
Abstract
Background:
Environmental copper has been implicated in the pathogenesis of Alzheimer’s disease based on evidence that: 1) brain copper levels increase with age, 2) copper promotes misfolding and toxicity of amyloid-β in vitro, 3) copper-modulating interventions reduce amyloid pathology in animal models. However, the effect of copper upon non-amyloid Alzheimer’s pathology is relatively under-explored.
Objective:
To determine if modulation of brain copper level affects brain tau pathology and/or associated cognitive impairment.
Methods:
We tested the hypothesis that brain copper modulates tau pathology by manipulating brain levels of copper in the PS19 transgenic mouse model of tau pathology. We treated PS19 and wild-type mice with oral zinc acetate, an established therapy for long term control of excess brain copper, and examined treatment effects upon brain copper, brain tau, NFT-like pathology, and spatial memory. We treated a second cohort of mice with exogenous dietary copper in order to evaluate whether excess environmental copper promotes brain tau pathology.
Results:
Copper-lowering with oral zinc attenuated spatial memory impairment in female but not male PS19 mice, without a significant effect upon tau pathology. Copper loading increased brain copper, but did not have an effect on brain tau pathology or spatial memory function.
Conclusion:
These findings suggest that a strategy to lower brain copper may be viable for symptomatic benefit in the setting of tau neuropathology, but unlikely to have robust effects on the underlying pathology. These findings are consistent with dietary or other exogenous copper being unlikely to promote tau pathology.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia and is expected to increase in future years as the population ages, so disease-modifying therapies are desperately needed. Two proteins contribute to pathological features of AD, amyloid-β (Aβ) which forms extracellular plaques and hyperphosphorylated tau which forms intracellular neurofibrillary tangles (NFT) [1]. While several risk factors for AD are not modifiable (e.g., age, genetics, and family history), metal dishomeostasis in the brain has been implicated as a potentially modifiable cause of AD [2–4].
Several lines of evidence implicate copper in the pathophysiology of AD. For example, copper and other metals are co-localized with Aβ plaques [4] in postmortem human brains. In another example, copper given in drinking water at trace levels (0.12 ppm) was sufficient to induce Aβ accumulation in a rabbit model of AD [5]. In a mouse model with Aβ plaques and hyperphosphorylated tau, memory deficits and pathological markers were worsened with the administration of copper [6–9]. We have also investigated copper lowering strategies in the Tg2576 mouse model of AD, finding reductions in insoluble Aβ and improvement in spatial memory [10, 11]. Clinical trials of copper-modulating therapies targeting Aβ in AD have shown promise [12, 13], but disappointing results with anti-amyloid trials has diminished enthusiasm for any agents which target Aβ, and increasing attention is now being directed to tau brain pathology as a therapeutic target.
While the effects of copper modulation are well-established in murine models of amyloid deposition, the effects in models of pathologic tau deposition are less clear. Copper administration has been shown to increase the accumulation of hyperphosphorylated tau in in vitro experiments [14, 15] but in vivo experiments are not as extensively reported. We found that a copper-lowering strategy in a transgenic mouse model of wild-type human tau reduced tau phosphorylation, but did not improve behavioral deficits [16]. This experiment was also limited by the fact that the transgenic mouse strain used in those experiments did not develop tau inclusions [16].
In this study, we consequently use the PS19 mouse model of robust tau accumulation [17, 18] to investigate whether oral zinc treatment can reduce copper accumulation and tau pathology, and attenuate the cognitive consequences of tau accumulation. PS19 mice express a transgene encoding human tau with the P301S mutation which is associated with frontotemporal dementia. These mice develop robust brain tau pathology and cognitive impairment. Lifespan is shortened by spinal cord tau pathology and dysfunction. They were selected for this translational study because they have been employed in translational studies by other groups [17–19], some of which have led to clinical trials in tauopathies [20]. Zinc acetate was selected as the intervention because it is currently available for clinical use for inherited disorders of copper overload (i.e., Wilson’s disease, in which zinc effectively lowers systemic copper by inducing metalloproteins which bind copper in the gastrointestinal tract [21]), so could be translated to clinical trials if indicated by the animal studies. We also investigated whether a trace dose (0.12 ppm as in the rabbit model cited above [5]) or a high dose of copper (the highest dose that mice would drink) was able to increase levels of hyperphosphorylated tau in the PS19 mouse model.
MATERIALS AND METHODS
Animals and diet
B6;C3-Tg(Prnp-MAPT*P301S)PS19Vle/J (PS19) mice were obtained from The Jackson Laboratory, where they were bred on a B6C3F1/J background to generate a colony of heterozygote transgene carriers and wild-type littermates. The mouse colony was maintained at the Veteran’s Administration Portland Health Care System (VAPORHCS). Following weaning, litters were genotyped and group housed (3–4 per cage) until old enough for experiments. Animals were housed in a climate-controlled facility with a 12 h light/12 h dark cycle. During experiments, animals were fed AIN-93M rodent diet (Dyets, Inc, Tehtlehem, PA). Animals were provided with water and diet ad libitum. All procedures were conducted in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals and were approved by the institutional Animal Care and Use Committee of the VAPORHCS.
Treatments
For the first experiment evaluating the copper-lowering effects of zinc acetate, male and female PS19 and wild-type mice were randomized to zinc acetate treatment (6.7 g/L or 2000 ppm zinc acetate in ad libitum deionized drinking water) or vehicle (deionized water to avoid trace metal levels) starting at the age of 3 months and continuing for 6 months (Fig. 1A). Mice were assigned to each group as follows: 50 wild-type mice (female vehicle n = 12, female zinc n = 12, male vehicle n = 12, male zinc n = 14) and 46 transgenic (female vehicle n = 12, female zinc n = 12, male vehicle n = 12, male zinc n = 10).
