
Abstract
Background:
Neuroinflammation plays an important role in Alzheimer’s disease (AD). During this process, activated microglia release pro-inflammatory cytokines such as interleukin (IL)-1α, IL-1β, IL-6, and tumor necrosis factor α (TNFα) that participate in neuron damage, but also anti-inflammatory cytokines (such as IL-10), which maintain homeostasis of immune response. Previous studies showed the association of IL-1α –889C/T (rs1800587), IL-1β–1473G/C (rs1143623), IL-6 –174C/G (rs1800795), IL-10 –1082G/A (rs1800896), and TNFα –308A/G (rs1800629) polymorphisms with AD.
Objective:
We aimed to investigate whether people with certain IL-1α, IL-1β, IL-6, IL-10, and TNFα genotypes in these polymorphisms are more prone to develop AD-related pathology, reflected by pathological levels of cerebrospinal fluid (CSF) AD biomarkers including amyloid-β1-42, total tau (t-tau), tau phosphorylated at Thr 181 (p-tau181), Ser 199 (p-tau199), and Thr 231 (p-tau231), and visinin-like protein 1 (VILIP-1).
Methods:
The study included 115 AD patients, 53 patients with mild cognitive impairment, and 11 healthy controls. The polymorphisms were determined using real-time polymerase chain reaction. Levels of CSF biomarkers were determined by enzyme-linked immunosorbent assay.
Results:
A significant increase in p-tau CSF levels was found in patients with the AA IL-10 –1082G/A and GG TNFα –308A/G genotypes, and in carriers of a G allele in IL-1β –1473C/G and IL-6 –174C/G polymorphisms. t-tau levels were increased in carriers of a G allele in IL-1β –1473C/G polymorphism. An increase in VILIP-1 levels was observed in patients with CG and GG IL-1β –1473C/G, GC IL-6 –174C/G, and GG TNFα –308A/G genotype.
Conclusion:
These results suggest that persons carrying certain genotypes in IL10 (–1082G/A), IL1β (1473C/G), IL6 (–174C/G), and TNFIα (–308A/G) could be more vulnerable to development of neuroinflammation, and consequently of AD.
INTRODUCTION
Inflammatory processes are enhanced in the brain of Alzheimer’s disease (AD) patients [1, 2]. Microglial cells become activated and produce high levels of cytokines. In early stages of AD, activated microglia phagocytize amyloid-β (Aβ) peptide, but when they are activated over extended periods [3], they can no longer clear Aβ, and the pro-inflammatory cytokines they release participate in propagation of pathological tau proteins and neuron damage [4, 5]. The main pro-inflammatory cytokines released from activated microglia are interleukin (IL)-1α, IL-1β, IL-6, and tumor necrosis factor α (TNFα) [6]. During sustained inflammation, anti-inflammatory cytokines (such as IL-10) are also released and maintain homeostasis of the immune response [6]. Single nucleotide polymorphisms (SNPs) in genes for IL-1α, IL-1β, IL-6, IL-10, and TNFα were previously associated with AD [7, 8]. It was shown that certain SNPs can influence gene transcription and consequently the amount of the produced cytokines [9–11]. Increase in the amount of pro-inflammatory cytokines and decrease in anti-inflammatory cytokines increases inflammation and favors development of AD. It is thought that IL-10 –1082 A genotype is risk genotype for AD development considering that production of IL-10 is significantly decreased in carriers of the IL-10 –1082 A genotype [12, 13]. In addition, IL-6 –174 C [10], IL-1α –889 C [14, 15], and IL-1β –1473 G genotypes are associated with decrease in levels of these pro-inflammatory cytokines and are considered to be protective against AD, while the reverse was observed for TNFα –308A/G [9, 16–18]. The association of these SNPs with AD has been mostly tested in epidemiological studies by comparison of genotype distribution between AD patients and healthy controls (HC). Only a few studies measured levels of cerebrospinal fluid (CSF) AD biomarkers in patients with IL-10 –1082G/A, IL-1β –1473C/G, IL-1α –889C/T, IL-6 –174C/G, and TNFα –308A/G genotypes [19, 20]. CSF AD biomarkers such as amyloid β1-42 (Aβ1-42), total tau (t-tau), tau phosphorylated at amino acids Thr 181 (p-tau181), Ser 199 (p-tau199), and Thr 231 (p-tau231), and visinin-like protein 1 (VILIP-1) serve as endophenotypes of AD, as they reflect AD-related pathology [21]. CSF Aβ1-42 [22] and phosphorylated tau proteins [23] are indicators of senile plaques and neurofibrillary tangles in the brain, respectively, while CSF VILIP-1 and t-tau reflect neurodegeneration [24, 25]. Here we assessed possible differences in the levels of CSF AD biomarkers (Aβ1-42, t-tau, p-tau181, p-tau199, p-tau231, and VILIP-1) among patients with different IL-10 –1082G/A, IL-1β –1473C/G, IL-1α –889C/T, IL-6 –174C/G, and TNFα –308A/G genotypes to test whether people carrying certain genotypes are more prone to develop AD-related pathology as reflected by their levels of CSF biomarkers.
MATERIALS AND METHODS
Study population
This study included 115 AD patients, 53 mild cognitive impairment (MCI) patients, and 11 HC (Table 1). All patients were recruited at the Clinical Hospital Center Zagreb. They gave informed consent for participation in this study and for lumbar puncture. Patients’ cognitive status was tested using a battery of neuropsychological tests, including the Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-Cog), Montreal Cognitive Assessment (MoCA), and Mini-Mental State Examination (MMSE) [26]. In addition to neuropsychological testing, complete blood tests (levels of folic acid (B9), vitamin B12, thyroid function test, serology for Lyme’s disease and syphilis) and a full neurological examination were done. Dementia due to AD was diagnosed by using the National Institutes on Aging–Alzheimer’s Association (NIA-AA) criteria of McKhann et al. [27], while for MCI diagnosis the criteria of Albert et al. [28] were used. All procedures were in accord with the Helsinki Declaration [29] and were approved by the Ethical Committee of the Clinical Hospital Centre Zagreb (case no. 02/21 AG, class 8.1-18/82-2 from April 24, 2018) and by the Central Ethical Committee of the University of Zagreb Medical School (case no. 380-59-10106-18-111/126, class 641-01/18-02/01 from June 20, 2018).
Levels of Aβ1-42, t-tau, p-tau181, p-tau199, p-tau231, and VILIP-1 in AD, MCI patients, and HC
Aβ1-42, amyloid β1–42 protein; AD, Alzheimer’s disease; HC, healthy control; MCI, mild cognitive impairment; p-tau181, tau protein phosphorylated at threonine 181; p-tau231, tau protein phosphorylated at threonine 231; p-tau199, tau protein phosphorylated at serine 199; SD, standard deviation; t-tau, total tau; VILIP-1, visinin-like protein 1.
