
Abstract
Background:
Accumulation of amyloid-β (Aβ) peptides, generated from amyloid-β precursor protein (AβPP) amyloidogenic processing, is one of the most salient disease hallmarks of Alzheimer’s disease (AD). Nicotine is able to promote α-secretase-mediated AβPP nonamyloidogenic processing and increase the release of sAβPPα and C-terminal fragment of 83 amino acids (C83). However, the potential molecular mechanism remains elusive.
Objective:
The aim of the present study was to investigate the effect of nicotine on AβPP processing in SH-SY5Y cells that stably express human Swedish mutant AβPP695 (SH-SY5Y-AβPP695).
Methods:
The expression of AβPP and its C-terminal fragments including C99, C89, and C83, was measured in SH-SY5Y-AβPP695 cells treated with nicotine for 6 h. Protein kinase C (PKC) antagonist Ro30-8220 or agonist PMA was used to determine the role of PKC in AβPP processing. Lentivirus-mediated shRNA targeting receptor for activated C-kinase 1 (RACK1) gene was added into the media to knockdown RACK1 expression, and then AβPP processing was examined.
Results:
The results showed that 6 h of nicotine exposure increased the expression of α-secretase (ADAM10) and C83 in a dose dependent manner. While the β-secretase (BACE1), AβPP amyloidogenic processing products C89 and C99 as well as Aβ peptides (including Aβ40 and Aβ42) remained unchanged. We also found that nicotine elevated the expression of phosphorylated PKC (P-PKC) and RACK1 on the cytomembrane. PKC antagonist Ro30-8220 treatment prevented the increase of ADAM10 and C83 by nicotine. Genetic knockdown RACK1 significantly inhibited P-PKC, and consequently abolished the increase of ADAM10 and C83 by nicotine.
Conclusion:
Taken together, these results indicate that nicotine effectively promotes AβPP nonamyloidogenic processing via RACK1-dependent activation of PKC in SH-SY5Y-AβPP695 cells and could be a potential molecule for AD treatment.
INTRODUCTION
Alzheimer’s disease (AD) is a chronic neurodegenerative disease leading to dementia, and so far there is no effective treatment to stop or reverse its progression [1]. By 2050, sporadic AD is expected to affect 150 million individuals worldwide, becoming an unsustainable socio-economical problem [2]. A growing body of evidence has shown that the imbalance between production and clearance of amyloid-β (Aβ) peptides causes Aβ accumulation, which may be the initiating factor in AD [3, 4]. Aβ originates from sequential proteolysis of the amyloid-β protein precursor (AβPP) by β-site AβPP-cleaving enzyme 1 (BACE-1) and γ-secretase, mainly presenilin 1 (PS1). Besides amyloidogenic processing, AβPP is able to be cleaved by α-secretase such as ADAM10 to generate secreted AβPP (sAβPPα) and C83, an 83-residue carboxy-terminal fragment. C83 is further cleaved by PS1 to generate extracellular p3 and the amyloid intracellular domain [5]. Further studies have revealed that α-secretase cleavage of AβPP mainly occurs at the plasma membrane [6], whereas the majority of secreted Aβ and intracellular Aβ are generated in the trans-Golgi network and the endoplasmic reticulum/intermediate compartment, respectively [7–9].
Nicotinic acetylcholine receptors (nAChR), composed of various combinations of α2-α6 subunits with β2-β4 subunits or homomeric pentamers formed from α7 to α10 subunits, are widely distributed in the central nervous system (CNS) [10]. It has been well documented that a consistent loss of various nAChR subtypes are observed in postmortem brain from AD patients [11, 12], which is significantly related to cognitive decline [10, 13]. In contrast, activation of nAChR by nicotine improves cognitive function in human beings and experimental animals [14–16]. Furthermore, a growing number of studies have revealed that nicotine is able to exert a neuroprotective effect against Aβ-induced cytotoxicity and increase the AβPP nonamyloidogenic processing to produce sAβPPα and C83 [17–19]. However, the potential molecular mechanism of nicotine-promoted AβPP nonamyloidogenic processing is still not very clear.
