trends were found in alpha diversity, beta diversity, phyla or genera quantification.

Microbiome Distribution of in Alzheimer’s Disease and Control Subjects in Study 1. a) Alpha diversity was estimated with total OTU counts (upper) and Shannon Index (lower), respectively. b) Beta diversity estimated with a weighted UniFrac analysis is represented as a PCoA plot to observe clustering between control and AD samples. c) Microbiome enumeration in control (blue) versus AD (red) samples are shown for the phyla (upper) and genera (lower) divisions. d) Individual representations of the phyla (left) and genera (right) are shown with the most abundant groups shown.
Microbiome abundances in AD versus control subjects in hippocampal and cerebellum samples in Study 2
Study 2 contains a more substantial data set and compared samples between the hippocampus, typically associated with AD pathology, and the cerebellum, which is not typically associated with AD. Within the hippocampal sections, there were no differences in the alpha diversity indices between control and AD subjects (Fig. 2a). There is a trend toward clustering of AD and control subjects in the weighted UniFrac PCoA analysis indicating that the quantities of the different microbial OTUs might be different (Fig. 2b). The same trend was not observed in the unweighted UniFrac PCoA analysis (not shown). Similar to Study 1, the most four most abundant phyla were Proteobacteria followed by Firmicutes, Actinobacteria, and Bacteriodetes, with no differences between the AD and control groups (Fig. 2c, upper; Supplementary Figure 1a). Interestingly, different genera abundances were observed in Study 2 hippocampi compared to Study 1. The most abundant genera in Study 2 include Bacillus, Staphylococcus, Pseudomonas, Acinetobacter, Enhydrobacter, Massilia, Escherichia-Shigella, Sphinogomonas, Roseomonas, and Stenotrophomonas. But again, there were no differences between the AD and control samples (Fig. 2c, lower; Supplementary Figure 1a). Comparing in parallel, the cerebellum samples in AD and control subjects showed no differences in alpha diversity (Fig. 2d) or weighted UniFrac PCoA analysis (Fig. 2e). Like the hippocampus, the cerebellum samples had the same distribution of phyla abundances Proteobacteria followed by Firmicutes, Actinobacteria, and Bacteriodetes (Fig. 2e, upper). There was again a unique distribution of genera with a relative abundance order of Pseudomonas, Acinetobacter, Escherichia-Shigella, Burkholderiaceae (OTU), Stenotrophomonas, Pedobacter, Staphylococcus, Bacillus, Corynebacteriaceae (OTU), and Burkholderiaceae (OTU) (Fig. 2f, lower) respectively. No differences in either phyla or genera between AD or control subjects were detected.
When directly comparing the control hippocampal to control cerebellar samples, no differences were observed in the alpha diversity (Fig. 3a), beta diversity (Fig. 3b), phyla, or genera (Fig. 3c). However, when comparing the hippocampal to cerebellar samples in AD subjects, albeit no differences in alpha diversity (Fig. 3d), there was a significant difference in the clustering of the weighted UniFrac analysis of the beta diversity (Fig. 3e; p < 0.05) indicating that there are potentially different quantities of the bacterial populations in the hippocampal versus cerebellar samples. Breaking these down into the quantification of the phyla and genera, no differences between specific phyla or genera were observed post FDR corrections.

Microbiome Distribution of in Alzheimer’s Disease and Control Subjects in Study 2. In the study 2 subjects, microbiome presence in AD and control subjects in the hippocampus identify (a) alpha diversity, (b) beta diversity, and (c) phyla (upper) and genera (lower) comparative enumeration. Microbiome presence also in the cerebellum of AD and control subjects are shown identifying (d) alpha diversity, (e) beta diversity, and (f) phyla (upper) and genera (lower) comparative enumeration.