DNA analysis
Plastic syringes with 1 ml of acid citrate dextrose as an anticoagulant were used for collection of venous blood. DNA was isolated from the peripheral blood by the salting-out method [30]. SNPs were determined using TaqMan SNP Genotyping Assays (Applied Biosystems) by ABI Prism 7300 Real Time PCR System apparatus (Applied Biosystems, Foster City, CA). Following polymorphisms were determined; IL-1α –889C/T (rs1800587), IL-1β –1473G/C (rs1143623), IL-6 –174C/G (rs1800795), IL-10 –1082G/A (rs1800896), and TNFα –308A/G (rs1800629).
Analysis of CSF biomarkers
CSF was collected by lumbar puncture between intervertebral spaces L3/L4 or L4/L5. CSF was centrifuged at 2,000 g for 10 min, aliquoted and stored at –80°C in polypropylene tubes. Levels of CSF biomarkers were determined by following enzyme-linked immunosorbent assays (ELISA): VILIP-1 (VILIP-1 Human ELISA, BioVendor, Brno, Czech Republic), t-tau (Innotest hTau Ag, Fujirebio, Gent, Belgium), Aβ1-42 (Innotest β-amyloid1–42, Fujirebio), p-tau181 (Innotest Phospho-Tau [181P], Fujirebio), p-tau199 (TAU [pS199] Phospho-ELISA Kit, Human, Thermo Fisher Scientific Waltham, MA), and p-tau231 (Tau [pT231] Phospho-ELISA Kit, Human, Thermo Fisher Scientific,).
Statistical analysis
Data normality was tested using the Kolmogorov–Smirnov test. However, because of the small number of subjects in some groups, non-parametric statistics were used regardless of the results of the test for normality. Levels of CSF biomarkers were compared among groups using the non-parametric Kruskal-Wallis test. Pairwise comparisons were done by post-hoc non-parametric test with calculation of the corrected p value. All statistical analyses were done in SPSS 19.0.1 (SPSS, Chicago, IL, USA). The level of statistical significance was set at α= 0.05. Comparison of the levels of CSF biomarkers between groups with different SNPs was conducted separately in AD subjects, MCI patients, a mixed group of AD, MCI, and HC subjects, as well as in a mixed group of AD patients and MCI patients with pathological levels of CSF biomarkers (pCSF MCI group) and also in mixed group of HC subjects and MCI patients with normal levels of CSF biomarkers (nCSF MCI group). Cut-off levels of CSF biomarkers are reported in Babić Leko et al. [31]. MCI patients with at least one pathological CSF biomarker were pooled into the pCSF MCI group. Only statistically significant associations were reported. Analysis of genotype and allele frequencies between groups was done using χ2-test.
RESULTS
There was no significant deviation from the Hardy–Weinberg distribution in subjects carrying any of analyzed genotypes [IL-1α –889 (χ2 = 0.120; df = 1; p = 0.729), IL-1β –1473 (χ2 = 0.150; df = 1; p = 0.699), IL-10 –1082 (χ2 = 0.597; df = 1; p = 0.439), IL-6 –174 (χ2 = 0.501; df = 1; p = 0.479), TNFα –308 (χ2 = 0.009; df = 1; p = 0.921)]. No association between IL-1α –889C/T (rs1800587) polymorphism and CSF biomarkers was detected in any of the analyzed groups.
Levels of CSF AD biomarkers (Aβ1-42, t-tau, p-tau181, p-tau199, p-tau231, and VILIP-1) in AD, MCI patients, and HC with different IL-1α –889C/T, IL-1β –1473C/G, IL-6 –174C/G, IL-10 –1082G/A, and TNFα –308A/G genotypes are presented in Figs. 1 and 2, and in Table 1.
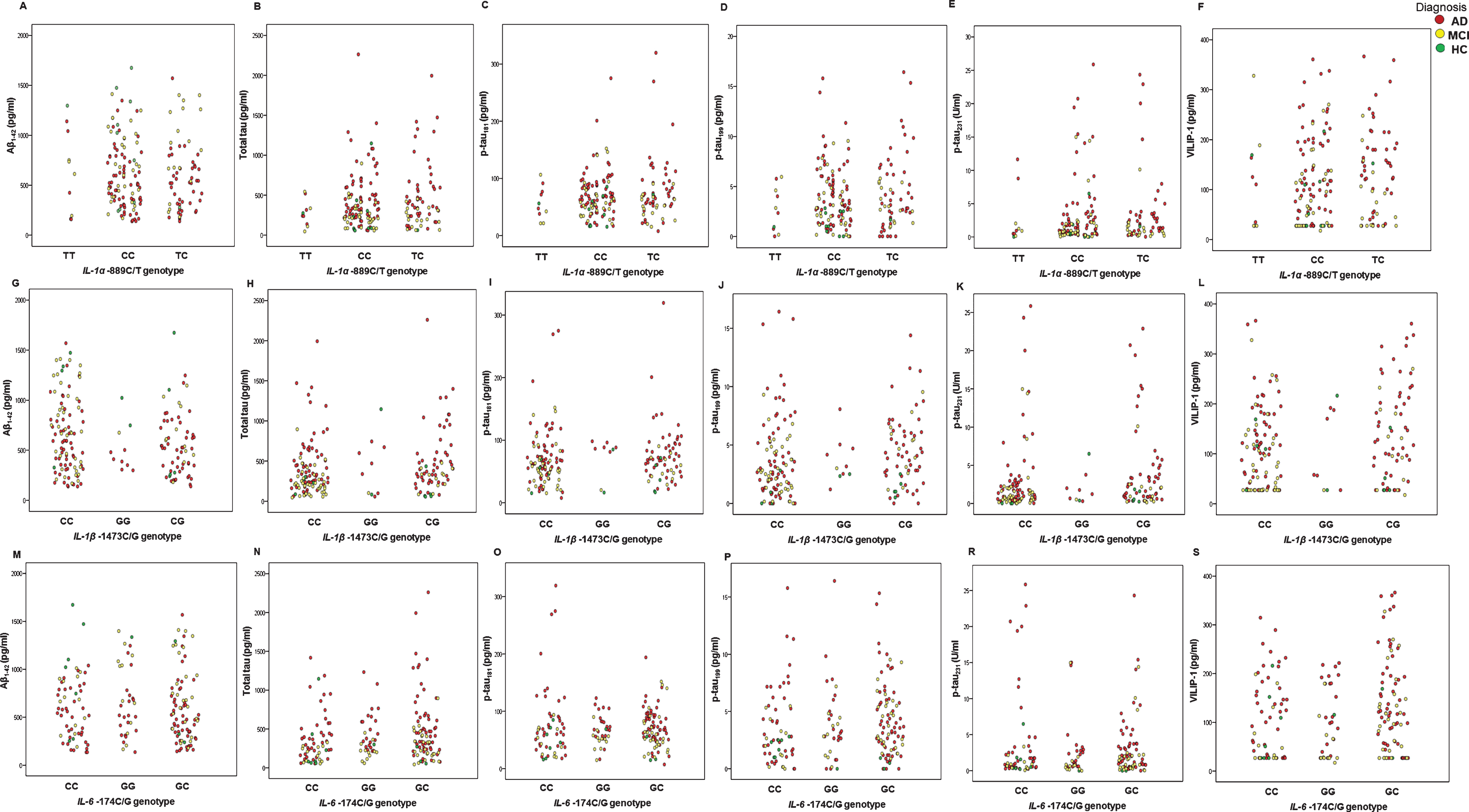
Levels of CSF AD biomarkers (Aβ1-42, t-tau, p-tau181, p-tau199, p-tau231, and VILIP-1) in AD, MCI patients, and HC with different IL-1α –889C/T, IL-1β –1473C/G, and IL-6 –174C/G genotypes.