Previous studies have shown that nicotine increases the activation of protein kinase C (PKC) in adrenal medullary cells, smooth muscle, and neurons [20–22]. PKC can activate α-secretase-mediated cleavage of AβPP by directly acting on α-secretase or indirectly through phosphorylation of extracellular-signal-regulated kinase [23]. Receptor for activated C-kinase 1 (RACK1), which served as an anchoring protein to translocate activated PKC from cytoplasm to membrane, plays an important role in PKC-mediated signal transduction pathways. Recent progresses have reported that RACK1 is significantly reduced in the brain of aging animals [24, 25] and AD patients [26]. The upregulation of RACK1 is related to neuroprotection [27], while the deficit of RACK1 contributes to the spatial memory impairment [28]. Nonetheless, contradictory results challenge these findings [29]. In the present study, we therefore introduced SH-SY5Y-AβPP695 model cell of AD, and investigated whether nicotine could promote AβPP nonamyloidogenic processing via RACK1/PKC signaling pathway by using a combination of pharmacological and genetic assessments.
MATERIALS AND METHODS
Cell culture and treatment
The SH-SY5Y-AβPP695 cells, stably transfected with human Swedish mutant AβPP695, was obtained from Professor Weihong Song (University of British Columbia, Vancouver, Canada) and cultured in DMEM (Invitrogen, CA, USA), supplemented with 10% fetal bovine serum (Invitrogen) and zeocin (Invitrogen) to a final concentration of 1μl/ml. Cells were maintained at 37°C in a 5% CO2 incubator.
Nicotine (Sigma, USA) was serial diluted and then added into the media for 6 h with the final concentration of 0 M, 10–7 M, 10–6 M, 10–5 M, 10–4 M, and 10–3 M. PMA (Sigma) or Ro31-8220 (Sigma) was added into the media 30 min before nicotine treatment with the final concentration of 10μM.
Lentivirus and transfection
Lentivirus with pLKD-CMV-eGFP-U6-shRNA plasmid (LVshRACK1) and the negative control (LVeGFP) was generated by Obio Technology (Shanghai, China). Three sequence of shRNA targeting RACK1 gene designed by Invitrogen BLOCK-iT™ RNAi Designer was as follows: LVshRACK1 - 1: 5’-CCAACAGCAGCAACCCTAT-3’, LVshRACK1 - 2: 5’-GCTGAAGACCAACCACATT-3’, LVshRACK1 - 3 5’-CCATGTTATGGGATCTCAA-3’. Cultured cells were infected by adding the viral suspension directly to the medium (1μl added to 4 ml culture medium) to produce the final concentration of MOI 20. After 72 h of infection, more than 90% cells appeared eGFP positive. The virus did not cause apparent toxicity to cells at least 4 days after infection.
Western blot assay
The cells were washed with ice-cold PBS and then lysed in RIPA lysis buffer containing a cocktail of complete protease inhibitors (Roche), and centrifuged (4°C, 10,000 rpm, 15 min) to collect the supernatants. Cytomembrane protein was extracted by Minute™ Plasma Membrane Protein Isolation and Cell Fractionation Kit (Invent, USA) according to the manufacturer’s instructions. The proteins were determined by BCA Protein Assay kit (Thermo, USA). The same amount of proteins (30μg) were loaded onto SDS-PAGE gels for electrophoresis and transferred onto PVDF membranes. To block nonspecific background, the membranes were incubated in 5% non-fat milk in TBST at 37°C for 1 h. The target proteins were immunoblotted with primary antibody overnight at 4°C to AβPP and its C-terminal fragments including C83, C89 and C99 (1 : 1000, obtained from professor Weihong Song in the University of British Columbia, Vancouver, Canada), PKC (1 : 1000, Abcam), P-PKC (1 : 1000, Cell Signaling Technology), RACK1 (1 : 1000, Abcam), ADAM10 (1 : 1000, Abcam), PS1 (1 : 1000, Abcam), and BACE1 (1 : 1000, CST), followed by incubation with corresponding horseradish peroxidase (HRP)-conjugated secondary antibody (1 : 3000; Abcam) at 37°C for 1 h. After washing 3 times with TBST (for 10 min each), the blots were imaged by the Bio-Rad Imager using enhanced chemi-luminescence (ECL). β-actin (1 : 3000, Sigma) was used as a loading control. The band intensity of each protein was quantified by the Bio-Rad Quantity One software.
Aβ ELISA
After being treated with nicotine, cell cultured medium was collected as described previously [30]. Secreted Aβ40 and Aβ42 were measured by a sandwich ELISA kit (R&D system, USA) according to the manufacturer’s instructions. Add 200μl of medium or control into the microplate coated with a monoclonal Aβ40 or Aβ42 antibody. The Aβ40 and Aβ42 Conjugate was added after 2 h of incubation. 2 h later, the Substrate Solution and STOP Solution was added before detection.
