
Abstract
Background:
The pathogenesis of Alzheimer’s disease (AD) involves various immune-related phenomena; however, the mechanisms underlying these immune phenomena and the potential hub genes involved therein are unclear. An understanding of AD-related immune hub genes and regulatory mechanisms would help develop new immunotherapeutic targets.
Objective:
The aim of this study was to explore the hub genes and the mechanisms underlying the regulation of competitive endogenous RNA (ceRNA) in immune-related phenomena in AD pathogenesis.
Methods:
We used the GSE48350 data set from the Gene Expression Omnibus database and identified AD immune-related differentially expressed RNAs (DERNAs). We constructed protein–protein interaction (PPI) networks for differentially expressed mRNAs and determined the degree for screening hub genes. By determining Pearson’s correlation coefficient and using StarBase, DIANA-LncBase, and Human MicroRNA Disease Database (HMDD), the AD immune-related ceRNA network was generated. Furthermore, we assessed the upregulated and downregulated ceRNA subnetworks to identify key lncRNAs.
Results:
In total, 552 AD immune-related DERNAs were obtained. Twenty hub genes, including PIK3R1, B2M, HLA-DPB1, HLA-DQB1, PIK3CA, APP, CDC42, PPBP, C3AR1, HRAS, PTAFR, RAB37, FYN, PSMD1, ACTR10, HLA-E, ARRB2, GGH, ALDOA, and VAMP2 were identified on PPI network analysis. Furthermore, upon microRNAs (miRNAs) inhibition, we identified LINC00836 and DCTN1-AS1 as key lncRNAs regulating the aforementioned hub genes.
Conclusion:
AD-related immune hub genes include B2M, FYN, PIK3R1, and PIK3CA, and lncRNAs LINC00836 and DCTN1-AS1 potentially contribute to AD immune-related phenomena by regulating AD-related hub genes.
INTRODUCTION
Alzheimer’s disease (AD) is a common type of neurodegenerative disease with memory decline and disability. It is histopathologically characterized by the deposition of amyloid-β, neurofibrillary tangles, and loss of synapses [1–4]; however, its etiology and pathogenesis have remained unclear for a century. Current treatments based on the amyloid cascade hypothesis have not been able to control AD progression. Therefore, it is essential to confirm the molecular basis of AD to design new pharmacological targets.
Numerous recent studies have reported that brain immune responses are closely associated with numerous aspects of AD pathogenesis. Immune genes including TREM2, CR1, CD33, and MS4A have been discovered through genome-wide association studies [5–10]. For instance, TREM2 promotes AD pathogenesis by altering the inflammatory state of microglia [11]. Furthermore, the immune response is believed to be not only a downstream phenomenon but also the initiating factor in AD [12]. Immunosenescence caused by environmental, genetic, and other factors may result in downstream alterations in AD pathophysiology. Acute and chronic inflammatory processes play important roles in regulating APP expression or tau transmission [13–15]. Activated microglia release an apoptosis-associated speck-like protein containing a CARD (ASC) protein, which acts as an inflammation-driven cross-seed for the amyloid-β pathology [16]. Furthermore, loss of NLRP3 inflammasome function reduces tau hyperphosphorylation and aggregation by regulating tau kinases and phosphatases [17].
Immune-related pathological phenomena result from the synergistic effects of interacting genes or RNAs [18–20]. Therefore, an integrated analysis of protein-encoding genes and noncoding RNAs would help reveal the underlying immune mechanisms [21]. Long non-coding RNAs (lncRNAs) are involved in numerous biological phenomena including cell differentiation, immune response, and development [22], and serve as competitive endogenous RNAs (ceRNAs), competing with microRNAs (miRNAs) to bind to mRNAs, thus reducing miRNA-mediated repression of target genes [23]. However, it remains unclear whether ceRNA contributes to AD-related immune responses.
This study aimed to explore the hub genes and to establish a competitive regulatory network of lncRNA-miRNA-mRNA for AD-related immune phenomena. Postmortem hippocampal tissue of AD patients was used and hub genes and lncRNAs were screened for their involvement in immune phenomena.
METHODS
Data acquisition and annotation
The GSE48350 dataset (Affymetrix Human Genome U133 Plus 2.0 Array: Platform GLP570) was downloaded from NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) [24]. The dataset contained 253 brain tissue samples, from which 62 hippocampal samples were analyzed, including those from 19 AD patients and 43 normal controls (NC). Pathological analysis was performed for AD and NC subjects. The dataset provided information regarding the age and sex of subjects in the two groups (age: AD, 83.05±8.48 years; NC, 62.48±25.39 years; female sex: AD, 52.63%, NC, 46.51%). mRNAs and lncRNAs in the expression profiles were re-annotated in accordance with the HUGO Gene Nomenclature Committee (HGNC) database (http://www.genenames.org/) [25]. Screening was performed in accordance with the following criteria: 1) empty probes not matching any mRNA and lncRNA should be deleted; 2) probes matching both mRNA and lncRNA should be deleted; 3) for different probes matching the same mRNA or lncRNA, the average value should be considered the expression level of mRNA or lncRNA.