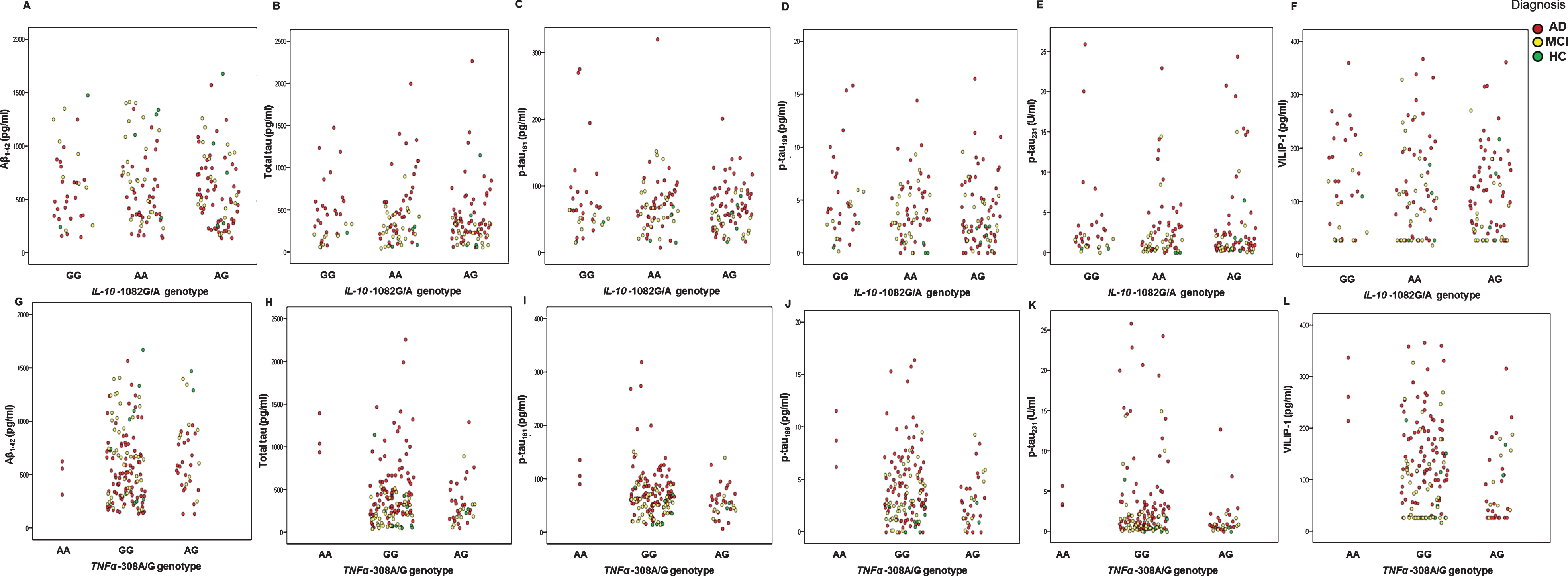
Levels of CSF AD biomarkers (Aβ1-42, t-tau, p-tau181, p-tau199, p-tau231 and VILIP-1) in AD, MCI patients, and HC with different IL-10 –1082G/A, and TNFα –308A/G genotypes.
Genotype and allele frequencies of these polymorphisms in AD, MCI patients, and HC are presented in Table 2. Even though we had a small cohort, we observed that the number of CC IL-1β –1473 homozygotes was significantly increased among MCI patients (p = 0.013).
Genotype and allele frequencies of IL-1α –889C/T, IL-1β –1473C/G, IL-6 –174C/G, IL-10 –1082G/A, and TNFα –308A/G gene polymorphisms in AD, MCI patients, and HC
AD, Alzheimer’s disease; HC, healthy control; IL, interleukin; MCI, mild cognitive impairment; N, number of patients; TNFα, tumor necrosis factor α. *p < 0.05.
IL-10 –1082G/A (rs1800896)
p-tau181 levels were significantly different between MCI patients with different IL-10 –1082 genotype (H test = 7.183, df = 2, p = 0.028). There was an increase in p-tau181 levels in MCI patients with AA compared to AG IL-10 –1082 genotype (Kruskal-Wallis [K-W]) post hoc p = 0.050) (Fig. 3). P-tau181 levels were also increased in patients with AA compared to GG and AG IL-10 –1082 genotype (MCI patients: U = 182, Z = –2.680, p = 0.007, Fig. 3).