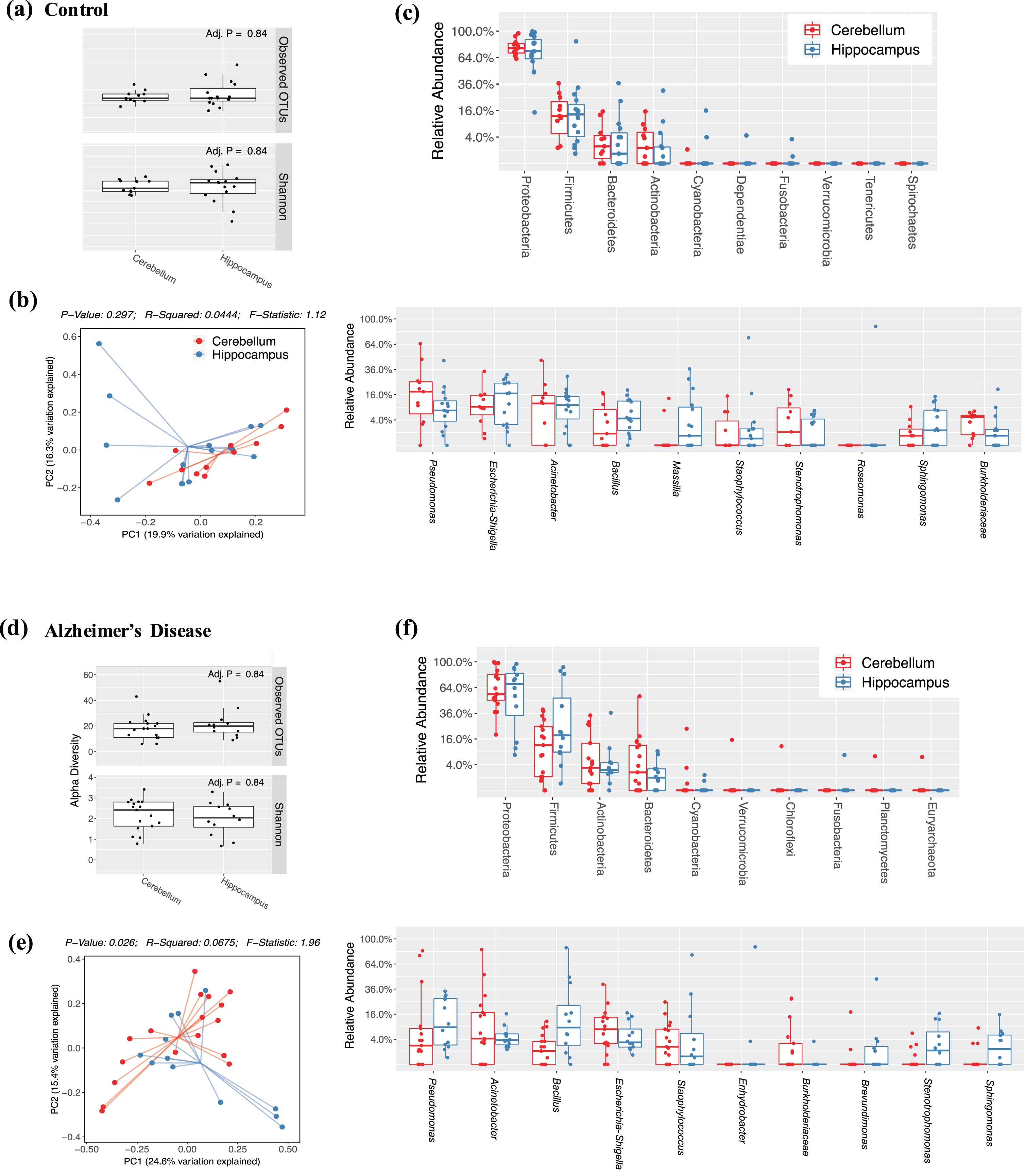
Hippocampal versus Cerebellar Microbiome Distributions in Study 2. Microbiome enumeration is compared between hippocampus and cerebellum samples in Study 2 among controls (a–c) and AD subjects (d-e). Alpha diversity in controls (a) and AD (d) are estimated with OTU counts and Shannon indices while beta diversity in controls (b) and AD (e) estimated with a weighted UniFrac analysis in PCoA plots are shown. Finally, phyla (upper) and genera (lower) quantifications are shown for control (c) and AD (f) subjects.