Differential expression analysis and acquisition of immune-related genes
Data analysis procedures are shown in Fig. 1. The robust multiarray averaging (RMA) method was used for background noise correlation and normalization. We used the R Limma package to screen differentially expressed RNAs (DERNAs) between AD and NC individual adjusted for age and sex. The screening thresholds were set at a false discovery rate (FDR) of <0.05 and |log2FC|>0.5. Genes involved in immune-related biological processes (BP) or pathways were identified using AmiGO2 (http://amigo.geneontology.org/amigo) and KEGG (https://www.kegg.jp/) databases, using the search term ‘immune’. [26, 27]
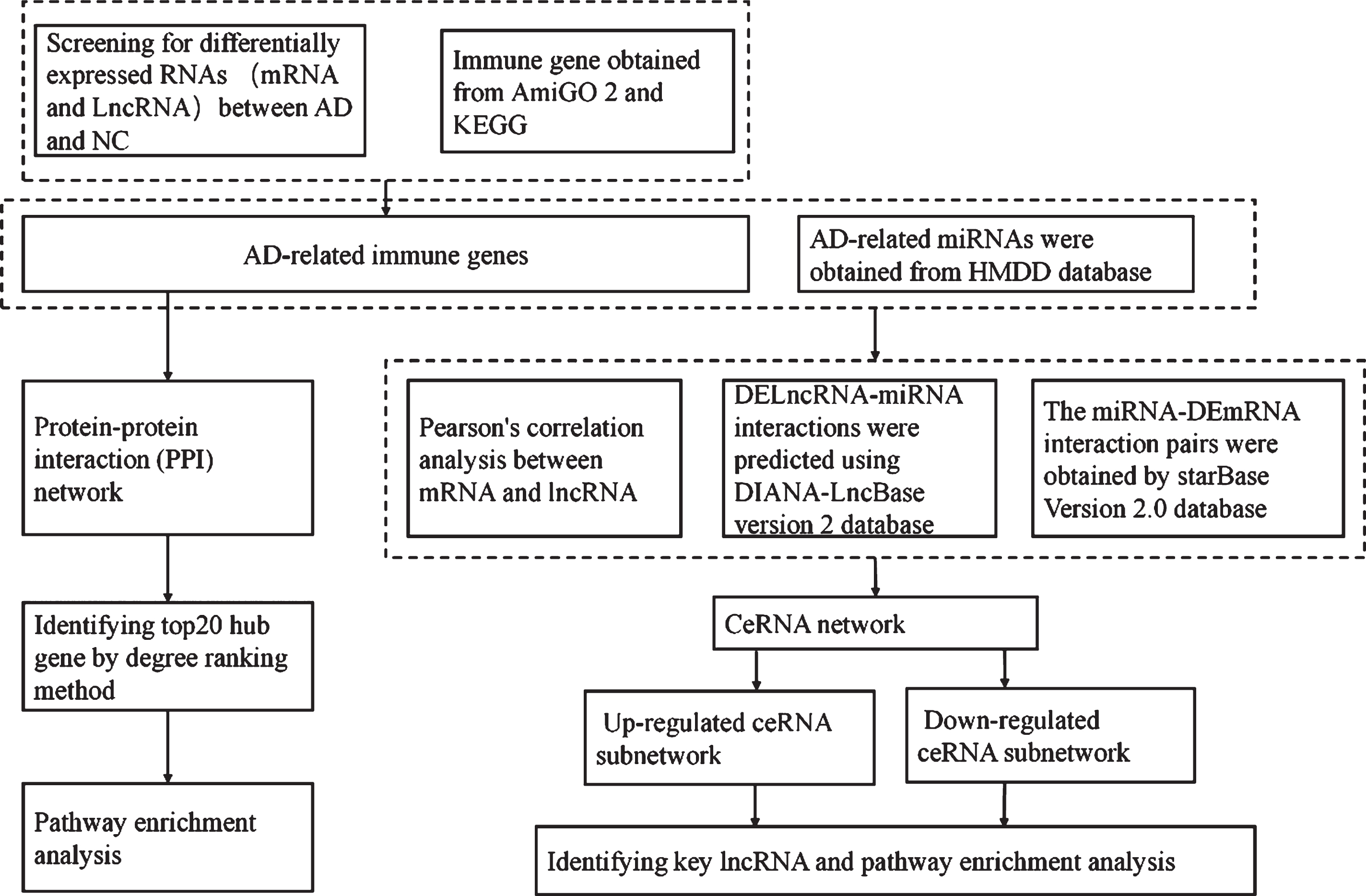
Schematic representation of the study protocol.
AmiGO2 is a web-based set of tools for searching and browsing Gene Ontology (GO) databases [26]. KEGG is a database resource for understanding high-level functions and utilities of biological systems [27]. The overlaps between DERNAs and these immune-related gene sets were considered to reflect AD-related immune genes.
Construction of the protein-protein interaction (PPI) network
We used the STRING database (http://string.embl.de/) to construct a PPI network [28]. STRING is a database of known and predicted protein-protein interactions. Interactions in STRING are derived from five main sources, including genomic context predictions, high-throughput lab experiments, (conserved) co-expression, automated text mining, and previous knowledge from databases. In this study, protein interaction pairs with a combined score of >0.9 were considered statistically significant and retained in the PPI network. The PPI network was visualized using Cytoscape v 3.6.0 software [29]. Furthermore, the top 20 hub genes were identified and ranked in accordance with their degree value, which, herein, refers to the number of nodes contacting other nodes in PPI networks, and the higher the degree value, the better the centrality of the node.
Pathway enrichment analysis
The Metascape is (http://metascape.org/) an online analytical tool used for gene annotation, functional enrichment, interactome analysis, and membership analysis [30]. In this study, Metascape was used for GO, KEGG, and reactome pathway analysis with an FDR of <0.01 as the cut-off value.