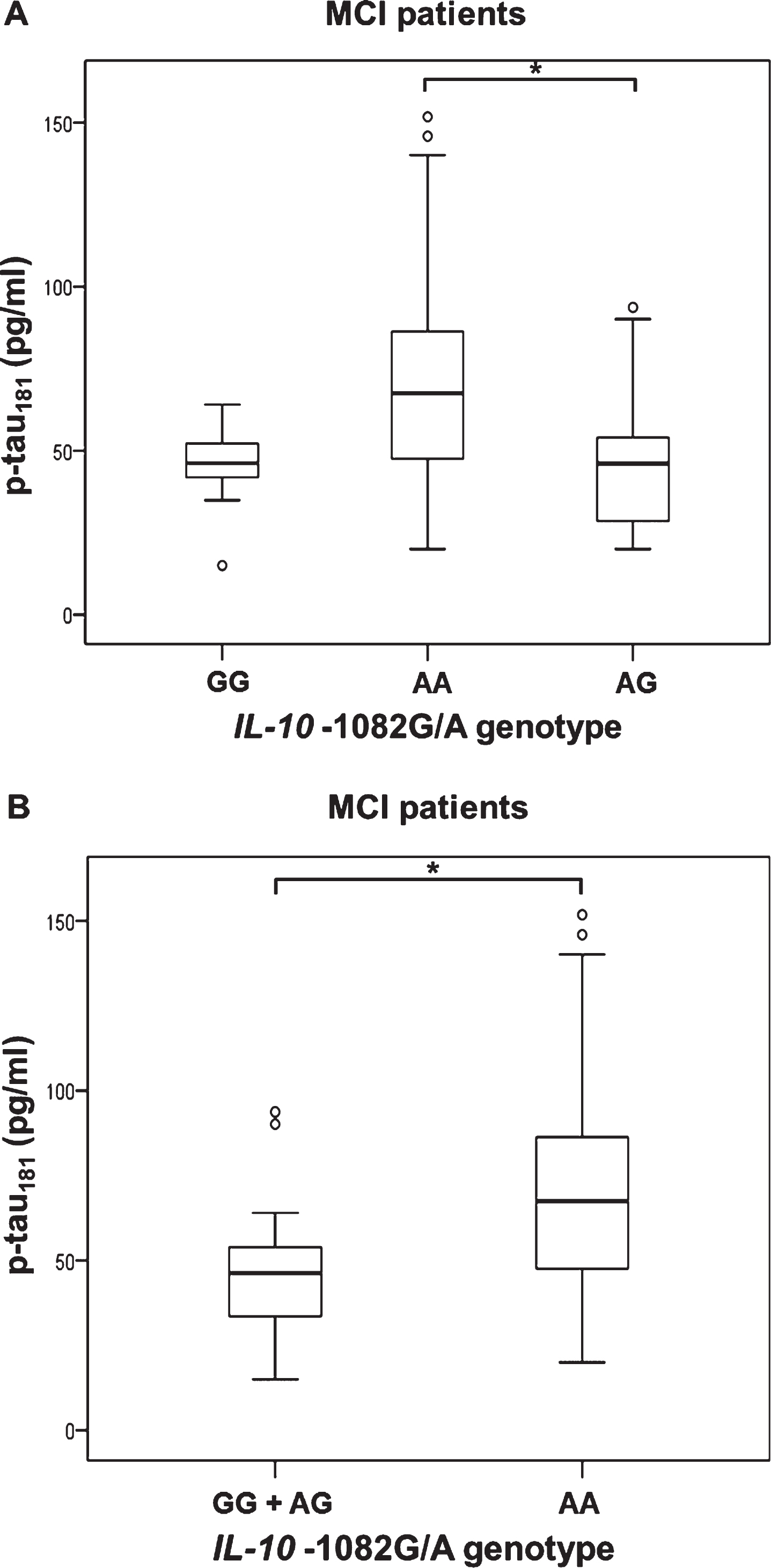
Levels of p-tau181 in (A, B) MCI patients with different IL-10 –1082G/A (rs1800896) genotypes; A) *p = 0.050, B) *p = 0.007.
IL-1β –1473C/G (rs1143623)
T-tau (U = 2272.5, Z=–2.324, p = 0.020) and VILIP-1 (U = 2177, Z = –2.150, p = 0.032) levels were significantly increased in AD and pCSF MCI patients with CG and GG genotype compared to patients with CC IL-1βI –1473 genotype (Fig. 4). P-tau199 levels were significantly increased in patients with CG and GG compared to CC IL-1β –1473 genotype (MCI patients: U = 150.5, Z = –2.177, p = 0.029; AD and MCI pCSF patients: U = 2253.5, Z = –2.297, p = 0.022, group of AD, all MCI patients and HC combined: U = 3099.5, Z = –2.248, p = 0.025, Fig. 4). p-tau231 levels were also significantly increased in patients with CG and GG compared to CC IL-1β –1473 genotype (AD, all MCI patients, and HC combined: U = 3046, Z = –2.087, p = 0.037; nCSF MCI patients and HC: U = 35, Z = –2.117, p = 0.035, Fig. 4).
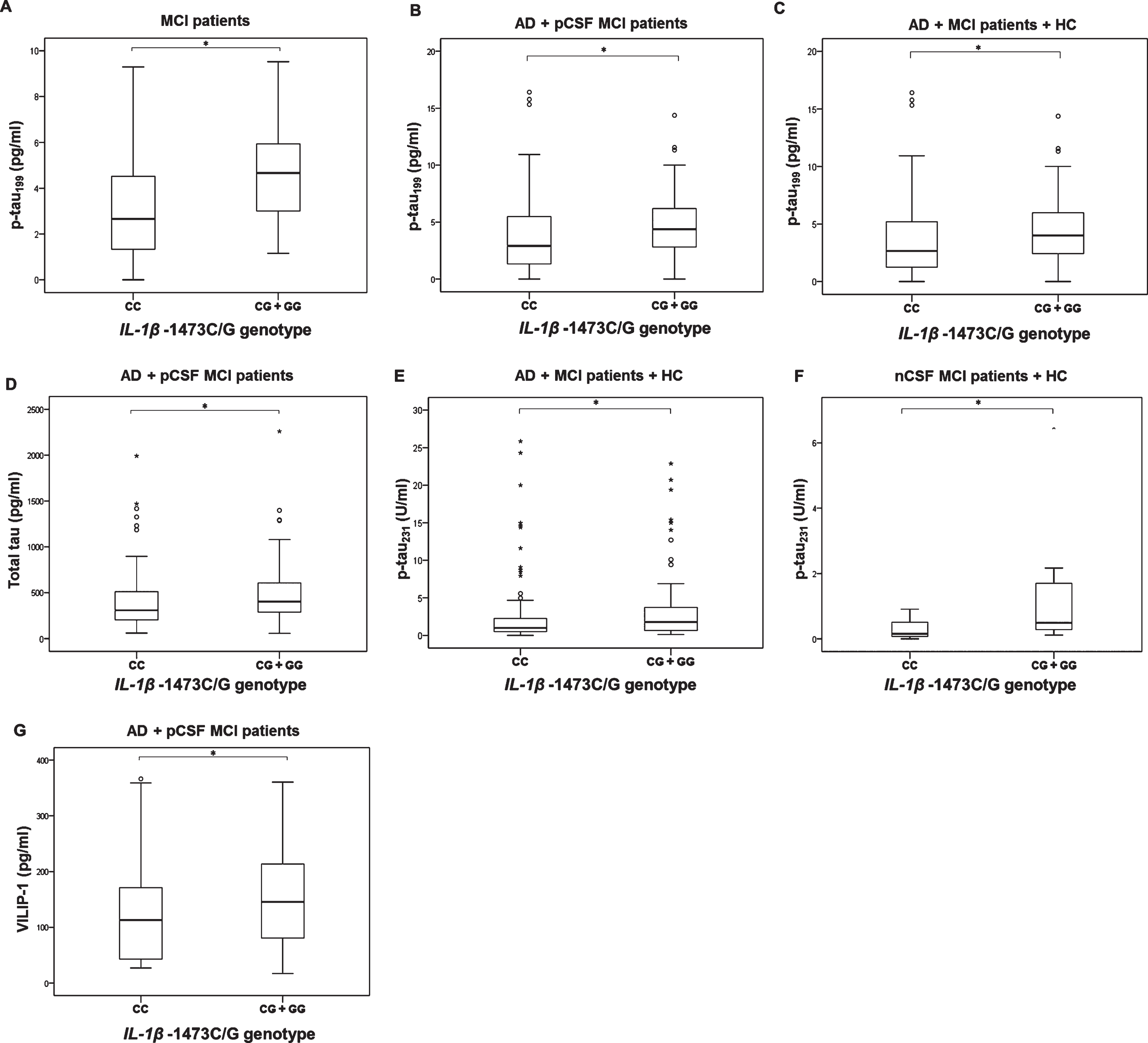
P-tau199 (A–C), t-tau (D), p-tau231 (E,F), and VILIP-1 (G) levels in subjects with different IL-1β –1473C/G (rs1143623) genotypes; A) *p = 0.029, B) *p = 0.022, C) *p = 0.025, D) *p = 0.020, E) *p = 0.037, F) *p = 0.035, G) *p = 0.032.