Time course of zinc and copper treatment experiments.
In the second experiment, evaluating the effects of copper loading, female animals were treated with vehicle, low dose copper (47.15 mg CuSO4/Liter or 0.12 ppm), or high dose copper (982.28 mg CuSO4/Liter 250 ppm) in ad libitum deionized water (Fig. 1B) starting at age 2 months and continuing for 6.5 months (Fig. 1B). Mice were assigned as follows: 36 wild-type mice (vehicle n = 12; low dose copper n = 11; high dose copper n = 13) and 35 transgenic mice (vehicle n = 11; low dose copper n = 12; high dose copper n = 12).
Behavioral analysis
To assess hippocampal-dependent spatial learning and memory, mice were trained in a standard Morris Water Maze task using the same apparatus and protocol used in our previous studies [11]. Briefly, a cylindrical tank (109 cm in diameter) constructed of seamless white high-density polyethylene was positioned in a testing room surrounded by various distinct extra maze cues. The tank was filled to a depth of 33 cm with water rendered opaque by the addition of non-toxic Tempera paint. A 13 cm diameter escape platform was constructed of clear plexiglass. All behavior was acquired by ANY-maze video tracking software. To verify that poor performance on the hidden platform task results from cognitive impairments rather than a deficit of vision, motivation, or locomotor function, mice first underwent visible platform training. Mice were trained to swim to a plexiglass platform positioned 1 cm above the water surface. The platform’s position changed every three trials. Mice were trained on the visible platform for 9 trials, three on the morning of day 1, three on the afternoon of day 1, and three more in the morning of day 2. 72 h after visible platform training, mice were spatially trained on the hidden platform water maze task. Extra-maze cues were positioned on floor to ceiling curtains. Mice were trained for 30 trials (6/day) to learn the location of an escape platform submerged 1 cm below the water surface in the center of the SE quadrant of the pool. The platform position remained fixed throughout training. During a given trial, the mouse was introduced into the pool, at one of three pseudo-randomly-chosen start points (N, S, W) and allowed 60 s to find the platform. If the mouse did not find the platform after 60 s, it was placed on the platform. The mouse remained on the platform for 15 s before being returned to a holding cage for an inter-trial interval minimum of 7 min. Escape latency (in seconds) was used as a measure of spatial learning. For spatial memory testing, a 60 s free swim probe test, in which the escape platform was not present in the pool, was conducted 2 h after every 6th trial at the end of the day, 24 h and 72 h after the final spatial training trial. Search behavior during the probe test was analyzed to determine 1) the percent dwell in each quadrant, 2) the number of crossings into a circular zone in the quadrant encompassing the previous location of the platform, and 3) the average distance of the mouse to the platform (in cm) during the probe test. A ratio of the number of crossings into the training zone quadrant to the total number of crossings into all four quadrants was generated. Search ratios greater than 25% indicate a bias towards the training quadrant, while a ratio less than 25% reflects a lack of preferential searching.
Euthanasia and tissue collection
Following behavioral analysis, animals were euthanized by CO2 inhalation and cervical dislocation. Blood and brain samples were collected and frozen. The anterior 4 mm of bilateral frontal cortex were dissected and frozen for metal analysis. The remaining right hemisphere was immersion fixed in 4% paraformaldehyde in phosphate buffered saline for histochemical analysis. The remaining left hemisphere was dissected into hippocampus, cortex, and deep gray were dissected, and each was frozen at –80°C until experimentation.
Hippocampus and cortex homogenization
Hippocampus and cortex tissue were separately homogenized in TBS containing Halt™ protease and phosphatase inhibitor cocktail (Thermo Scientific, Rockford, IL), then centrifuged at 100,000×g for 30 min at 4°C. The resulting pellet was collected and homogenized in AT+ buffer (Ultrapure Tris (Invitrogen, Camarillo, CA), ethylene glycol-bis(β-amino-ethyl ether)-N,N,N’,N’-tetraacetic acid(Sigma), Dithothreitol (Sigma), 10% sucrose (Sigma), and 1% Triton X-100 (Roche, Indianapolis, IL)) containing Halt™ protease and phosphatase inhibitor cocktail, then centrifuged at 100,000×g for 30 min at 4°C. The supernatant from this step was labeled “soluble fraction”. The resulting pellet was collected, and homogenized in AT+ buffer containing 1% N-Lauroylsarcosine sodium salt (Sarcosyl) (Sigma) then centrifuged at 1000,000×g for 30 min at 4°C. The pellet was collected and washed in TBS containing protease and phosphatase inhibitors and re-centrifuged. The supernatant was discarded and the final pellet was sonicated for 30 s at 4°C in 70% formic acid (Sigma), incubated 1 h at room temperature, and centrifuged at 100,000×g 30 min. The resulting supernatant was collected and labeled “insoluble fraction”. Total human tau (HuTau) and phosphorylated T231 (pT231) were measured in the soluble and insoluble fractions using commercial ELISA (Invitrogen) according to the manufacturer’s instructions.
Immunohistochemistry
Forty-micron frozen coronal sections were cut on a freezing microtome. Sections were incubated with agitation in blocking buffer (100 mM TBS, pH 8.0, 2 mg/ml bovine serum albumin, 2% horse serum, 0.5% Triton X-100) for 2 h, then incubated overnight with primary antibody diluted 1:1000 in blocking buffer (biotinylated mouse polyclonal antibodies directed against phosphorylated tau Ser202/Thr205 (AT8) (Thermo Scientific). Sections were then incubated for 2 h with an avidin-linked peroxidase complex (ABC, Vector Labs), then developed with diaminobenzidine (DAB, Sigma) in PBS. Sections were washed, dehydrated, mounted in Permount (Fisher Scientific, Pittsburg, PA) and cover slipped. Protein expression was quantified in at least three coronal sections from each mouse, representing anterior, middle and posterior hippocampus and cortex. Hippocampal and cortical areas were traced using a computerized stage and stereo investigator software (Image J, Wayne Rasband, NIH, USA). Phospho tau levels were expressed as percentage of hippocampus or cortex occupied by these proteins. Mean values for each parameter were calculated from at least three sections per animal. Total area for hippocampus over three sections were quantified for each animal.