Construction of a ceRNA network
A ceRNA network reflects the associations among lncRNA, mRNA, and miRNA. Interaction pairs, including lncRNA-mRNA, lncRNA-miRNA, and miRNA-mRNA pairs, are necessary for constructing the network. Herein, we constructed a correlation matrix containing Pearson correlation coefficients of all lncRNA-mRNA pairs. AD-related miRNAs were identified from the Human MicroRNA Disease Database (HMDD; http://www.cuilab.cn/hmdd), which contains a number of miRNA-disease association entries from literature [31]. DELncRNA-miRNA interactions were predicted using DIANA-LncBase version 2 database (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php), which provided miRNA recognition elements (MREs) on lncRNAs supported by experimentally and in silico predicted evidence [32]. Finally, the starBase Version 2.0 database (http://starbase.sysu.edu.cn/), which records regulatory interaction networks among multiple classes of RNAs, was used to predict the target genes for AD-related miRNAs; thus, the miRNA-DEmRNA interaction pairs were ob-tained [33]. Overall, DElncRNA-miRNA-DEmRNA regulatory network was constructed and visualized using Cytoscape v 3.6.0 software.
RESULTS
Identification of AD immune-associated DEmRNAs and DElncRNAs
Eighteen DElncRNAs (nine significantly downregulated and nine significantly upregulated) and 1,334 DEmRNAs (889 significantly downregulated and 445 significantly upregulated) were identified with |log2FC|>0.5 and FDR < 0.05 (Fig. 2A, B and Supplementary Table 1). In total, 3,255 genes involved in immune-related phenomena were derived from the AmiGO2 and KEGG databases, respectively. Upon overlapping with DERNAs, 552 immune-related differentially expressed genes were identified (Fig. 2C).
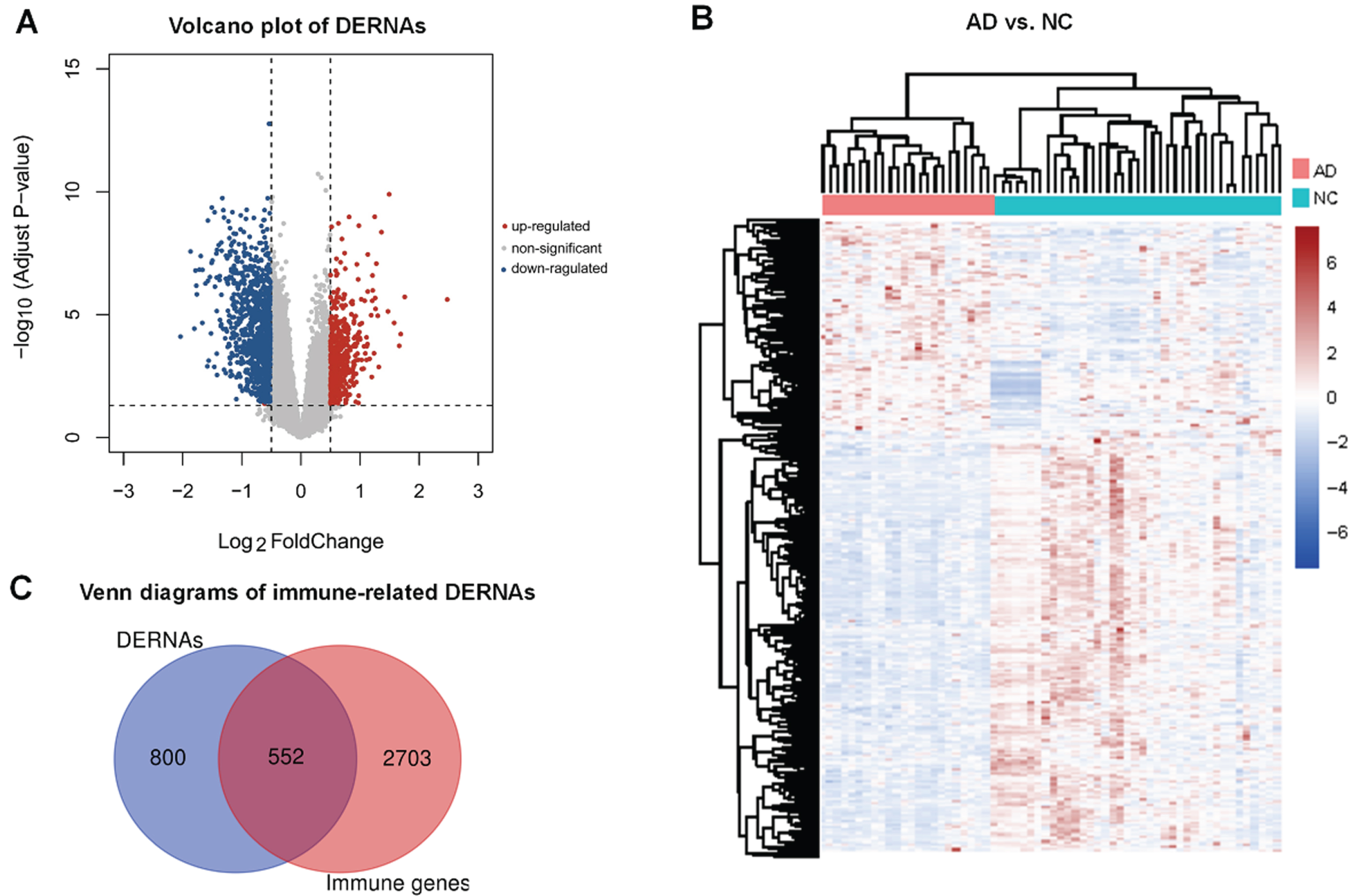
Analysis of differentially expressed RNAs (DERNAs). A) Volcano plot of DERNAs among Alzheimer’s disease (AD) patients and normal controls (NC); B) Hierarchical clustering heatmap of DERNAs; C) Venn diagrams of overlapping genes from DERNAs and immune-related genes derived from AmiGO2 and KEGG databases.
