
Abstract
Alzheimer’s disease and related dementias lack effective treatment or cures and are major public health challenges. Risk for Alzheimer’s disease and related dementias is partially attributable to environmental factors. The heavy metals lead, cadmium, and manganese are widespread and persistent in our environments. Once persons are exposed to these metals, they are adept at entering cells and reaching the brain. Lead and cadmium are associated with numerous health outcomes even at low levels of exposure. Although manganese is an essential metal, deficiency or environmental exposure or high levels of the metal can be toxic. In cell and animal model systems, lead, cadmium, and manganese are well documented neurotoxicants that contribute to canonical Alzheimer’s disease pathologies. Adult human epidemiologic studies have consistently shown lead, cadmium, and manganese are associated with impaired cognitive function and cognitive decline. No longitudinal human epidemiology study has assessed lead or manganese exposure on Alzheimer’s disease specifically though two studies have reported a link between cadmium and Alzheimer’s disease mortality. More longitudinal epidemiologic studies with high-quality time course exposure data and incident cases of Alzheimer’s disease and related dementias are warranted to confirm and estimate the proportion of risk attributable to these exposures. Given the widespread and global exposure to lead, cadmium, and manganese, even small increases in the risks of Alzheimer’s disease and related dementias would have a major population impact on the burden on disease. This article reviews the experimental and epidemiologic literature of the associations between lead, cadmium, and manganese on Alzheimer’s disease and related dementias and makes recommendations of critical areas of future investment.
INTRODUCTION
Dementia is characterized by impairment in at least one cognitive domain, including memory, language, perception, attention, social cognition, and executive function [1, 2]. Globally, 50 million people are currently estimated to have dementia, and this number is expected to reach 152 million in 2050 [3]. Dementia involves a heterogeneous cluster of disorders including frontotemporal lobe dementia, vascular dementia, and Lewy body dementia. Alzheimer’s disease (AD) is a prevalent form of dementia, implicated in 70% of dementia cases [4]. Despite urgent need and tremendous research efforts, pharmacological trials for AD have struggled [5]. Treatment is complicated by neuropathologic changes observed many years prior to symptom onset [6]. Particularly when treatment is challenging, risk factor characterization is essential [7]. Risk for AD and related dementias is attributable to genetic and environmental factors [8].
Identification of modifiable environmental risk factors can substantially impact prevention and treatment for AD and related dementias. Many environmental chemicals are long known to be neurotoxic [9], particularly in laboratory models and in humans during neurodevelopment. In human populations, assessment of likely environmental factors during the risk window before disease manifestation is challenging due to the potentially long latency period of disease (Fig. 1). Environmental AD and related dementia studies often examine exposures at the time of clinical symptom onset, though relevant exposures may have occurred years or decades prior, or possibly even during early life. Evidence for environmental chemical neurotoxicity in older adults is accumulating and furthering our “understanding (of) the impact of the environment to advance disease prevention” is a major component of the National Institute of Aging’s key strategic plan to treat and prevent AD by 2025 [10].