Tissue metal assays
Metal analysis of serum and copper was identical to analysis previously published [10]. Briefly serum and brain zinc and copper were measured using inductively coupled plasma mass spectroscopy (ICPMS). Tissue was digested in 1 mL of concentrated nitric acid (∼69.5%), diluted in distilled water, and then diluted again with 2% nitric acid to be in the linear absorption range of the calibration curve. Twenty μl of each sample was injected up to 3 times depending on the standard deviation of measurements. A comparison of the samples absorption with a standard curve for each element derived concentrations for zinc and copper.
Enzyme-linked immunosorbent assay (ELISA)
Commercial ELISAs for human total tau, and phospho-tau 231 were run on the “soluble fractions” and “insoluble fractions” of hippocampus according to manufacturer’s instructions (Thermo Scientific). Briefly standards were created from lyophilized standards included in the kits and serial diluted with standard diluent buffer. Hippocampus samples were diluted with same buffer to fall within the standard range of the assay and incubated for 3 h with equal volumes of detection antibody at room temperature with shaking. At this time, samples were diluted and run in a BCA assay. Plates were washed and incubated for 30 min with secondary antibody. Placed were washed again and incubated for 30 min in the dark after stabilized chromagen was added to each well. After 30 min an equal volume of stop solution was added and the plate was read at 450 nm. Values were calculated as ng of target protein divided by mg of total protein from the Bicinchoninic acid assay (BCA). Values were also calculated as ng phosphor-tau 231 divided by ng of total tau.
Bicinchoninic acid assay
Albumin standard (Thermo Scientific) was serial diluted in ELISA standard diluent buffer. Samples were diluted in same buffer to be within range of the assay. Samples and standards were incubated for 30 min at 37°C in Pierce BCA Protein Assay Reagent (Thermo Scientific). Results were read at 520 nm and used to calculate values in ELISA assays.
Statistics
Data were analyzed using GraphPad Prism 5 (GraphPAD Software Inc) using one- or two-way analysis of variance followed by the Bonferroni post-hoc test (for comparisons between two samples at a time).
RESULTS
Copper-lowering intervention
Zinc treatment and brain zinc and copper levels
Zinc acetate administration increased serum zinc in both wild-type and PS19 female mice (Fig. 2A, p < 0.01 and p < 0.05, respectively) and in male wild-type mice (Fig. 2B, p < 0.01). Zinc acetate administration also lowered serum copper in wild-type female mice (Fig. 2E, p = 0.01). Brain zinc was not significantly increased in any of the zinc-treated mice (Fig. 2C, D). Brain copper was marginally lower in zinc-treated female PS19 mice (Fig. 2G; p = 0.048) but the difference was not significant after Bonferroni correction.

Serum and brain copper zinc and copper levels in mice treated with zinc acetate or vehicle. Figures show means and SEM. *p < 0.05 in two-way ANOVA after Bonferroni correction; **p < 0.01 in two-way ANOVA after Bonferroni correction. As expected, serum zinc is increased in all animals treated with zinc acetate (A, B). However, brain zinc is not affected by zinc treatment (C, D). Serum copper levels are significantly lower in zinc-treated female wild type mice (E) and trends lower in other zinc-treated mice (E, F). Brain copper is marginally lower in zinc treated compared to vehicle treated female PS19 (p = 0.048) but the difference is not significant after Bonferroni correction.
Zinc treatment promoted survival and mobility in female PS19 mice
PS19 mice exhibit early mortality due to paralysis in the tail and hindquarters and eventually the forelimbs, with feeding difficulty ultimately leading to death, sometimes by reaching criteria for euthanasia. Ability to swim is also impaired in aging PS19 mice, as tail paralysis occurs before hindquarter paralysis, and tails are the primary source of mobility for swimming in mice. Mice that had difficulty in swimming or staying afloat were excluded from the Morris Water Maze. In both female and male mice, fewer PS19 mice survived and were able to swim in the Morris Water Maze (Fig. 3A–D). With zinc treatment, more female mice survived to participate in the Morris Water Maze (Fig. 3A, D, p = 0.0175). There was no effect of zinc treatment in male mice.

Survival of female PS19 mice to Morris Water Maze testing is reduced by zinc treatment. Fewer PS19 mice than wild type mice survive to be able to participate in Morris Water Maze testing (A–D). Female PS19 mice receiving zinc acetate are more likely to survive to testing compared to female PS19 receiving vehicle (C).
Zinc treatment attenuates spatial memory impairment in female PS19 mice in the Morris water maze behavioral assay
There were no differences in any of the treatment groups in ability to find the platform in the visible platform (sensorimotor control) trials (Fig. 4A, B). In the hidden platform (memory acquisition) portion of the Morris Water Maze, female but not male PS19 mice showed a non-significant trend toward impairment (Fig. 4C, D).

Zinc acetate attenuates spatial memory impairment in female PS19 mice. Figures show means and SEM. There is no significant difference between genotypes or treatments in the visible platform control condition (A, B) or the hidden platform memory acquisition phase (C, D) of the Morris Water Maze. However, in the 72 h probe trial evaluating memory retention, the preference for the target quadrant is significantly reduced in vehicle-treated female PS19 mice compared to wild type mice (E; p < 0.05). The zinc-treated PS19 mice are not significantly different from wild type mice (E). There is no genotype or treatment effect in male mice (F).