PPI network analysis
To delineate the interactions among the DEmRNA-coding proteins, we constructed a PPI network from the STRING database. In total, 365 nodes and 1,826 edges were included in the network; the nodes represented DEmRNAs, and the edges represented the interactions among DEmRNAs (Fig. 3). We determined the degree value for each mRNA in the PPI network and selected the top 20 mRNAs as hub genes (Fig. 4A).
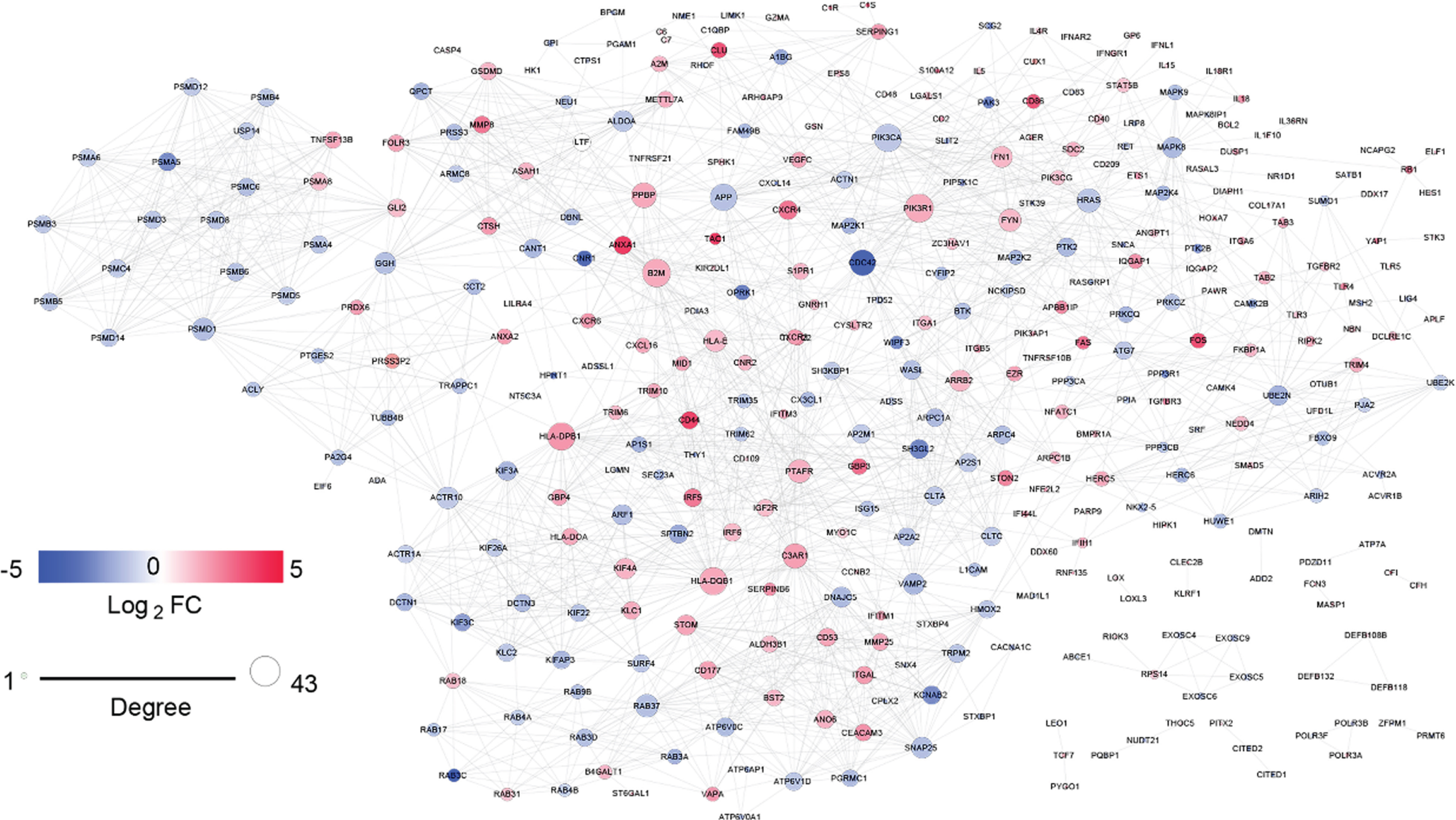
Protein-protein interaction networks of immune-related differentially expressed mRNAs (DEmRNAs) in Alzheimer’s disease. Nodes denote the protein-coding genes corresponding to DEmRNAs and edges denote the interactions among genes. The expression levels of DEmRNAs are indicated using colors. Red and blue circles indicate upregulated and downregulated DEmRNAs, respectively.