IL-6 –174C/G (rs1800795)
P-tau199 levels were increased in MCI patients (U = 156.5, Z = –2.050, p = 0.040) with GG and GC compared to CC IL-6 –174 genotype (Fig. 5). VILIP-1 levels were also significantly different in MCI patients with different IL-6 –174 genotype (H test = 6.695, df = 2, p = 0.035). There was an increase in VILIP-1 levels in MCI patients with GC compared to GG IL-6 –174 genotype (K-W post hoc p = 0.039; Fig. 5).
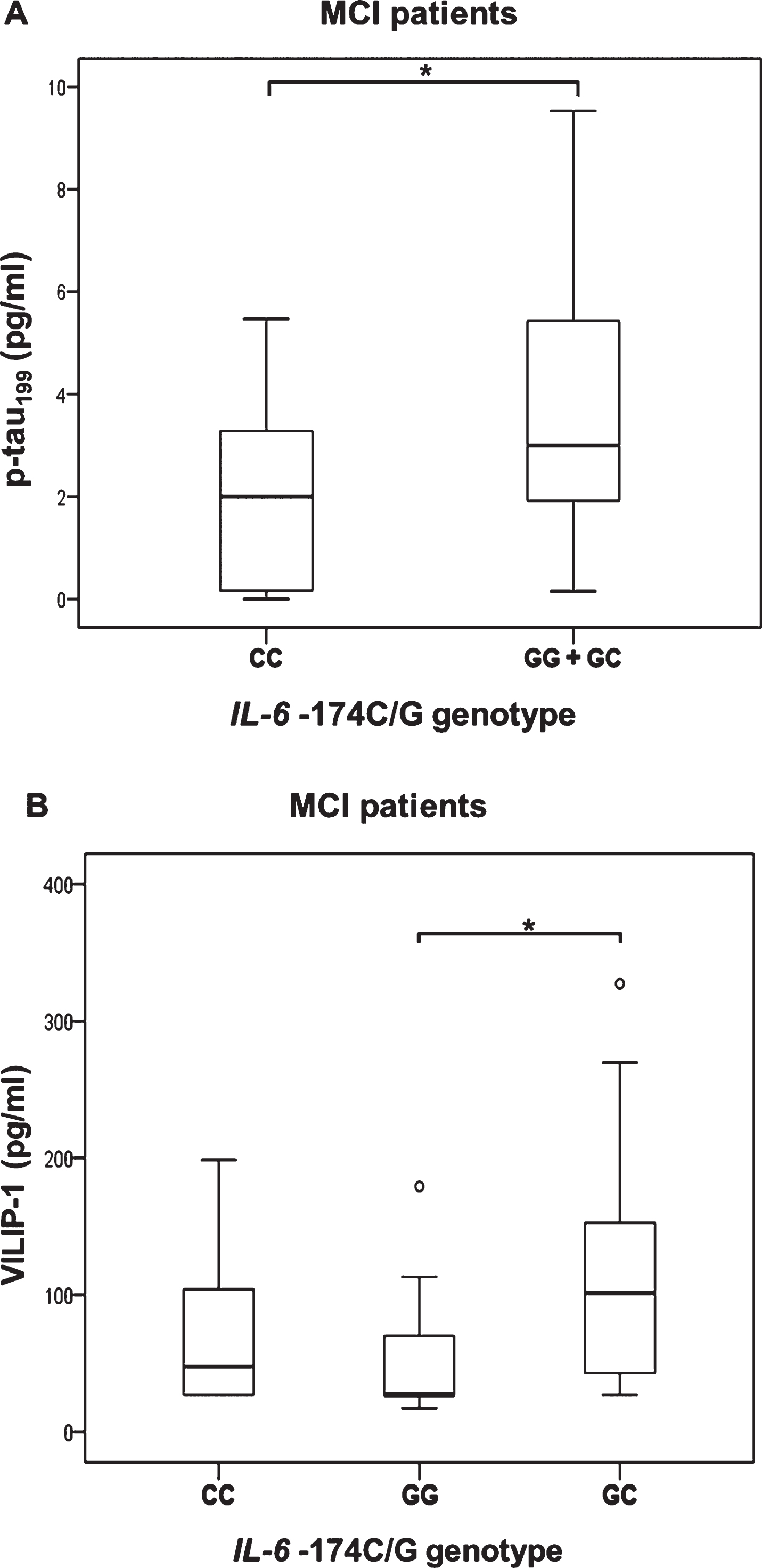
P-tau199 (A) and VILIP-1 (B) levels in subjects with different IL-6 –174C/G (rs1800795) genotypes; A) *p = 0.040, B) *p = 0.039.
TNFα –308A/G (rs1800629)
As only three AD patients were carriers of AA TNFα –308 genotype (Table 2), these patients were grouped together with carriers of AG TNFα –308 genotype. P-tau231 (U = 805.5, Z = –2.220, p = 0.026) and VILIP-1 (U = 762.5, Z = –2.517, p = 0.012) levels were significantly increased in AD patients with GG compared to AA and AG TNFα –308 genotype (Fig. 6). Additionally, p-tau231 levels were significantly increased in patients with GG compared to AG TNFα –308 genotype (in AD, MCI patients, and HC combined, K-W post hoc p = 0.038), in AD and pCSF MCI patients (K-W post hoc p = 0.024), and in AD patients (K-W post hoc p = 0.015*); Fig. 7, Table 3). VILIP-1 levels were also significantly increased in AD patients with GG compared to AG TNFα –308 genotype (K-W post hoc p = 0.002; Fig. 7, Table 3). Levels of t-tau, p-tau181, p-tau199, p-tau231, and VILIP-1 were significantly increased in patients with AA compared to AG TNFα –308 genotype in all patients (when all subjects were grouped together, in AD, MCI patients, and HC combined, in AD and pCSF MCI patients combined, and in AD patients), while levels of t-tau and VILIP-1 were increased in patients with AA compared to GG TNFα –308 genotype (when all subjects were grouped together and in AD, MCI patients and HC; Table 3, Fig. 8). The three AD patients who were carriers of AA TNFα –308 genotype, could not be evaluated separately and should be validated in a larger of population.