Statistical analysis
All data are expressed as mean±SEM. T-test and one-way ANOVA, followed by Tukey’s post-test (Prism 7, GraphPad Software, Inc) were used to analyze the data. The significance level was set at p < 0.05.
RESULTS
Nicotine promotes AβPP nonamyloidogenic processing in SH-SY5Y-AβPP695 cells
To determine the effects of nicotine on AβPP processing, different concentrations of nicotine (from 10–7 to 10–3 M) were used to treat SH-SY5Y-AβPP695 cells for 6 h. The protein levels of AβPP and its C-terminal fragments including C83, C89, and C99, as well as AβPP-cleaving enzymes such as ADAM10, BACE1, and PS1, were measured by western blotting assay. The results showed that AβPP remained unchanged after treatment with nicotine (Fig. 1a; n = 4). The expression of C83 was significantly increased in a dose-dependent manner (n = 4 in each group; 10–7 M: 145.07±13.18% relative to 0 M, p = 0.336 versus 0 M; 10–6 M: 172.49±19.87% relative to 0 M, p = 0.036 versus 0 M; 10–5 M: 175.15±7.55 % relative to 0 M, p = 0.028 versus 0 M; 10–4 M: 187.52±22.23% relative to 0 M, p = 0.009 versus 0 M; 10–3 M: 230.78±19.76% relative to 0M, p < 0.001 versus 0 M; Fig. 1b, c). Notably, nicotine exposure did not affect the expressions of C89 (n = 4 in each group; Fig. 1b, d) and C99 (n = 4 in each group; Fig. 1b, e). Given the main components of senile plaques are Aβ peptides, we next directly measured the secreted Aβ40 and Aβ42 in cell cultured media by using ELISA. We found that the secreted Aβ40 (n = 3; Fig. 1f) and Aβ42 (n = 3; Fig. 1 g) in media also remained unchanged compared with the control.
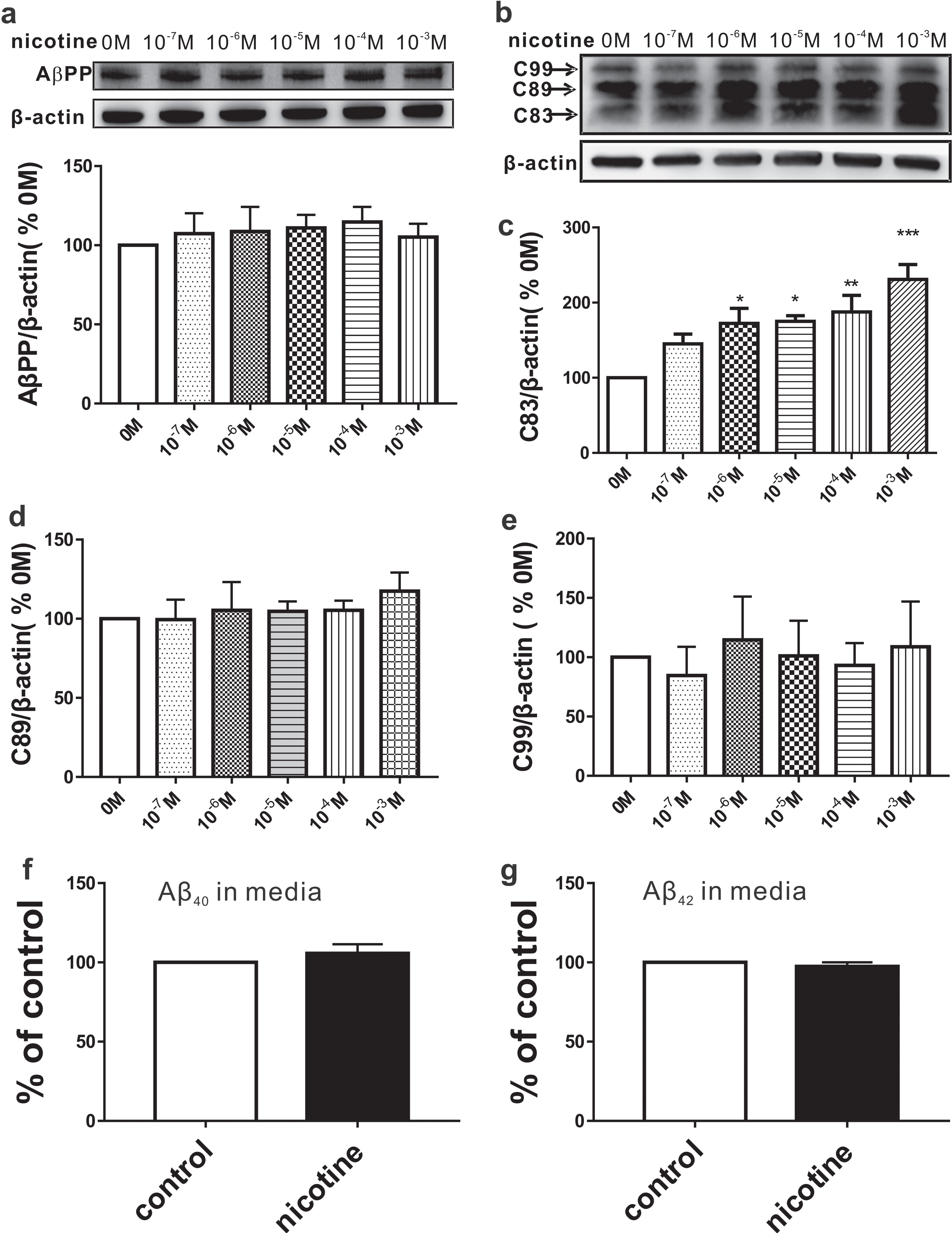