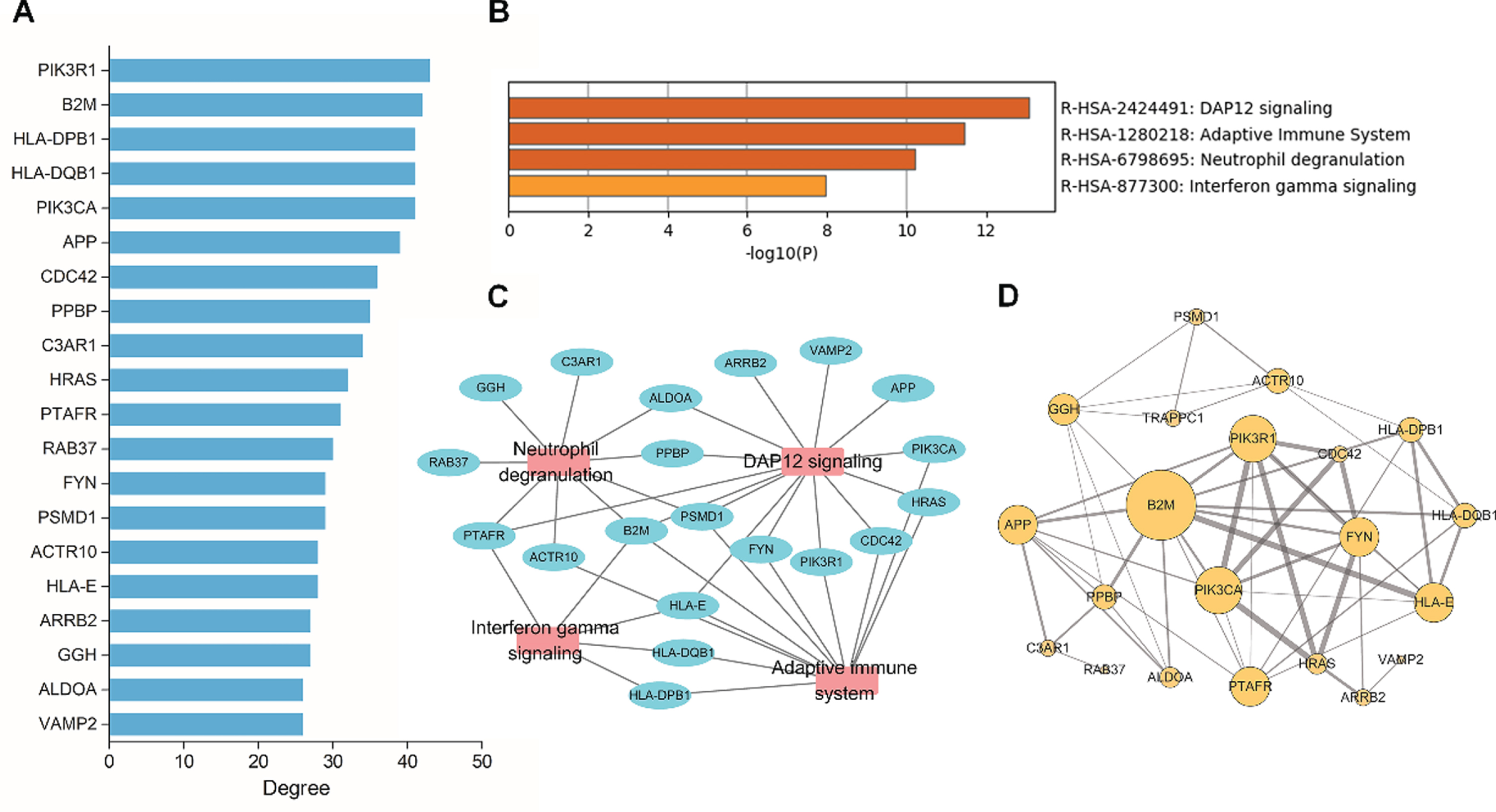
Hub genes and pathway enrichment analysis based on the protein-protein interaction (PPI) network. A) The top 20 hub genes were identified by calculating the degree values in the PPI network. B) Pathway enrichment analysis of hub genes. C) The connection network for hub genes and its enriched pathway. D) PPI subnetwork constructed on the basis of the 20 hub genes. The size of the circle is associated with the degree value. The thicker the edge, the stronger the interaction between two hub genes.
Differentially expressed hub genes are listed in Table 1. Furthermore, we performed pathway analysis and constructed a subnetwork for the 20 hub genes (Fig. 4B-D), which revealed that these genes were enriched in “DAP12 signaling,” “adaptive immune system,” “neutrophil degranulation,” and “interferon gamma signaling” pathways.
Differential expression of the top 20 hub genes and key lncRNAs
Construction of ceRNA network
To clarify the mechanism underlying immune-related phenomena in AD, we constructed a regulatory ceRNA network based on DElncRNA-miRNA-DEmRNA interactions. In total, 499 DElncRNA-DEmRNA interaction pairs were selected through a Pearson’s correlation coefficient of >0.6. Eighty-four DELncRNA-miRNA interaction pairs obtained from the DIANA-LncBase database with a miRNA target gene score (miTG-score) >0.8 were retained. Subsequently, we identified the target mRNAs for AD-related miRNAs, using the starBase database. After overlapping these targeted mRNAs and DEmRNAs, 719 miRNA-mRNA regulatory pairs were obtained. Together, a ceRNA network was generated, comprising 10 lncRNAs, 19 miRNAs, and 201 mRNAs (Fig. 5).