However, in the probe test of memory retention, female PS19 mice performed significantly worse than wild-type mice at 72 h after training (Fig. 4E, p < 0.05), and trended worse at 24 h. This spatial memory impairment was attenuated by zinc treatment, so that the zinc-treated PS19 mice were not different from wild type (Fig. 4E). Male PS19 mice, in contrast, did not show a spatial memory impairment when compared to wild-type mice (Fig. 4F).
Zinc treatment does not significantly affect tau neuropathology and hippocampal volume
Pathologic forms of tau were measured by immunohistochemistry, using the AT8 antibody for p-tau. Consistent with the sex-dependent differences in spatial memory, female PS19 mice had more p-tau pathology than male PS19 mice, and there was a non-significant trend to reduced p-tau pathology with zinc treatment in female mice (Fig. 5). There were no significant changes in the ratio of p-tau to total tau in the soluble (p = 0.2352) or insoluble fractions (p = 0.1961) of hippocampal tissue from zinc-treated female PS19 mice compared to vehicle-treated mice.
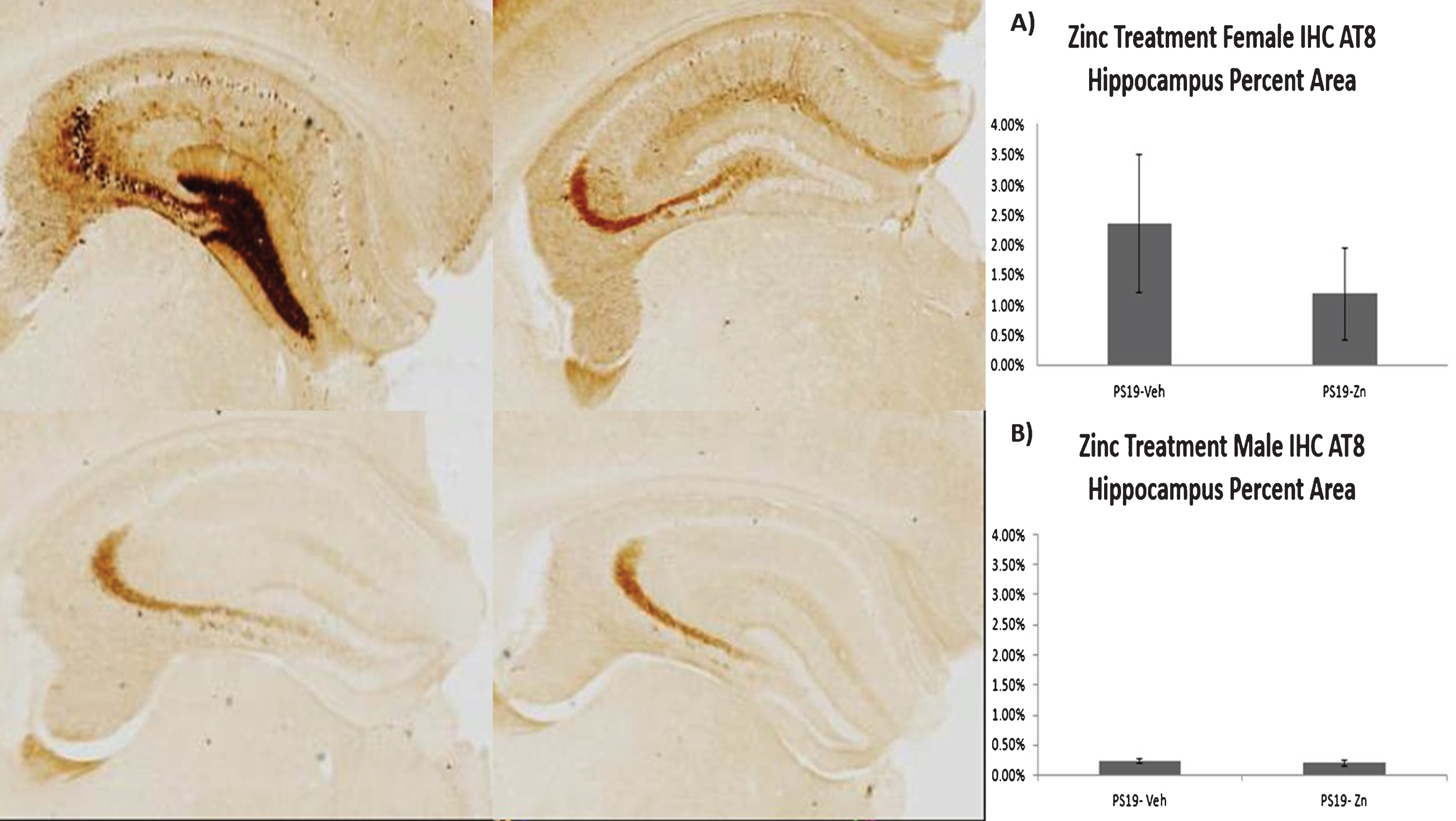
The micrographs show phosphorylated tau (p-tau) pathology in hippocampus labeled with AT8 antibody. The upper micrographs are female PS19 mice and the lower micrographs are male PS19 mice. The figures on the left are vehicle-treated, on the right are zinc-treated. While these photomicrographs emphasize the gender difference in p-tau burden, it should also be noted that there is substantial animal-to-animal variability as indicated by the large error bars in the quantified data in A. Panels A and B represent quantification of p-tau/AT8 pathology in groups of female (A) and male (B) PS19 mice, expressed as percent of hippocampal area occupied by AT8 immunohistochemical labeling in representative sections as detailed in Methods. Figures show means and SEM. The difference between males and females is self-evident. A trend to lower p-tau abundance in zinc treated (compared to vehicle-treated) female mice is also evident but is not statistically significant.