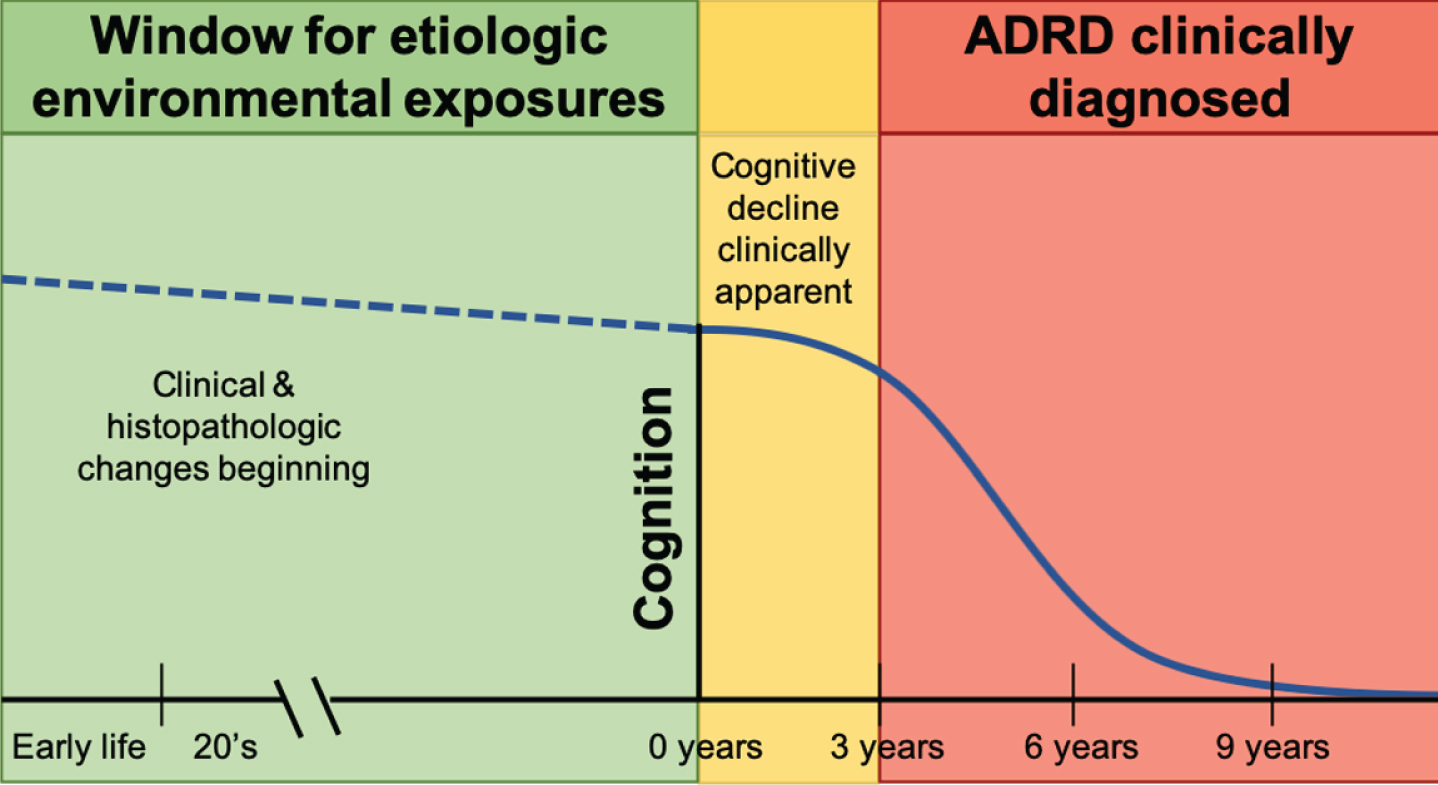
Etiologic window for environmental exposures linked to Alzheimer’s disease and related dementias (ADRD).
Among environmental factors, the roles of heavy metals such as lead, cadmium, and manganese are particularly of interest, given widespread population exposure. Lead and cadmium are notable metals for their neurotoxic effects even at low levels of exposure encountered in the general population. Manganese is an essential trace metal required for normal physiological functions including neuronal health, but it is toxic at low levels or in excess. Understanding the roles of these heavy metals in the etiology of AD and related dementias is critical. In the current article, we review the experimental and epidemiologic literature of lead, cadmium, and manganese and their associations with AD and related dementias. We focus on these three metals in this review because neurotoxicity of these metals is attributed to environmental contamination. Reviews for other potential candidate elements, such as zinc, iron, and copper, can be found elsewhere [11, 12].