Effects of nicotine on AβPP processing. SH-SY5Y-AβPP695 cells were exposed to various concentrations of nicotine (0 – 10–3 M) for 6 h. The relative protein levels of AβPP (a), C83 (b, c), C89 (b, d), and C99 (b, e) are normalized by 0 M. One way ANOVA: F (5,18) = 0.964, p = 0.465 for AβPP; F (5,17) = 7.351, p < 0.001 for C83; F (5,18) = 1.446, p = 0.256 for C89; F (5,18) = 0.604, p = 0.698 for C99. The secreted Aβ40 (f) and Aβ42 (g) measured by ELISA. Student’s t-test: p = 0.063 for Aβ40; p = 0.062 for Aβ42. Data are expressed as mean±SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
AβPP can be cleaved by either ADAM10 to generate C83 or BACE1 to generate C89 and C99. Therefore, we next wanted to determine whether nicotine-promoted AβPP nonamyloidogenic processing is attributed to an increase in ADAM10. The results showed that nicotine exposure dramatically increased the expression of ADAM10 in a dose-dependent manner (n = 6 in each group; 10–7 M: 123.00±4.55% relative to 0 M, p = 0.300 versus 0 M; 10–6 M: 140.62±7.34% relative to 0 M, p = 0.009 versus 0 M; 10–5 M: 150.33±11.86% relative to 0 M, p < 0.001 versus 0 M; 10–4 M: 162.90±7.93% relative to 0 M, p < 0.001 versus 0 M; 10–3 M: 152.46±8.48% relative to 0 M, p < 0.001 versus 0 M; Fig. 2a). While BACE1 (n = 4; Fig. 2b) and PS1 (n = 4; Fig. 2c) displayed no obviously difference after nicotine treatment. Taken together, these results indicate that nicotine exposure increases α-secretase, and consequently promotes AβPP nonamyloidogenic processing, without affecting AβPP amyloidogenic processing.

Effects of nicotine on AβPP processing secretase. The relative protein levels of ADAM10 (a), BACE1 (b), and PS1 (c) are normalized by 0 M (n = 4– 6 in each group). One way ANOVA: F (5,30) = 9.110, p < 0.001 for ADAM10; F (5,18) = 0.258, p = 0.930 for BACE1; F (5,18) = 0.352, p = 0.874 for PS1. Data are expressed as mean±SEM, **p < 0.01, ***p < 0.001.