Hippocampal volumes, estimated from serial sections of brain tissue, were lower in both male and female PS19 mice compared to wild-type, and zinc treatment attenuated these effects, though none of these findings reached statistical significance (Fig. 6A, B).
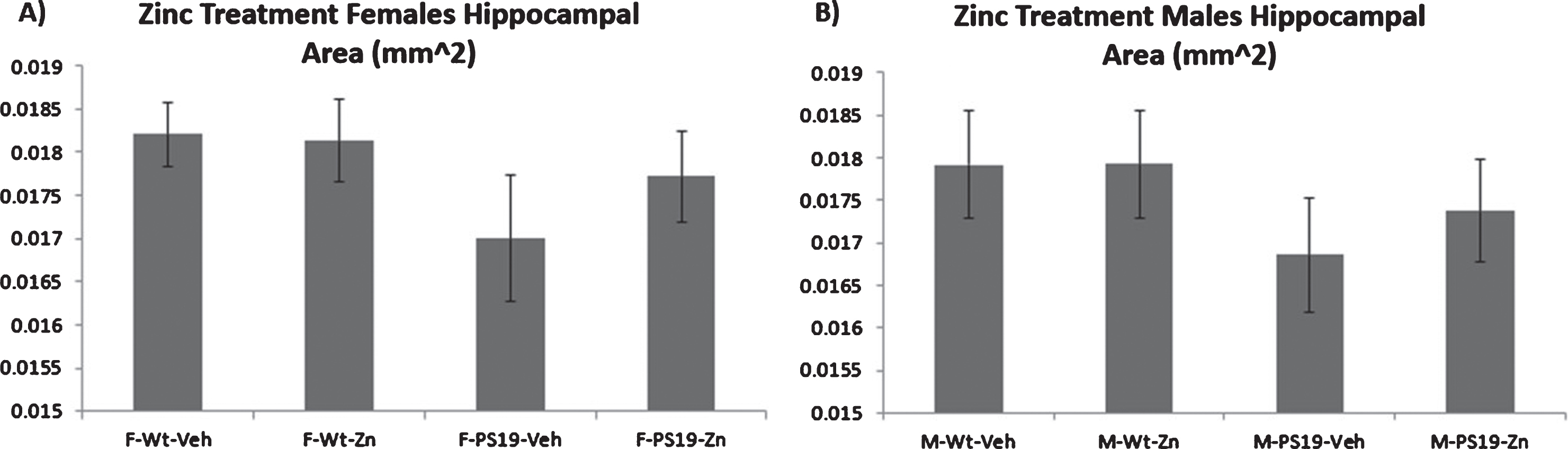
Hippocampal atrophy is estimated from the total area of selected hippocampal sections from treated and untreated wild type and PS19 mice. Figures show means and SEM. Trends to hippocampal atrophy in untreated PS19 mice are evident for both males and females but these did not reach statistical significance.
Copper-loading intervention
Copper loading increases brain levels of copper in female wild-type and PS19 mice with no effect on survival
Because both tau pathology and spatial memory impairment were more robust in female than in male PS19 mice, copper-loading experiments were confined to female mice. Although the copper-loading treatments did not significantly change serum levels of copper, chronic administration of both low and high dose copper increased brain levels of copper. Low dose copper significantly raised brain copper levels in both wild-type (p = 0.011 for low dose and p = 0.0022 for high dose) and PS19 mice (p = 0.0178 for high dose) when compared to vehicle treatment (Fig. 7).
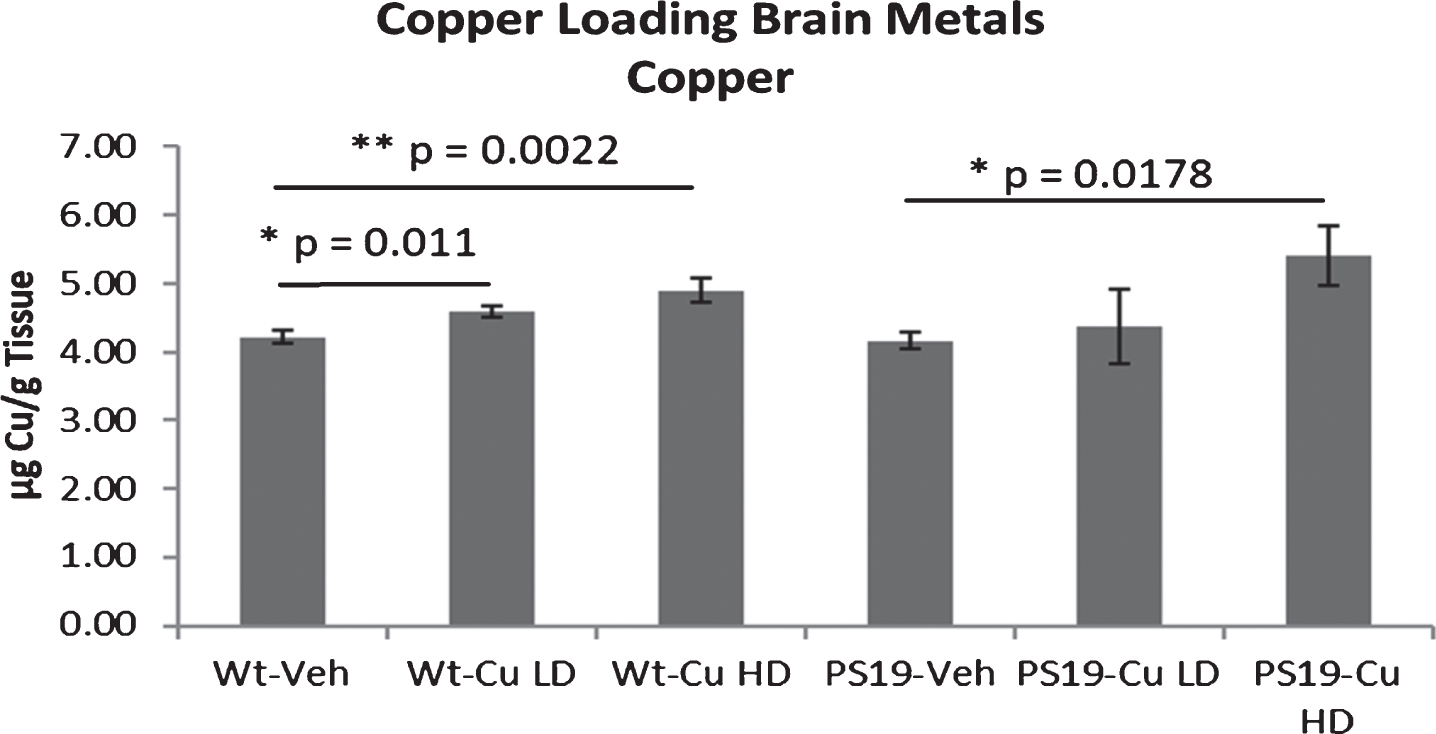
Brain copper levels are increased by dietary copper in female mice. Figures show means and SEM. Both low dose of copper (Cu LD) and high dose of copper (Cu HD) produced a statistically significant increase in brain copper levels in wild type (Wt) mice, and high dose copper produced a statistically significant increase in brain copper in PS19 mice. Note that only female mice are studied in the copper-loading experiments.