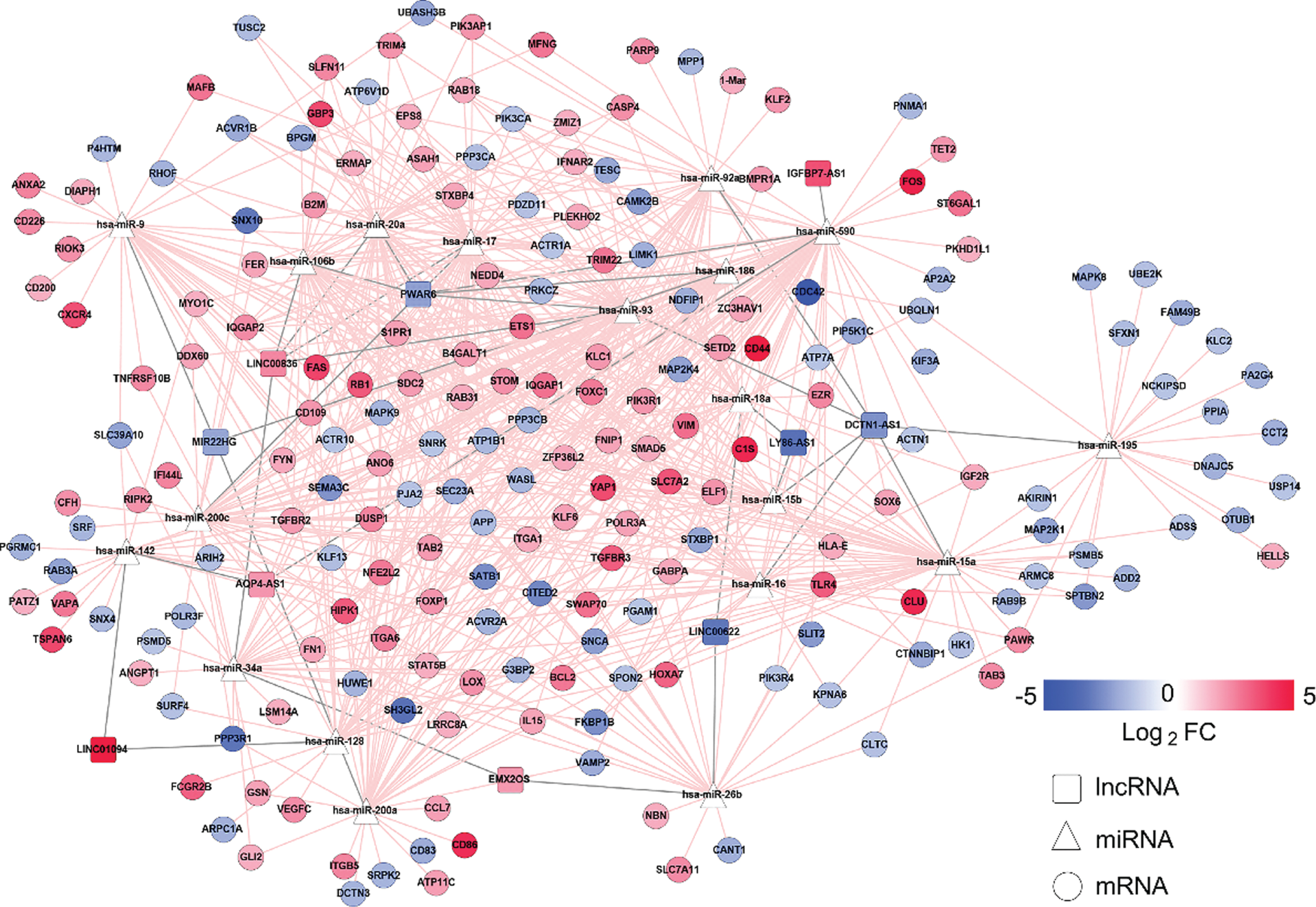
Construction of the ceRNA network. The pink and blue nodes denote the upregulated and downregulated RNAs, respectively. Gray edges represent lncRNA-miRNA interactions, whereas pink edges represent other interactions between RNAs. LncRNAs, miRNAs, and mRNAs are denoted by squares, triangles, and circles, respectively.
Analysis of upregulation and downregulation ceRNA subnetworks
Based on lncRNA and mRNA expression levels, we generated upregulation and downregulation ceRNA subnetworks (Fig. 6A, B). The upregulation ceRNA network comprised 5 lncRNAs, 9 miRNAs, and 83 mRNAs; downregulation ceRNA network, 5 lncRNAs, 16 miRNAs, and 86 mRNAs. We determined a number of mRNAs linked with each lncRNA in the upregulation and downregulation ceRNA subnetworks, and the lncRNA linking most mRNAs was identified as the key lncRNA. Upregulated lncRNAs may have upregulated mRNAs in the upregulation subnetwork comprising five lncRNAs including LINC00836, AQP4-AS1, EMX2OS, LINC01094, and IGFBP7-AS1. Particularly, LINC00836 linked the most mRNAs, and was considered the key lncRNA. Interestingly, hub genes B2M, PIK3R1, and FYN in the PPI network were competitively regulated by LINC00836 through four miRNAs, including hsa-miR-106b, hsa-miR-17, hsa-miR-20a, and hsa-miR-93. Moreover, DCTN1-AS1 was identified as the key lncRNA in the downregulation subnetwork. Hub genes CDC42, ACTR10, and PIK3CA may have been downregulated by DCTN1-AS1. Furthermore, in order to clarify the potential biological functions of mRNA regulated by the key lncRNA, we selected all mRNAs linked to LINC00836 in the upregulated network and those linked to DCTN1-AS1 in the downregulated network for pathway enrichment analysis (Fig. 6C, D). Differential expression profiles of LINC00836 and DCTN1-AS1 are summarized in Table 1.