Study bias in brain microbiome samples
Considering that there, unexpectedly, few differences in the AD subjects compared to the control subjects, we wanted to determine if there was an implicit study biases as each study was sourced from separate locations and importantly, bacterial DNA was extracted in different ways. In control subjects, there were significant variations in the alpha (Fig. 4a; p < 0.01) and beta diversity (Fig. 4b; p < 0.01) between studies with Study 2 having reduced bacterial richness compared to Study 1. Looking at the individual phyla, there was a significant reduction of the phyla Actinobacteria and Deinococcus-Thermus compared to Study 1 controls (Fig. 4c, upper) with no variations in the genera between studies. Similarly, AD subjects had significant variations in alpha diversity (Fig. 4d; p < 0.01) and beta diversities (Fig. 4e; p < 0.01), again with Study 2 demonstrating lower bacterial richness compared to Study 1. Comparing the phyla and genera abundances in AD subjects, no significant variations between phyla or genera were observed (Fig. 4f), likely due to the increased variability in the Study 2 sample cohort and a very conservative FDR. When assessing for biomarker significance, there is an increased risk of accepting false positives as a greater number of tests are conducted based on random chance alone. To combat against accepting a high number of false positives, FDR corrections, as described in the statistical methods, were applied to lower the rate of accepting positives. The FDR correction method is considered a moderate method for false positive corrections.
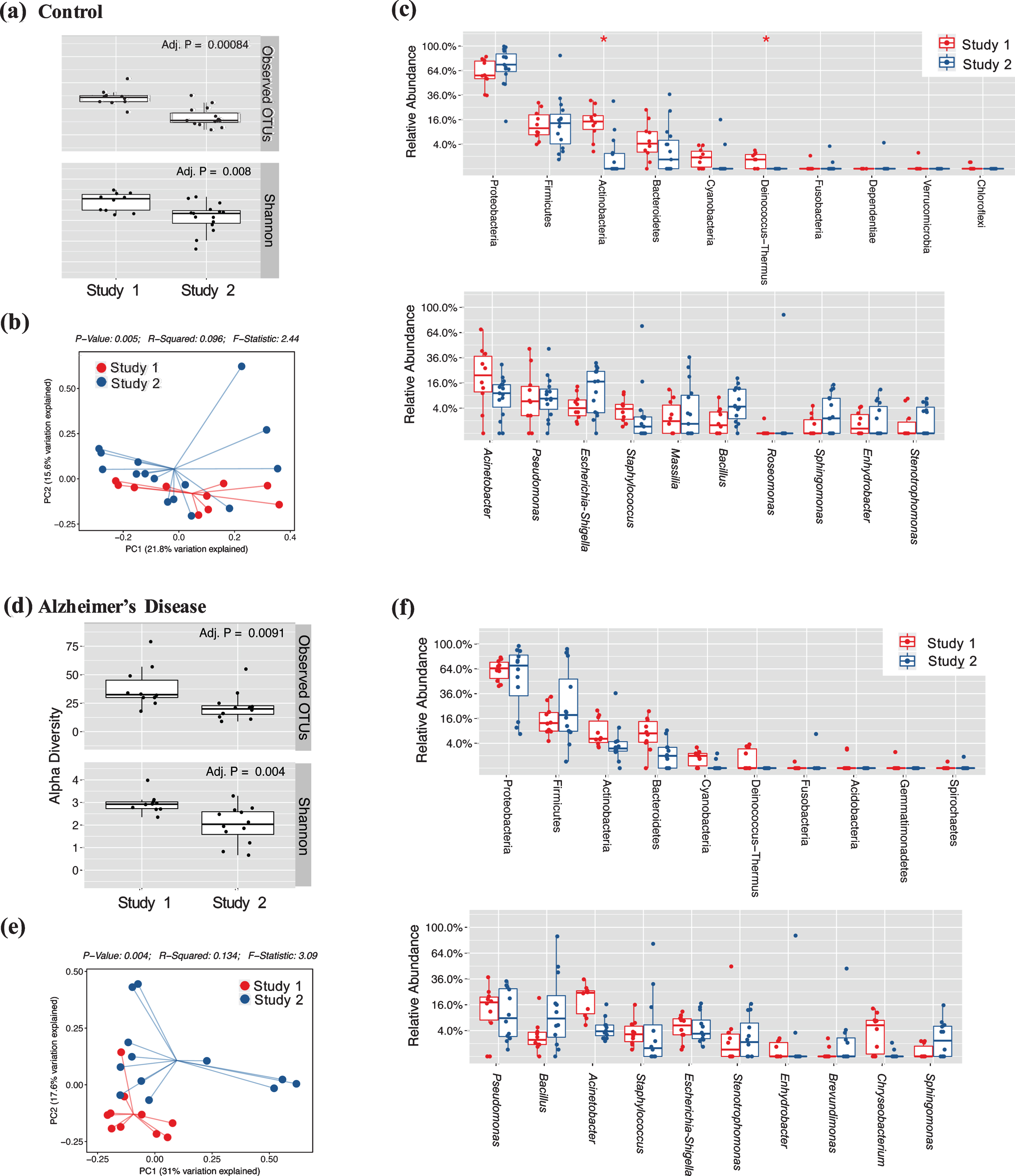
Study Bias between Microbiome Cohorts of Independent Studies. Measures of microbiome differences are directly compared between Study 1 and 2. Alpha diversity in controls (a) and AD (d) are estimated with OTU counts and Shannon indices while beta diversity in controls (b) and AD (e) estimated with a weighted UniFrac analysis in PCoA plots are shown. Finally, phyla (upper) and genera (lower) quantifications are shown for control (c) and AD (f) subjects. Significant differences are represented (*) is p < 0.05.