Despite the effect on brain copper, there was no difference in survival in the copper-treated PS19 mice compared to the untreated PS19 mice (data not shown).
Copper loading did not worsen spatial memory, tau pathology, or hippocampal atrophy in PS19 mice
As in the zinc experiments, no differences were observed in visible platform and hidden platform performance between treatment and genotypes (Fig. 8A, B), but female vehicle-treated PS19 mice performed significantly worse than wild-type mice during the 24 h (p = 0.028) and 48 h (p = 0.017) probe trials (Fig. 8C). Copper treated PS19 mice, however, did not perform any differently than vehicle-treated PS19 mice. Copper loading treatments also had no effect on hippocampal AT8 pathology (Fig. 9).
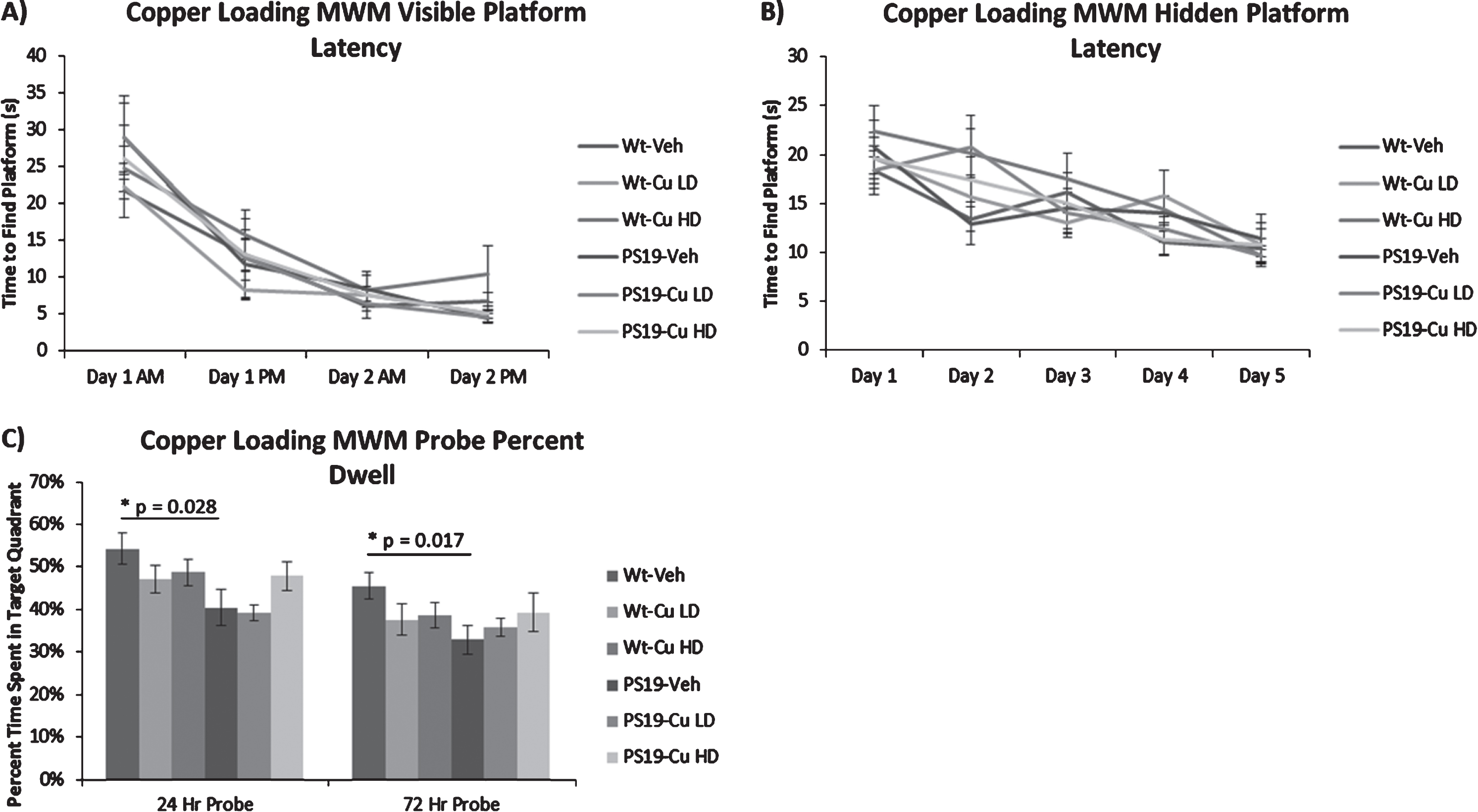
Copper loading does not worsen spatial memory deficit in female PS19 mice. Figures show means and SEM. PS19 mice are not impaired in the visible platform sensorimotor control test (A), nor in the hidden platform memory acquisition portion (B) of the Morris Water Maze, but they are impaired in the spatial memory retention Probe test portion of the Morris Water Maze at both 24 and 72 h (C). Copper loading does not worsen the deficit at either low (CuLD) or high dose (CuHD). Note that only female mice are studied in the copper-loading experiments.