Nicotine-promoted AβPP nonamyloidogenic processing is dependent on RACK1/PKC pathway
Given that nicotine can activate PKC [20–22], which promotes α-secretase-mediated cleavage of AβPP [23], we next examined the activation of PKC after nicotine exposure. Since nicotine exposure from 10–6 M to 10–3 M displayed similar effects on AβPP nonamyloidogenic processing, we chose the lowest concentration of nicotine (10–6 M) in the following experiments. The results showed that the expression of PKC was not altered by nicotine exposure at the concentration of 10–6 M (n = 4; 94.06±2.32% relative to control, p = 0.084 versus control; Fig. 3a), but phosphorylated PKC (P-PKC), an activated form of PKC (n = 7; 137.21±6.88% relative to control, p = 0.002 versus control; Fig. 3b), was significantly increased on the cytomembrane. To further confirm the relationship between PKC and AβPP nonamyloidogenic processing, we examined ADAM10 and C83 in SH-SY5Y-AβPP695 cells treated with PKC agonist PMA or co-treated with nicotine and PKC antagonist Ro30-8220 (Nicotine+Ro). The results showed that PMA treatment significantly increased the expression of ADAM10 (n = 3; 175.70±7.10% relative to control, p = 0.001 versus control; Fig. 3c) and C83 (n = 4; 203.91±34.52% relative to control, p = 0.011 versus control; Fig. 3d). More importantly, Ro30-8220 administration 30 min before nicotine markedly inhibited the nicotine-induced increase of ADAM10 (n = 3; 85.31±14.71% relative to control, p = 0.63 versus control, p = 0.004 versus nicotine; Fig. 3c) and C83 (n = 4; 50.20±4.80% relative to control, p = 0.304 versus control, p = 0.016 versus nicotine; Fig. 3d).
Since RACK1 plays an important role in translocating activated PKC to cytomembrane, we next examined the influence of nicotine exposure on RACK1 expression. The results showed that the total RACK1 displayed no difference in SH-SY5Y-AβPP695 cells exposed to nicotine (n = 4; 104.71±7.64% relative to control, p = 0.581 versus control; Fig. 4a). However, the expression of RACK1 on the cytomembrane was significantly increased after nicotine treatment (n = 5; 136.20±10.05% relative to control, p = 0.023 versus control; Fig. 4b). To further determine the involvement of RACK1 in nicotine-induced AβPP nonamyloidogenic processing, lentivirus carrying RACK1 shRNA (LVshRACK1) was added into the media to knockdown RACK1 expression. The results showed that LVshRACK1 succeeded in reducing the expression of RACK1 (n = 4; 55.26% ±5.74% relative to control, p = 0.017 versus control, p = 0.005 versus LVeGFP; Fig. 4c), compared with control virus (LVeGFP). Importantly, the administration of LVshRACK1 (LVshRACK1 + nicotine) 3 days before nicotine treatment obviously inhibited the nicotine-induced increase of P-PKC (n = 4; 60.81±10.04% relative to control, p = 0.038 versus control, p < 0.001 versus LVeGFP+nicotine; Fig. 4d), ADAM10 (n = 4; 95.25 % ±9.83% relative to control, p = 0.987 versus control, p = 0.027 versus LVeGFP+96 versus control, p = 0.035 versus LVeGFP+nicotine; Fig. 4f). Notably, the decrease of P-PKC (LVshRACK1 +nicotine versus LVshRACK1, p = 0.997; Fig. 4d), ADAM10 (LVshRACK1 +nicotine versus LVshRACK1, p = 0.243; Fig. 4e) and C83 (LVshRACK1 +nicotine versus LVshRACK1, p = 0.534; Fig. 4f) induced by LVshRACK1 remained unchanged after nicotine exposure. Collectively, these data demonstrate that nicotine exposure increases cytomembrane RACK1, and subsequently activates PKC and elevates the production of ADAM10 and C83, whereas RACK1 knockdown can abolish these effects, suggesting that nicotine may promote AβPP nonamyloidogenic processing via RACK1/PKC signal pathway.