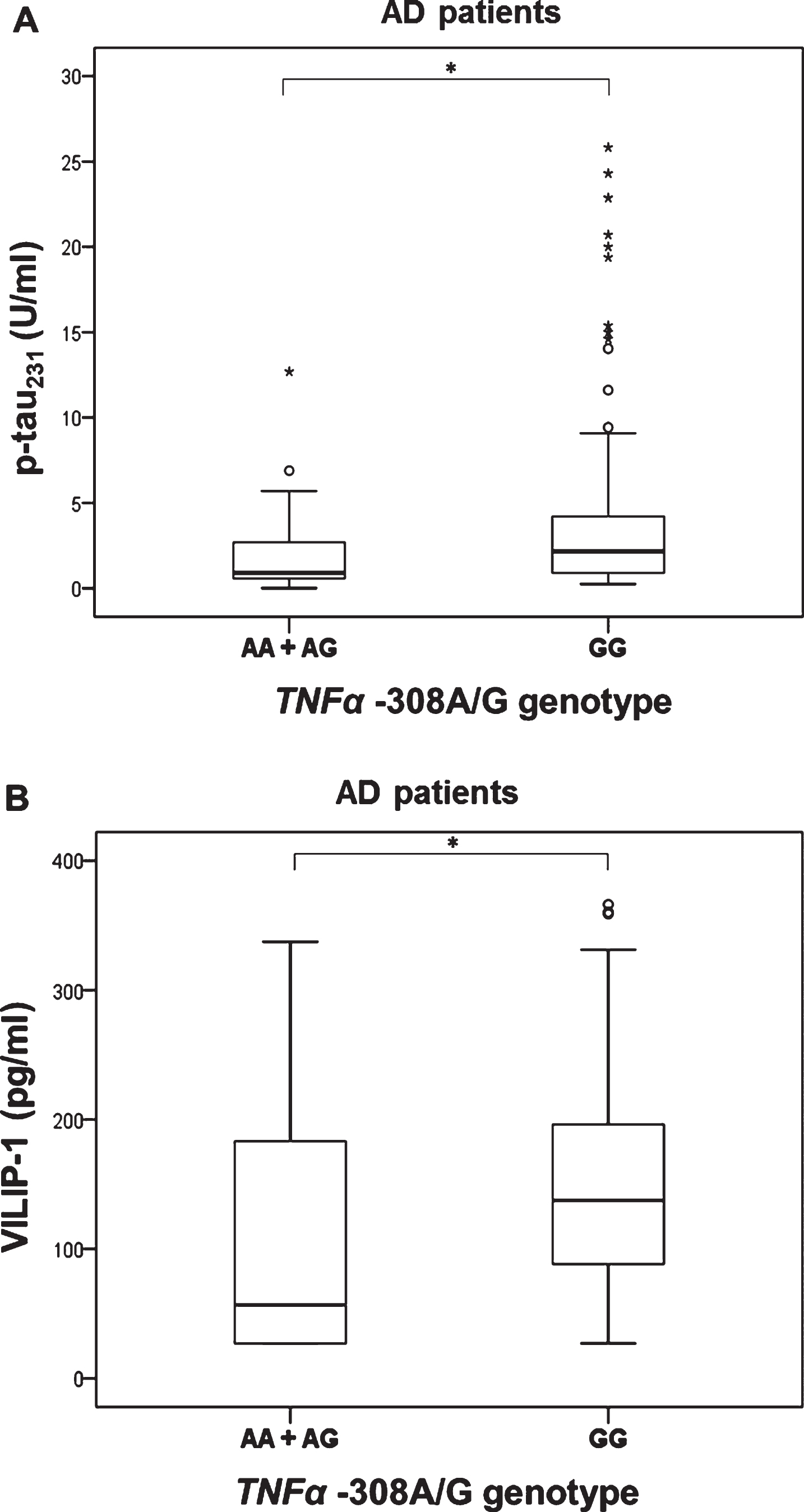
Levels of p-tau231 (A) and VILIP-1 (B) in AD patients with different TNFα –308A/G (rs1800629) genotypes; A) *p = 0.026, B) *p = 0.012.
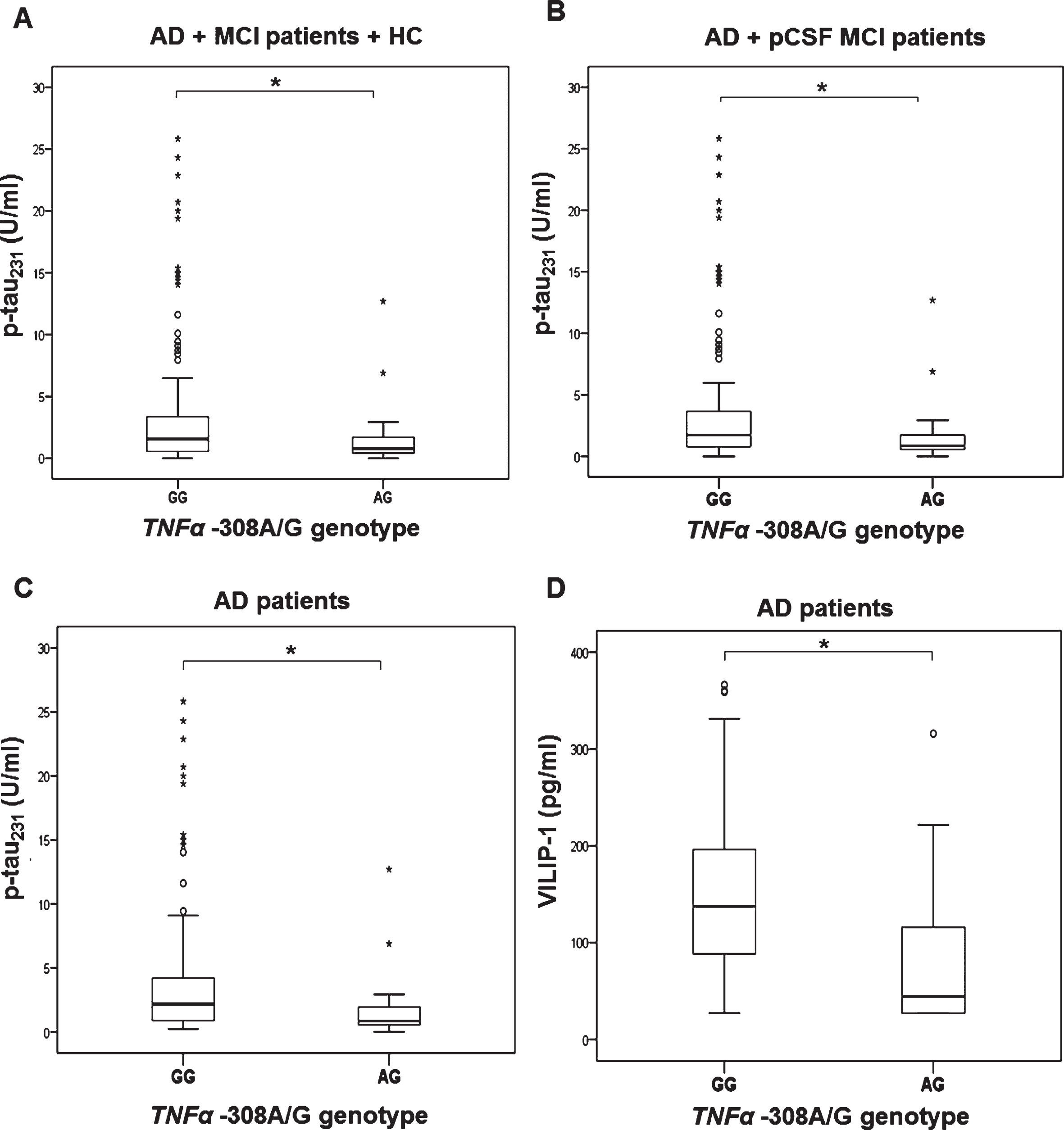
Levels of p-tau231 (A–C) and VILIP-1 (D) in subjects with different TNFα –308A/G (rs1800629) genotypes; A) *p = 0.038, B) *p = 0.024, C) *p = 0.015, D) *p = 0.002.