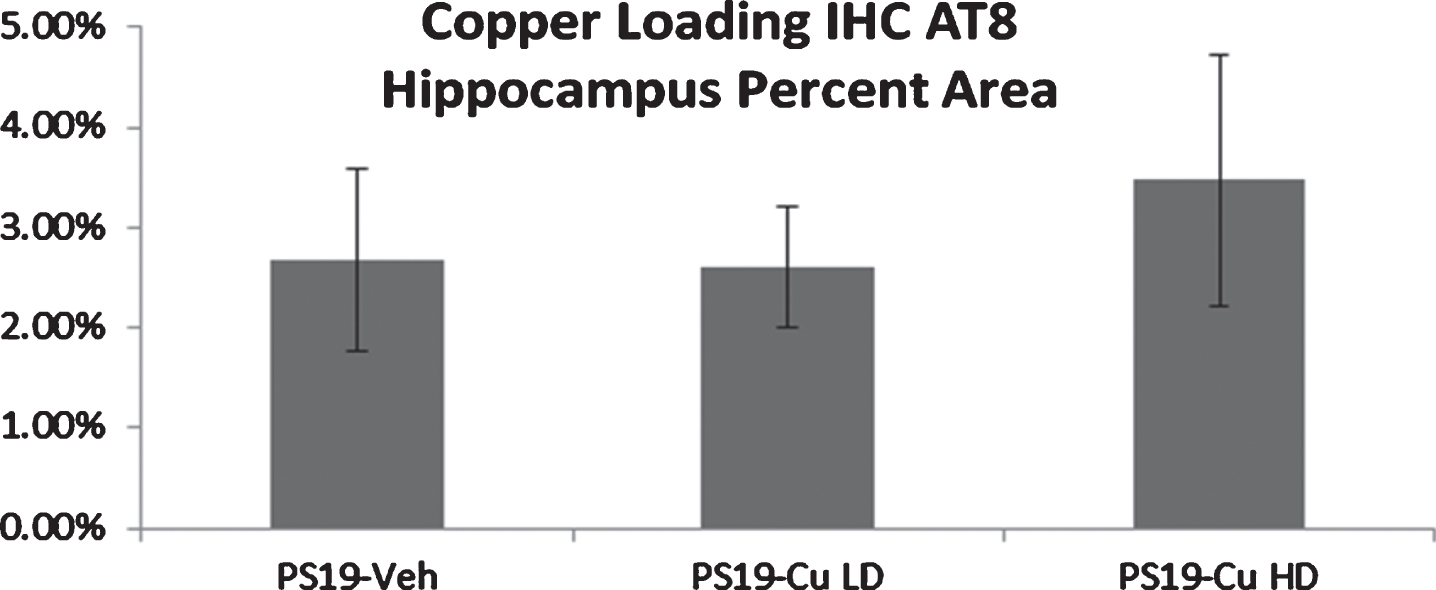
Phosphorylated tau pathology was measured with AT8 immunohistochemistry as in the zinc experiments in Fig. 5. Figures show means and SEM. Despite an increase in brain copper levels, copper loading did not increase hippocampal p-tau pathology. Note that only female mice are studied in the copper-loading experiments.
Vehicle treated wild-type PS19 mice had significantly less hippocampal volume than vehicle treated wild-type mice (Fig. 10, p = 0.04), but neither low dose nor high dose copper had an effect on hippocampal volume (Fig. 10).