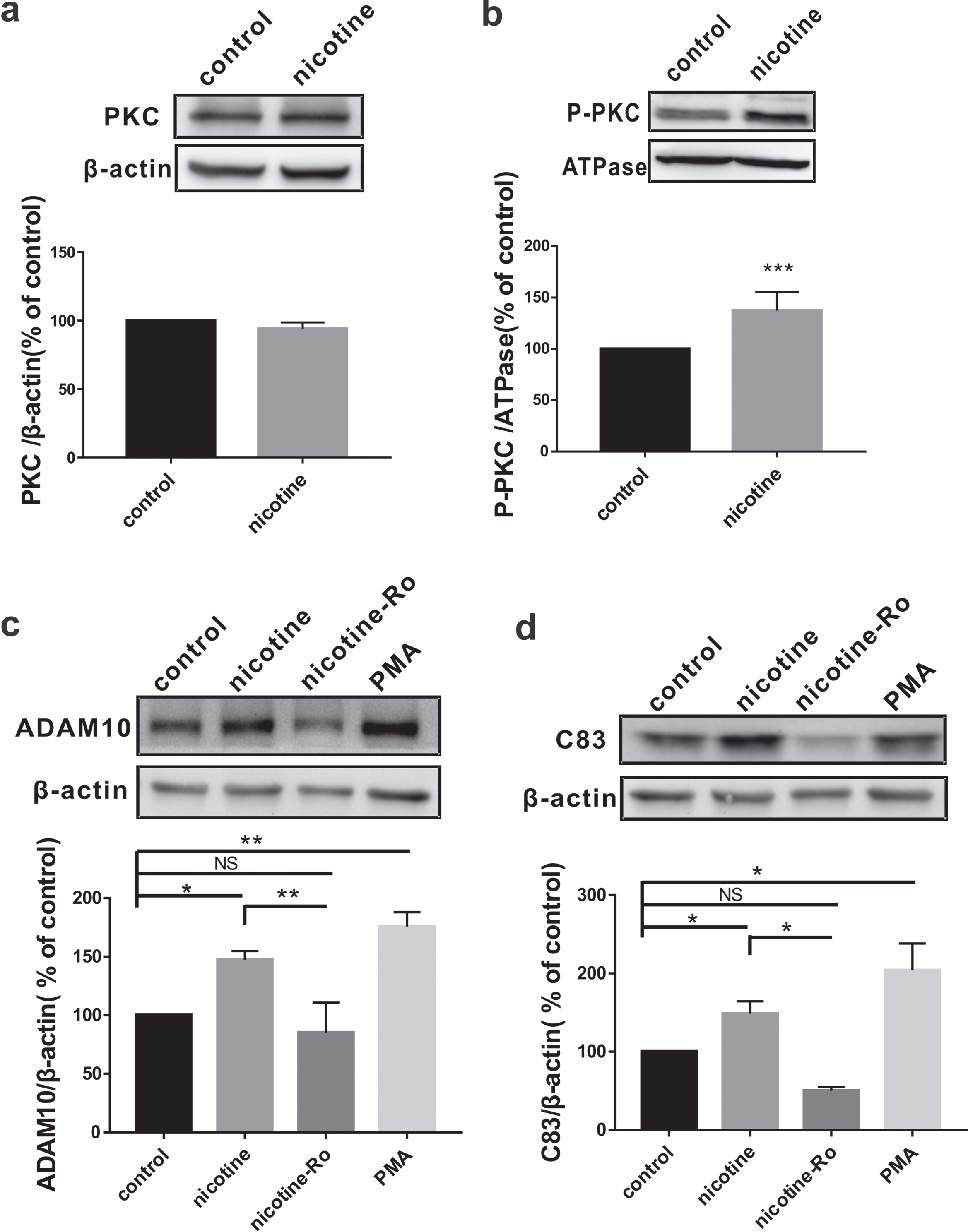
Nicotine promotes AβPP nonamyloidogenic processing by activating PKC. The relative protein levels of total PKC (a) and P-PKC (b) assessed by western blot in SH-SY5Y-AβPP695 cells without (control) or with nicotine treatment. Student’s t-test: p = 0.084 for PKC; p < 0.001 for P-PKC. The relative protein levels of ADAM10 (c) and C83 (d) assessed by western blot in SH-SY5Y-AβPP695 cells with PKC agonist PMA or antagonist Ro30-8220 treatment. One way ANOVA: F (3,8) = 24.399, p < 0.001 for ADAM10; F (3,12) = 11.775, p < 0.001 for C83. Data are expressed as mean±SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
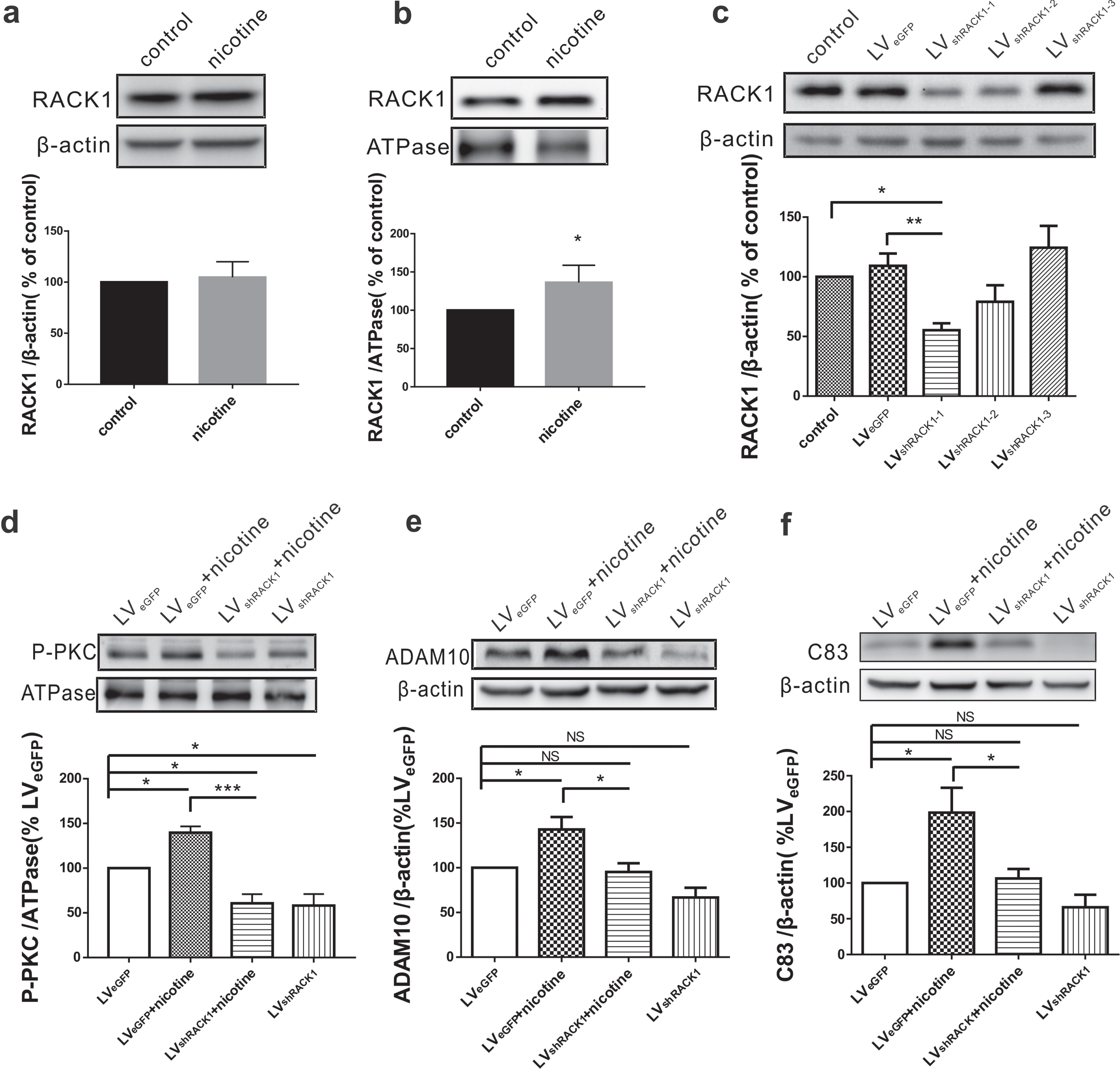
Nicotine-promoted AβPP nonamyloidogenic processing is dependent RACK1 translocation to cytomembrane. The relative protein levels of total RACK1 (a) and cytomembrane RACK1 (b) assessed by western blot in SH-SY5Y-AβPP695 cells without (control) or with nicotine treatment. Student’s t-test: p = 0.581 for RACK1; p = 0.023 for cytomembrane RACK1. c) The relative protein levels of RACK1 assessed by western blot in SH-SY5Y-AβPP695 cells treated with LVshRACK1. The relative protein levels of P-PKC (d), ADAM10 (e), and C83 (f) assessed by western blot in SH-SY5Y-AβPP695 cells after cotreatment with nicotine and LVshRACK1. One way ANOVA: F (3,12) = 18.751, p < 0.001 for P-PKC; F (3,12) = 9.625, p = 0.002 for ADAM10; F (3,12) = 7.597, p = 0.004 for C83. Data are expressed as mean±SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
In the present study, we find that nicotine exposure promotes AβPP nonamyloidogenic processing in SH-SY5Y-AβPP695 cells by increasing α-secretase ADAM10. Furthermore, nicotine-induced AβPP nonamyloidogenic processing is dependent on the RACK1/PKC signaling pathway. The production of AβPP nonamyloidogenic processing sAβPPα is considered as a neuroprotective factor for synaptic plasticity, synaptogenesis, and memory [31, 32]. Genetic overexpression of sAβPPα in aged mice results in a significant reduction in soluble Aβ species and plaque formation [33]. Thus, nicotine could be a potential molecule for AD treatment through increasing sAβPPα.