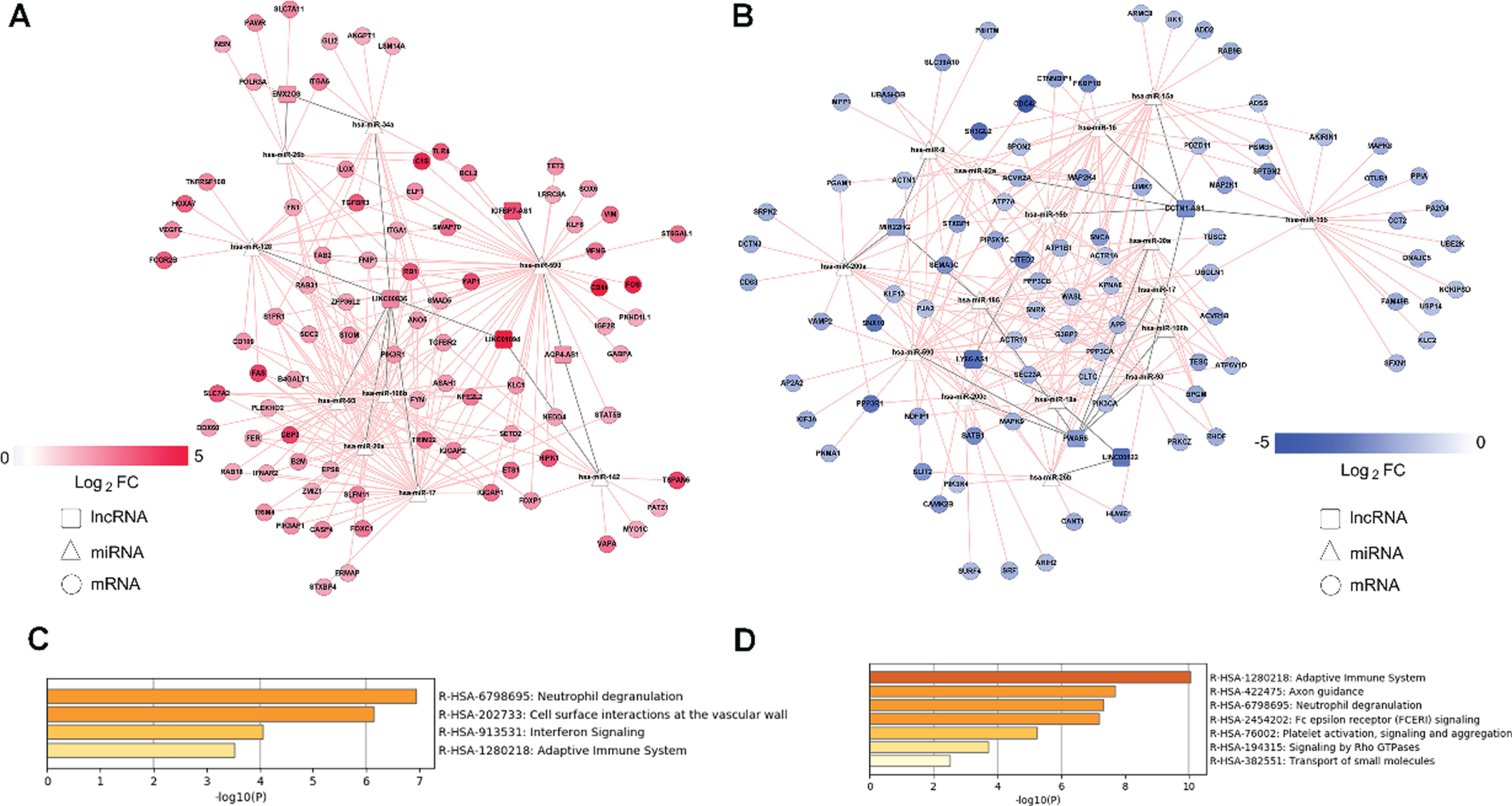
Upregulation and downregulation subnetworks derived from ceRNA networks and pathway enrichment analysis for two key lncRNAs LINC00836 and DCTN1-AS1. A) Upregulation subnetworks. B) Downregulation subnetworks. The pink and blue nodes denote upregulated and downregulated RNAs, respectively. Gray edges represent lncRNA-miRNA interactions, whereas pink edges represent other interactions between RNAs. LncRNAs, miRNAs, and mRNAs are denoted by squares, triangles, and circles, respectively. C) Enriched pathways of mRNAs associated with LINC00836 in the upregulation ceRNA subnetwork. D) Enriched pathways of mRNAs associated with LINC00836 in the downregulation ceRNA subnetwork.
DISCUSSION
This study focused on immune-related hub genes involved in AD pathogenesis. We identified 20 hub genes through PPI network analysis and constructed a ceRNA network to explore the regulatory mechanisms among lncRNA, miRNA, and mRNA interactions.
Since this was a retrospective study including all samples, it was not necessary to determine the sample size. On analyzing the differentially expressed genes between the AD and NC groups, we identified 18 DElncRNAs and 1334 DEmRNAs. On PPI network analysis for the DEmRNAs, the top 20 hub genes were identified from degree values. B2M was characterized as a hub gene, encoding β2-microglobulin, a serum protein related to the major histocompatibility complex (MHC) class I heavy chain on the surface of almost all nucleated cells [34, 35]. Previous studies detected elevated soluble B2M in cerebrospinal fluid and blood in patients with AIDS dementia complex, and AD [36, 38]. Vanni reported that B2M was significantly upregulated in the brain tissue in Creutzfeldt-Jacob disease patients and slightly upregulated in AD patients [39]. B2M upregulation in the AD brain was correlated with viral infection, thus potentially supporting the hypothesis that viral infection is associated with AD pathogenesis. FYN was another key gene identified herein, belonging to the Src tyrosine kinases family. Aβ oligomer can bind with the cellular prion protein receptor on the neuronal surface with high affinity, thus inducing FYN-mediated pathological cascades and leading to the deterioration of synaptic plasticity and long-term potentiation [40–44]. Aβ activates immune and adhesion molecules on the microglial surface, resulting in FYN recruitment, activation of extracellular signal-related protein kinase (ERK) and mitogen-activated protein kinase (MAPK) pathways, and the subsequent elevation in cytokine and chemokine levels. These pathological changes lead to neurotrophic and synaptic loss [45–47]. FYN-related inhibitors including Saracatinib (AZD0530) have been used for developing AD drugs [48]. An ongoing Phase IIa clinical trial on saracatinib for AD has reported that reductions in brain FYN levels in AD patients may be a promising therapy for AD. Furthermore, herein, both PIK3R1 and PIK3CA were identified as hub genes belonging to class IA PI3K. Zhang reported that Aβ stimulates PI3K to induce superoxide production and cause neurotoxicity [49]. PIK3R1 interferes with the insulin signaling pathway in the brain of AD patients [50]. Aberrant PI3K signaling can lead to immune deficiency and immune dysregulation; however, further studies are required to investigate whether PI3K mediates immune processes in AD.