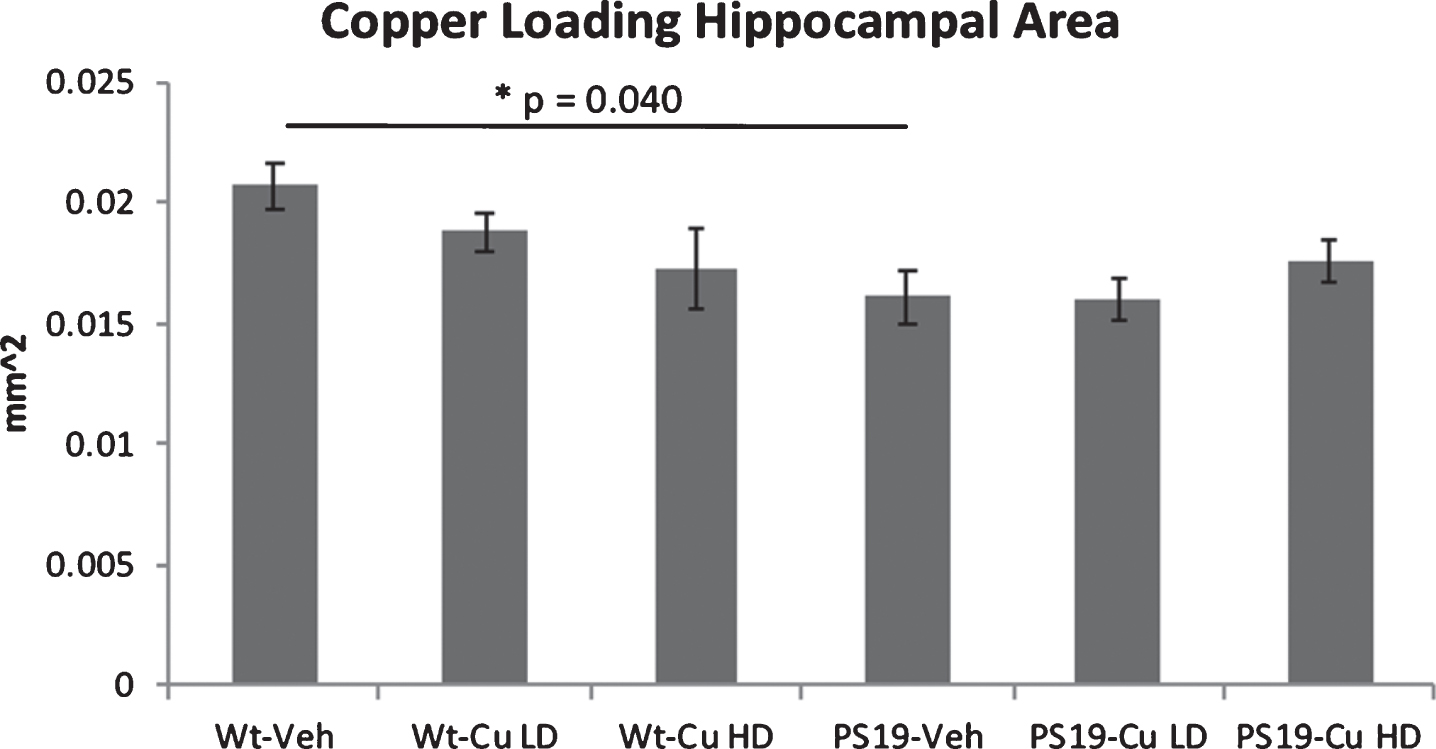
Hippocampal volume was estimated from select sections as in Fig. 6. Figures show means and SEM. A significant reduction in hippocampal volume was observed when comparing untreated PS19 mice with wild type mice. Copper-loading had no effect on hippocampal volume, as the copper-treated PS19 mice did not differ from the untreated PS19 mice. Note that only female mice are studied in the copper-loading experiments.
DISCUSSION
These experiments show that lowering brain copper exerts a beneficial symptomatic effect in female mice with NFT-like brain pathology, but increasing brain copper does not exacerbate pathology or memory impairment in these mice. This suggests that although environmental and brain copper levels may have a potential role in the pathogenesis of brain Aβ pathology, they do not have a potent direct effect on tau pathology. This is not inconsistent with a prior study [11] which reported an exacerbation of tau pathology with copper loading, as those experiments were conducted in mice that expressed both amyloid and tau pathology, so tau effects in that study may be “downstream” of Aβ. Our findings consequently may diminish enthusiasm for copper-modulators in “pure” tauopathies. This should not, however, diminish interest in copper chelators in AD per se, since some have shown promising results in clinical trials [12, 13].
The gender-specific effects of copper modulation in the zinc experiments were unexpected. They may simply be due to gender difference in abundance of AD pathology, as increased levels of AD pathology in females have been previously reported in both mouse models [22–24] and human postmortem brain [25]. In the PS19 strain, gender differences in tau burden have been reported [19, 27], but not to this extreme and sometimes in the opposite direction [26, 27]. In the present study, however, the concordance of gender differences in hippocampal tau pathology and in spatial memory argues that the gender differences in effects of copper modulation are best explained by these sex differences in brain pathology. Alternatively, these findings may reflect gender-dependent differences in copper metabolism, as we [28] and others [29] have observed higher plasma and brain copper levels in females, with other studies indicating that these higher levels are due to effects of estrogen [30, 31]. Finally, this may reflect an inadvertent difference in dosing, as male mice are larger than females, so receive a lower mg/kg dose when administered the same concentration of drug in drinking water. In any case, the gender difference in tau pathology will be important to consider in future translational work with this mouse model.
Interpretation of these findings must also consider the possibility that zinc itself may have a direct effect on brain pathology or function in treated mice, as zinc is critically involved in several biological functions in the brain [32, 33], including synaptic plasticity, ischemic neuronal death, intraneuronal signaling [32] and age-associated cognitive decline [33]. Zinc may also play a pathogenic role in AD, by way of interactions with Aβ [34, 37], glial mediated zinc homeostasis [35], or synaptic function [36]. Based on these types of observations, studies of zinc supplementation in transgenic mice with tau brain pathology have been reported previously, but findings have been inconsistent. In one case, zinc supplementation was associated with worsening of tau pathology [38], while in a second case, zinc supplementation reduced tau pathology and improved cognition [39]. These prior studies also studied mice with combined amyloid and tau pathology, while the present study examined mice with only tau pathology, in order to avoid the possibility of indirect effects mediated by changes in Aβ. Although a direct effect of zinc upon cognitive function cannot be excluded in the present study, the lack of change in brain zinc levels among zinc-supplemented mice reduces the likelihood that the behavioral changes seen in this experiment are direct effects of changes in brain zinc. It is also important to note that the zinc dosages used here are based on our prior experience with using zinc to lower brain copper [10, 16], and are much higher than others have used in zinc supplementation studies in murine models of AD [38, 39].
In summary, these findings provide little support for targeting tau pathology with copper-lowering strategies, and may also reduce concern about adverse health effects of environmental copper.