It has been well documented that α4 (known as α4β2) and α7 nAChRs are predominantly expressed in the CNS. Activation of nAChRs can regulate neurotransmitter release and hippocampal plasticity, and consequently improves cognitive function under both physiological and pathological conditions [13, 34]. For example, both acute and chronic treatment with nicotine can improve learning and memory processing and long-term potentiation (LTP) [16, 36]. Nicotine exposure decreases accumulation of Aβ in the cortex and hippocampus of AD model mice [37] since Aβ42 may bind with extracellular domain of nAChRα7 [38], which is believed to contribute to anti-inflammatory and neuroprotective effects [39]. Thus, acetylcholinesterase inhibitors such as tacrine, donepezil, galanthamine, and rivastigmine, are clinically used in the treatment of AD by enhancing the intrinsic action of nAChRs. Consistent with these findings, we here reported that activation of nAChRs by nicotine markedly increases AβPP nonamyloidogenic processing production C83 and α-secretase ADAM10. However, contradictory reports challenge these findings. For example, recent studies demonstrate that prenatal or postnatal nicotine exposure impairs spatial cognitive and hippocampal synaptic plasticity in adolescent male rats [40]. Furthermore, nicotine treatment exacerbates tau phosphorylation and cognitive impairment in the AD rat model by hippocampal injections of Aβ25 - 35 [41]. These discrepancies may be accounted for by different animal and cell models, length of the Aβ peptides, specific brain areas investigated, and other differences in experimental conditions. Given this controversy, determining the exact role and potential mechanism of nicotine exposure in AD treatment is essential in the future study.
There is a growing body of evidence has shown that Aβ production and deposition lead to an increase in intracellular Ca2 + concentration [42] by enhancing the activity of ligand-gated calcium channels or voltage-dependent calcium channels [43, 44]. Increased Ca2 + can trigger calcium-sensitive cascades, which subsequently increases transmitter release, tau protein phosphorylation, and neuronal death. Nicotine exposure can reduce the generation of Aβ [18, 21], and may consequently exert neuroprotective effects against Aβ-induced neurotoxicity by inducing a reduction in intracellular Ca2 + concentration and neural excitation [45, 46]. However, we did not find any alteration of Aβ40 and Aβ42 after nicotine exposure for 6 h in SH-SY5Y-AβPP 695 cells. One possible explanation is that 6 h treatment with nicotine was not long enough to induce a reduction in Aβ40 and Aβ42 significantly. Alternatively, given that SH-SY5Y-AβPP695 cells have been highly expressed human mutant AβPP and are able to produce an abundance of Aβ peptides, it may mask nicotine-induced decrease of Aβ40 and Aβ42. Although Aβ peptides remains unchanged after nicotine exposure, AβPP nonamyloidogenic processing production C83 is significantly increased, indicating an increase in the ratio of AβPP nonamyloidogenic and amyloidogenic processing, which may have a beneficial effect on AD treatment.
In summary, this study shows that acute nicotine exposure promotes AβPP nonamyloidogenic processing in a RACK1/PKC dependent manner, indicating that nAChRs agonists, such as nicotine, may serve as potential therapeutics for treating the learning and memory deficits associated with both patients with AD and aged populations. However, we here only examined the effects of nicotine on AβPP processing in AD model cells, future studies to determine the influence of nicotine exposure on memory decline in vivo, especially in various transgenic animal models of AD, need to be further explored.
Footnotes
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (No. 81622015, 91749116, and 81571042), the Science and Technology Research Program of Chongqing Municipal Education Commission (No. KJZD-K201900403) and Innovation Research Group at Institutions of Higher Education in Chongqing (No. CXQTP19019019034).