Levels of Aβ1-42 (A), t-tau (B), p-tau181(C), p-tau199 (D), p-tau231 (E), and VILIP-1 (F) in AD patients with different TNFα –308A/G (rs1800629) genotypes; A) *p = 0.014, B) *p = 0.029, C) *p = 0.017, D) *p = 0.015, E) *p = 0.003, 0.002.
Comparison of Aβ1-42, t-tau, p-tau181, p-tau199, p-tau231 and VILIP-1 levels in different groups of patients with TNFα –308A/G (rs1800629) genotypes
Aβ1-42, amyloid β1–42 protein; AD, Alzheimer’s disease; CSF, cerebrospinal fluid; HC, healthy control; KW, Kruskal-Wallis test; MCI, mild cognitive impairment; PH KW, Kruskal-Wallis post hoc; p-tau181, tau protein phosphorylated at threonine 181; p-tau231, tau protein phosphorylated at threonine 231; p-tau199, tau protein phosphorylated at serine 199; pCSF, pathological CSF; TNFα, tumor necrosis factor alpha; VILIP-1, visinin-like protein 1. *p < 0.05.
DISCUSSION
Few studies have investigated whether levels of CSF AD biomarkers differ among patients with different IL-10 –1082G/A, IL-1β –1473C/G, IL-1α –889C/T, IL-6 –174C/G, and TNFα –308A/G genotypes that were previously associated with AD [19, 20]. We compared the levels of six AD CSF biomarkers (Aβ1-42, t-tau, p-tau181, p-tau199, p-tau231, and VILIP-1) among patients with aforementioned genotypes. This study gave several notable findings. Levels of t-tau were increased in carriers of G allele in IL-1β –1473C/G polymorphism. p-tau levels were significantly increased in patients with AA IL-10 –1082G/A and GG TNFα –308A/G genotype, and in carriers of G allele in IL-1β –1473C/G and IL-6 –174C/G polymorphisms. Levels of VILIP-1 were increased in patients with CG and GG IL-1β –1473C/G, GC IL-6 –174C/G, and GG TNFα –308A/G genotype.
SNPs in genes for IL-1α, IL-1β, IL-6, IL-10, and TNFα can influence transcription and consequently the amount of the produced cytokines [9–11]. Decrease in the amount of anti-inflammatory cytokines and increase in pro- inflammatory cytokines results in increased inflammation, favoring the development of AD [32]. In that way certain genotypes in these SNPs (IL-10 –1082G/A, IL-1β –1473C/G, IL-1α –889C/T, IL-6 –174C/G, and TNFα –308A/G) can make some people more vulnerable to the development of neuroinflammation and consequently the development of AD. Given that the production of IL-10 is significantly decreased in carriers of the IL-10 –1082 A genotype [12,13, 12,13], a decrease in anti-inflammatory cytokine IL-10 levels could result in increased inflammation, favoring the development of AD [32]. It was found that the C IL-6 –174 allele is associated with decrease in IL-6 plasma levels [10] so this genotype could be protective against AD. TNFα being a main pro-inflammatory cytokine, its higher production is associated with increased inflammation and AD progression. TNFα inhibitors have been suggested as potential therapeutics for AD [33]. The influence of TNFα –308 polymorphism on TNFα protein production, however, remains unclear. Most studies reported that the A TNFα –308 allele is associated with increased production of TNFα [9, 17], while some studies did not find differences in TNFα protein levels in patients with different TNFα –308 genotypes [18]. Regarding polymorphisms in additional pro-inflammatory cytokines IL-1α and IL-1β that were also tested in this study, it was showed that T allele in the IL-1α –889 polymorphism was associated with increased transcriptional activity in IL-1α gene and overexpression of IL-1α protein [14, 15], while G allele in IL-1β –1473 polymorphism was associated with weaker promoter activity [34]. Our results support most of these studies, because we observed pathological levels of CSF AD biomarkers in carriers of A allele in IL-10 –1082 polymorphism, carriers of G allele in IL-6 –174 polymorphism and carriers of A allele in TNFα –308 polymorphism. However, regarding polymorphisms in genes for IL-1α and IL-1β, our results differed from aforementioned studies. CSF AD biomarkers did not differ between patients with different IL-1α –889 genotypes, while levels of CSF AD biomarkers were pathological in carriers of G allele in IL-1β –1473 polymorphism.
IL-10 –1082G/A (rs1800896), IL-1β –1473C/G (rs1143623), IL-1α –889C/T (rs1800587), IL-6 –174C/G (rs1800795), and TNFα –308A/G (rs1800629) polymorphisms were previously associated with AD in epidemiological studies. Studies on association of IL-10 –1082G/A polymorphism and AD yielded inconsistent results. Associations between the A allele in IL-10 –1082 polymorphism and increased risk for AD or the G allele and decreased risk for AD have been reported [11, 35–41]. However, other investigators found no association between IL-10 –1082 polymorphism and AD [42–50] or showed GG IL-10 –1082 genotype to be significantly increased in AD patients [51] and AA IL-10 –1082 genotype to decrease the risk for AD [52]. Meta-analyses revealed an association between IL-10 –1082 AA and AG genotype and increased risk for AD [53], and an association between IL-10 –1082 GG genotype and reduced risk for AD [54]. However, the meta-analysis of Mun et al. found no association between IL-10 –1082 polymorphism and AD risk [8]. Our results agree with studies showing association between IL-10 –1082 A genotype and increased risk for AD [11, 35–41].
Cytokine IL-1β is likely involved in cognitive decline related to inflammation [55]. As such, polymorphisms in IL-1β were studied to assess possible association with AD (for example, IL-1β –511, IL-1β –31, and IL-1β+3953 polymorphisms [8, 56–58]). Association of IL-1β –1473G/C polymorphism with AD was assessed in only two studies. There was no significant difference in distribution of IL-1β –1473 genotypes between AD patients and controls [59, 60]. In contrast to these studies, we observed levels of various CSF AD biomarkers to be altered in subjects with different IL-1β –1473 genotypes. Our results indicate that IL-1β –1473 polymorphism may represent a susceptibility marker of AD and that the frequency of IL-1β –1473 genotypes should be further tested on larger AD and MCI cohorts.