CDC42 encodes a cell cycle protein and is activated in Aβ42-treated hippocampal neurons [51]. CDC42 can modify the morphology of dendritic spines by regulating the polymerization state of actin [52, 53]. GWAS identified one SNP (rs1140317) in HLA-DQB1, which was significantly correlated with entorhinal cortical thickness, serving as an AD neuroimaging biomarker [54]. Complement C3a receptor 1 (C3AR1) is an allergic toxin released during complement activation. Deletion of C3AR1 in PS19 mice alleviates tau pathologies, neuroinflammation, synaptic defects, and neurodegeneration [55]. Pro-Platelet basic protein (PPBP) is primarily involved in the regulation of mitosis, cAMP intracellular aggregation, and DNA synthesis. PPBP is reportedly differentially expressed in the sera of AD patients compared with normal controls [56]. B-Arrestin-2 (ARRB2) primarily regulates G-protein-coupled receptors. Variations in ARRB2 significantly increase the AD risk among Han Chinese individuals [57]. Vesicle-associated membrane protein 2 (VAMP2) is a vesicular membrane protein contributing to neuronal signal transduction and hormone release. Aβ42 disrupts the interaction between VAMP2 and synaptophysin, thus hindering angiogenesis and finally affecting baseline neural activity in the brain [58]. Besides the well-recognized pathogenic gene APP, the role of other hub genes in AD has not yet been reported.
On pathway enrichment analysis based on the top 20 hub genes, four hub genes, B2M, FYN, PIK3R1, and PIK3CA, contributed to the DAP12 signaling pathway. DAP12 is an adapter for TREM2 signal transduction. TREM2-DAP12 signaling stimulates certain downstream TREM2 pathways, including the phosphorylation of spleen tyrosine kinase (SYK) and glycogen synthase kinase 3β (GSK3β) [59, 60]. Furthermore, microglial activation by Aβ in vitro can enhance TREM2-DAP12 interaction [61]. Although numerous studies have emphasized the importance of the DAP12 pathway in AD, it is still an interesting question how these hub genes contribute to this pathway.
The immune-related ceRNA regulatory network constructed herein comprises 10 lncRNAs. LY86-AS1 is reportedly negatively correlated with the Braak stages of AD [62], and the other nine lncRNAs have been reported herein for the first time. LINC00836 and DCTN1-AS1 were the key lncRNAs in the up- and downregulation subnetworks, respectively. Notwithstanding a clear functional annotation, these genes are highly and specifically expressed in the brain [63]. Enrichment analysis indicated that the mRNAs connected with the two key lncRNAs in ceRNA subnetwork were enriched in the “adaptive immune system” and “neutrophil degranulation” pathways. Adaptive immunity may be potentially involved in AD pathogenesis. The number of CD3 + T cells is greater in AD patients than in healthy controls, and a higher number of hippocampal CD3+ T cells is associated with tau-related pathologies [64, 65]. It remains unclear whether neutrophil degranulation is involved in AD pathogenesis. Among other pathways involving the key lncRNAs, LINC00836 may regulate the “interferon signal pathway,” while DCTN1-AS1 may regulate “MHC class II antigen presentation,” “Fc epsilon receptor (FCERI) signaling,” “fc-gamma receptor (FCGR)-dependent phagocytosis,” “platelet activation, signaling, and aggregation,” and other immune-related pathways independently, suggesting that LINC00836 and DCTN1-AS1 can regulate multiple immune-related response in AD. Another interesting finding is that the key lncRNAs potentially regulate the hub genes revealed in the PPI network. Upregulated LINC00836 may attenuate the inhibitory effect of hsa-miR-106b, hsa-miR-17, hsa-miR-20a, and hsa-miR-93 on B2M, PIK3R1, and FYN. In contrast, downregulated DCTN1-AS1 may enhance the inhibitory effects of hsa-miR-93, hsa-miR-15a, hsa-miR-16, hsa-miR-15b, and hsa-miR-92a on APP, CDC42, ACTR10, and PIK3CA. Together, the key lncRNAs can regulate immune-related phenomena through different aspects, potentially through their regulation of immune-related hub genes.
In conclusion, this study is the first to identify hub genes based on differentially expressed RNAs associated with immune function in AD. Twenty hub genes, including B2M, FYN, PIK3R1, and PIK3CA, were identified herein. Furthermore, the present results show that LINC00836 and DCTN1-AS1 are the key lncRNAs in the ceRNA network, suggesting the potential significance of lncRNAs in modulating AD-related immune phenomena. Although the potential function of these hub genes and lncRNAs warrant further in-depth investigation, the present study furthers the current understanding of AD-related immune processes and provides novel insights into the development of new immunotherapies for AD.
Footnotes
ACKNOWLEDGMENTS
This study was supported by the Key Project of the National Natural Science Foundation of China (81530036); the National Key Scientific Instrument and Equipment Development Project (31627803); Mission Program of Beijing Municipal Administration of Hospitals (SML20150801); Beijing Scholars Program; Beijing Brain Initiative from Beijing Municipal Science & Technology Commission (Z161100000216137); Project for Outstanding Doctor with Combined Ability of Western and Chinese Medicine; and Beijing Municipal Commission of Health and Family Planning (PXM2019_026283_000003).