Correlation of Variations in Gut Microbiota Populations with Alzheimer’s disease Metadata
AD is defined along a spectrum of pathologies including the accumulation of amyloid-β plaques, tau tangles, and levels of dementia with the confound of age and PMI. In particular, the PMI has been shown to be a major issue for the integrity and quality of the tissue sections and could influence not only the macromolecule (protein, DNA, RNA, etc.) reads, but also the brain microbiome [16]: factors which can be exasperated by the diseased state in AD. Nevertheless, several interesting correlations were observed in both studies (Table 1). Interestingly, the genera Corynebacteriaceae (OTU) was found to be correlated in Study 1 positively to PMI and negatively to age, amyloid-β plaque, and Clinical Dementia Rating (CDR); however, the significance of these differences were lost post FDR correction. In Study 2, Corynebacteriaceae (OTU) negatively correlated to PMI, and positively to amyloid-β, Braak, and CDR in the cerebellum, but not the hippocampus, while following FDR correction, only the correlation between Corynebacteriaceae (OTU) and PMI remained significant. Considering correlations in difference phyla, Proteobacteria were negatively associated with age in Study 1 (insignificant post FDR correction), while total OTUs and Actinobacteria were negatively correlated to PMI in the hippocampus of Study 2 (significant post FDR correction) and positively correlated to Braak (insignificant post FDR correction). In the cerebellum, the adjusted p value but not post FDR correction, Enterococcus was negatively while Staphyloccocus was positively associated to Braak. Notably, the positive correlations to Braak in the hippocampi of Study 1 and 2 were different, with the genus Kocuria and fungal mitochondria having a positive correlation in Study 1 and Actinobacteria, Lawsonella and Methylobacterium in Study 2. The variations in populations correlated either positively or negatively with the aforementioned metrics of AD support the study-specific bias in 16S sequencing of brain microbiome samples.