The association of IL-6 –174C/G polymorphism with AD is ambiguous. Some studies found an association between a C allele in IL-6 –174 polymorphism and decreased risk for AD [61–68], while others found no association between the IL-6 –174 polymorphism and AD [45, 69–78]. Additionally, some studies found the C allele in the IL-6 –174 polymorphism to be associated with increased risk for AD [36, 79–81]. Meta-analyses testing association of IL-6 –174 polymorphism with AD also returned inconsistent results. Dai et al. [82] and Qi et al. [83] showed the CC IL-6 –174 genotype to be associated with decreased risk for AD, while Mun et al. showed that the IL-6 –174 polymorphism is not associated with AD [8]. Our results support studies showing that the CC IL-6 –174 genotype is associated with a decreased risk for AD [61–68, 83].
Studies on association of pro-inflammatory IL-1α cytokine brain overexpression with AD [84] showed that the presence of a T allele in the IL-1α –889 polymorphism is associated with an increased risk for AD [85–94]. Other studies, however, did not report an association between this polymorphism and AD [46, 95–114]. Yet, meta-analyses demonstrated that an association between the IL-1α –889 polymorphism and AD exists [8, 115–117]. Our study found no association between this polymorphism and CSF biomarkers in any of the analyzed groups.
Variable results were also obtained from investigations of the association between the TNFα –308A/G polymorphism and AD. Several studies indicate that presence of the A allele in the TNFα –308 polymorphism increases the risk for AD [44, 118–120], while others found no association between this polymorphism and AD [18, 121–126]. Other authors suggested that the A allele in the TNFα –308 polymorphism is protective against AD [20, 128]. Meta-analyses also gave inconsistent results. Furthermore, Di Bona et al. [129] did not confirm the association between TNFα –308 polymorphism and AD. The meta-analysis of Lee et al. [7] showed that the A allele in the TNFα –308 polymorphism may be a risk factor for AD in East Asians, but not in Middle Easterners and Europeans. Wang [130] confirmed that the A allele increases risk for AD in Asians but decreases risk in Northern Europeans. Our study included only three AD patients with the AA TNFα –308 genotype. These three patients had pathological levels of all examined CSF AD biomarkers, except for Aβ1–42 (Table 3). This result, however, remains inconclusive due to the small sample. We also detected pathological levels of CSF AD biomarkers in patients with the GG TNFα –308 genotype. The levels of CSF AD biomarkers in patients with different TNFα –308 genotypes were also investigated by Sarajärvi et al. [20] and Laws et al. [19]. Although the genetic analysis of Sarajärvi et al. [20] showed that A allele carriers are less susceptible for AD than GG homozygotes, their analysis of biomarkers in patients with different TNFα –308 genotypes revealed that levels of Aβ1–42 were pathological in carriers of an A TNFα –308 allele compared to GG homozygotes [20]. This contrasts with our study as we detected pathological CSF levels of p-tau231 and VILIP-1 in GG homozygotes in comparison to carriers of an A TNFα –308 allele, and we found no differences in CSF Aβ1–42 levels between patients with different TNFα –308 genotype. The findings of Laws et al. support our results [19]. Although the results of our previous genetic study [126] showed no significant difference in distribution of TNFα –308 genotypes between AD patients and HC, in the present study we detected pathological levels of CSF p-tau231 and VILIP-1 in AD patients with the GG compared to AG TNFα –308 genotypes. Other groups also did not detect a difference in distribution of TNFα –308 genotypes between AD patients and HC, but observed difference in distribution of haplotypes (that include the TNFα –308 polymorphism) between AD patients and HC [128, 131]. Thus, the scope of our next study should be analysis of TNFα haplotypes’ distribution between AD patients and HC. Our study suggests that heterozygosity in TNFα –308 polymorphism could be protective against AD, as pathological levels of CSF AD biomarkers were detected in both AA and GG TNFα –308 homozygotes. This deserves further validation.
IL-1α, IL-1β, IL-6, IL-10, and TNFα were also studied as potential biomarkers of AD. However, the results on measurement of these and other inflammatory markers in body fluids were inconsistent [132]. Thus, recently a lot of meta-analyses were conducted with purpose to determine the potential of inflammatory markers as biomarkers of AD. The increase in IL-6 was associated with all-cause dementia, but not AD in meta-analyses of Darweesh et al. [133] and Koyama et al. [134]. Additional meta-analyses observed increase in peripheral IL-6, IL-1β [135–137], and TNF-α [137] in AD patients compared to HC. However, meta-analyses of Saleem et al. [138] and Su et al. [136] observed no significant difference in inflammatory markers between MCI patients and HC. Brosseron et al. [132] divided inflammatory markers measured in body fluids into three groups by involvement in the disease: 1) cytokines unchanged during disease (like IL-1α), 2) cytokines that increase slightly but steadily during disease (like IL-1β, IL-6, and TNF-α), and 3) cytokines that have a peak when MCI converses to AD.
In conclusion, our study reveals altered levels of CSF AD biomarkers in carriers of different genotypes in IL-10 –1082A/G, IL-1β –1473C/G, IL-6 –174C/G, and TNFα –308A/G polymorphisms, while CSF AD biomarkers did not differ between patients with different IL-1α –889C/T genotypes. These polymorphisms as potential genetic biomarkers of AD should be further compared with CSF AD biomarkers on bigger cohort of patients and comparison with neuroimaging AD biomarkers should be also made. Additionally, it should be assessed whether different genotypes in these polymorphisms are the cause of the observed inconsistencies in the levels of these cytokines measured in body fluids, as well as their relationship to inflammasome and microglial activation [5]. Finally, the results of this study suggest a potential for AD therapeutics with emphasis on personalized medicine. As some genotypes in these SNPs (IL-10 –1082G/A, IL-1β –1473C/G, IL-6 –174C/G, and TNFα –308A/G) make some people more vulnerable to the development of neuroinflammation and of AD-related pathology, such patients represent potential candidates for targeted anti-inflammatory therapies in AD. As our sample was relatively small, statistical comparison of frequency of IL-1α –889C/T, IL-1β –1473C/G, IL-6 –174C/G, IL-10 –1082G/A, and TNFα –308A/G genotypes and alleles between AD, MCI patients, and HC should be further validated on a larger cohort.
Footnotes
ACKNOWLEDGMENTS
This work was funded by The Croatian Science Foundation grants IP-2014-09-9730 (“Tau protein hyperphosphorylation, aggregation, and trans-synaptic transfer in Alzheimer’s disease: cerebrospinal fluid analysis and assessment of potential neuroprotective compounds”) and IP-2019-04-3584 (“Role of blood-brain barrier, innate immunity, and tau protein oligomerization in the pathogenesis of Alzheimer’s disease“) to GŠ and by the Scientific Centre of Excellence for Basic, Clinical and Translational Neuroscience CORE-NEURO (“Experimental and clinical research of hypoxic-ischemic damage in perinatal and adult brain”; GA KK01.1.1.01.0007 funded by the European Union through the European Regional Development Fund), and in part by the NIH grant P50 AG005138 to PRH.