Correlation of Brain Microbiome Quantification to Patients Metadata for Alzheimer’s Disease Markers
Significant correlations between all features (phyla, genera, alpha and beta diversities) are shown for instances where p < 0.05. Positive/negative signs in parentheses indicate directionality of the r-value from the Spearman rho correlation. PMI, postmortem interval; CDR, clinical dementia rating.
DISCUSSION
Dysbiotic gut microbiota has become over the last decade a well-known comorbid condition of AD [17, 18]. In clinical studies with AD patients, reported microbiome profiles contain many variabilities; however, reports are generally consistent with showing that AD patients have a reduction in microbiome diversity, abundance, and butyrate-producing species. Many postulate that this dysbiotic microbiome is a major driving force to the onset and progression of AD [1, 20] through mechanisms involving gut-brain-axis communication. Notably, the inflammatory destabilization created by a dysbiotic gut microbiome is considered a major factor in the pathogenesis of AD. One of the consequences of the inflammatory response to dysbiotic bacteria in AD is an opening of the epithelial barriers in the gut and the brain allowing increased infiltration of bacterial products and cells from the GI lumen into the brain [21, 22]. Based on this notion, several groups have begun a systematic exploration of the presence of a gut microbiota in the brain, with the potential increased incursion or altered diversity in AD patients.
In parallel with other studies [5, 9], an enrichment of Proteobacteria, Firmicutes, Actinobacteria, and Bacteriodetes phyla were observed in both the control and AD subjects in both Study 1 and 2. However, unlike these studies, there were no significant differences in the composition of phyla between controls and AD in either the hippocampus or cerebellum, or in the higher resolution assessment of the genera. This is in contrast to a previous study which showed a clear increase in bacterial load in the brain of AD subjects compared to controls in both frozen and formalin-fixed samples, but in the prefrontal cortex [5]. One reason could be the sample extraction protocol, which was different than the current study. In the study of Emery et. al., a phenol/chloroform method was used to extract DNA from the frozen tissue segments while the Qiagen Formalin-fixed, Paraffin Embedded (FFPE) kit was used for the formalin-fixed samples.
Although there were no differences in bacterial population or diversity (alpha and beta diversities) observed between the AD and control subjects in Studies 1 and 2, the results remain significant since the taxa reported are not typically exclusively associated with the skin, thus can be assumed to be derived from the GI tract [23]. In general, the human skin microbiome is composed primarily of Staphylococcus epidermidis, Corynebacterium, and Propionibacterium acnes, which are distinct from the significant phyla and genera observed in the current study. In addition, there were significant variations in the genera between individuals within a single group (AD or control), allowing the assumption to be made that these reads are not artifacts of contamination but rather representative of the actual variability in the brain sample cohorts. In addition, groups have shown unequivocally that several fungal and bacterial proteins can be found in the corpora amylacea of AD patients [25]. This is an interesting observation as the corpora amylacea, spherical bodies composed of polyglucans and proteins, accumulate gradually in the CNS of aging persons, but to a greater extent in patients with neurodegenerative disorders and AD. This indicates that fungi and bacteria may be present in the body for a significant amount of time, and hence not merely an artifact of postmortem tissues. This group additionally showed using immunohistochemistry with anti-peptidoglycan and Clostridium antibodies and imaging with confocal microscopy that fungal or prokaryotic structures were present in the brains of AD patients confirming that microbial products are present in the brain of AD patients [8].
In addition to assessing the composition of the brain microbiome, the current study correlated variations in the brain microbiome and its complexity to the patient metadata for measures of AD. One potential confound based on the patient metadata is the variation in PMI. Previously, the gut microbiota was predicted to be an indicator of PMI, as determined through a random forest correlation in cases when the time of death was uncertain [26, 27]. Interestingly, between Studies 1 and 2, there was a different trend between positive and negative correlation of bacterial populations with PMI. Study 2 consistently showed a negative correlation between bacterial groups and total OTU counts in the hippocampus and cerebellum while Study 1 showed a mix of positive and negative correlations. This indicates that there is not a consistent bias for PMI with microbiome populations in the brain; however, it must be considered a potential factor based on the results from other studies. In addition, the average and range of PMI is very different between the two studies. In Study 1, the PMI ranges from 125 to 1,437 hours with a median of 265 hours, whereas in Study 2, the range is much smaller from 1.5 to 29.6 hours with a median of 14.2 hours. It has been suggested that studying multiple brain regions in the same subject can determine if PMI is actually a factor in the sample integrity and in this case, bacterial richness. As such, in Study 2, the alpha diversity, observed OTUs, levels in hippocampus was negatively associated with PMI indicating that PMI may be a factor in the richness and/or diversity of the brain microbiome. Interestingly, we found a limited number of correlations between different genera and PMI. Corynebacteriaceae (OTU) had a negative correlation with PMI in the cerebellum whereas Chryseobacterium and Peptoniphilus were negatively correlated with PMI in the hippocampus.
An obvious population of interest in the correlative study is the presence of the genera Corynebacteriaceae (OTU) and its correlation with multiple AD metadata in both Studies 1 and 2. As noted before, bacterial species in the family Corynebacteriaceae are also found on the skin as well as in the GI tract. In general, bacteria in the family Corynebacteriaceae are generally pathogenic causing a range of serious infections in humans and animals including diphtheria, endocarditis, and lymphadenitis [28], although some non-pathogenic strains have also been identified [29]. In Study 1, Corynebacteriaceae in the hippocampus correlated negatively to age, amyloid-β, Braak, and CDR while retaining a positive correlation to PMI; however, the significance of these results was lost post FDR correction. In contrast, in Study 2, there were no correlations of Corynebacteriaceae to AD metrics in the hippocampus while in the cerebellum, Corynebacteriaceae was positively associated to amyloid-β, Braak, and CDR and negatively to PMI again outlining the Study-specific bias in studying the brain microbiome.
The most interesting correlation observed was the positive correlation between the phyla Actinobacteria and Braak in the hippocampus samples of Study 2 and the positive association between Staphylococcus and Braak in the cerebellum. Despite there being no significant differences in the control versus AD populations of these phyla, the trending changes could be a significant driver of AD pathogenic markers in humans.
Another important fact that should be acknowledged regarding brain infection and the pathogenicity of AD is that there is a commensal fungal population specifically in AD CNS tissue, which is not present in controls [30]. One group determined fungal material in both intra- and extracellular tissue in multiple brain regions including the cerebellar hemisphere and the hippocampus. This group also identified evidence of several fungal species in these regions and in the vascular tissue [31]. Similarly, immunohistochemical assessment of fungal components were observed in the cytoplasm of approximately 10% of AD patient’s frontal cortex tissue, but not control subjects confirming the intracellular localization [32]. These results have been confirmed by other groups where the most abundance fungal genera were identified as Alternaria, Botrytis, Candida, and Malassezia, which were distinctly different from healthy control subjects [6]. The colonization of fungi and bacteria in the brain could contribute to the abnormalities in the innate immune system in both the periphery and the brain: a widely accepted part of AD’s pathophysiology [30]. In contrast, the disrupted innate immune response consequent of possibly other underlying factors may facilitate the incursion of fungi particles or bacterial particles into the brain, exasperating the AD phenotype. Regardless, infection of the aging adult brain may be a significant source of the AD pathophysiology opening novel avenues for therapeutic approaches.
Footnotes
ACKNOWLEDGMENTS
We thank the NIH NeuroBioBank for obtaining postmortem brain tissue in this study. GMP coordinated sample procurement and study design. SW interpreted NGS analysis and wrote the manuscript. DD conducted the NGS analysis and produced the figures. DNA extraction of Study 1 was conducted by Mr. Braithwaite and Dr. Frolinger while DNA extraction of Study 2 samples was outsourced. 16S sequencing, alignment, and analysis was conducted by Diversigen Inc. (Houston, TX). Financial support of this project was from discretionary funding to GMP.