LEAD (Pb) AND ALZHEIMER’S DISEASE
Lead introduction
Lead overall health effects
Lead is responsible for approximately 1% of the global burden of disease [13], including permanent effects on childhood intelligence and behavioral problems [14], although there is evidence that this is underestimated [15]. In US children under 5 years of age, there are annually 22,947,450 intelligence quotient points lost due to lead exposure at an estimated cost of $50 billion [16]. In older adults, lead exposure is associated with an increased risk of amyotrophic lateral sclerosis [17], Parkinson’s disease [18], hearing loss [19], age-related cataracts [20], glaucoma [21], and other chronic conditions. Specific to this review, lead exposure is associated with accelerated cognitive decline and dementia.
Current lead exposure levels
Lead exposure is a historic and current problem. Lead toxicity was observed as early as 370 BC [22], and more recently in the Flint, Michigan community [23]. The US Centers for Disease Control and Prevention (CDC) established a reference level of 5μg/dL blood lead for children and pregnant women; however, a safe level of blood lead has not been identified and evidence-based levels of concern have continued to lower [16]. Approximately 500,000 children ages 1–5 years in the US have levels exceeding the reference [24], particularly concentrated in cities and low socioeconomic areas [25].
Lead exposure sources
The removal of lead from paint and gasoline is a major public health success [26], though lead’s persistence in soil, dust, and built environments make abatement from our lives and environments difficult [27]. Despite US legislative efforts to minimize lead exposure, lead is still used in multiple industrial applications, including automobile lead-acid storage batteries [27]. Common lead exposure sources vary by age and geographic location. Housing build prior to 1970 may have paint containing lead, contributing to house dust, which adults and children inhale [28]. Local residents have higher body burden of lead due to contamination of air and soil [29]. Globally, high lead levels are associated with electronic waste recycling, lead mining, and smelting [30]. Children ingest lead dust due to frequent hand to mouth behavior. Older homes may also have leaded pipes or solder in their plumbing, which adults and children ingest through water. Industrial lead smelters and trash incinerators release lead into the local atmosphere as a by-product. Lead exposure remains widespread world-wide and domestically, with the primary routes of exposure being through inhalation or ingestion.
In older adults, the primary source of lead exposure can be endogenous. Excretion of lead is relatively slow, and accumulation is common [31]. During early and middle life, lead is sequestered in the bones, where it replaces calcium in the hydroxyapatite structure [32]. The skeleton contains 70–95% of the body burden of lead where lead can remain for decades [32], which can be exploited for exposure assessment research. Adults experiencing loss of bone mass via osteoporosis release lead into the bloodstream. In older adults, 40–70% of blood lead can be attributed to previous body stores [32]. Lead that entered the body during previous periods of high exposure can become biologically active decades later.
Lead transport to the brain and into neuronal cells
Lead absorption into the bloodstream and travel to the brain
Once lead enters the body, it is absorbed into cells and tissues. Inhaled lead particles cause local damage in the lungs. Depending on particle size, 30–40% can be absorbed into the bloodstream [31]. Adults only absorbs 10–15% of ingested lead, though pregnant women and children absorb 50% of ingested lead [31]. Individual level factors, such as diet (low iron, calcium, phosphorus, or zinc) and genetic polymorphisms (delta-aminolevulinic acid dehydratase and hemochromatosis genes) influence the intestinal absorption rate [33]. Organic lead is absorbed by the skin, and this route is most often observed in occupational settings [31]. Lead primarily enters the bloodstream through absorption at the lungs, gastrointestinal tract, or dermal surfaces (Fig. 2).
Lead transport into cells

Transport of lead (Pb), cadmium (Cd) and manganese (Mn) to the brain. Lead, cadmium and manganese enter the body through the gut and lung and are distributed in the bloodstream and transported to the brain. Cadmium and manganese also reach the brain through the olfactory nervous system. Lead crosses the blood-brain barrier and accumulates in the brain. All three metals can accumulate in the choroid plexus, a component of the blood-CSF (cerebrospinal fluid) barrier. The image was created in the Mind the GRAPH (https://mindthegraph.com/).
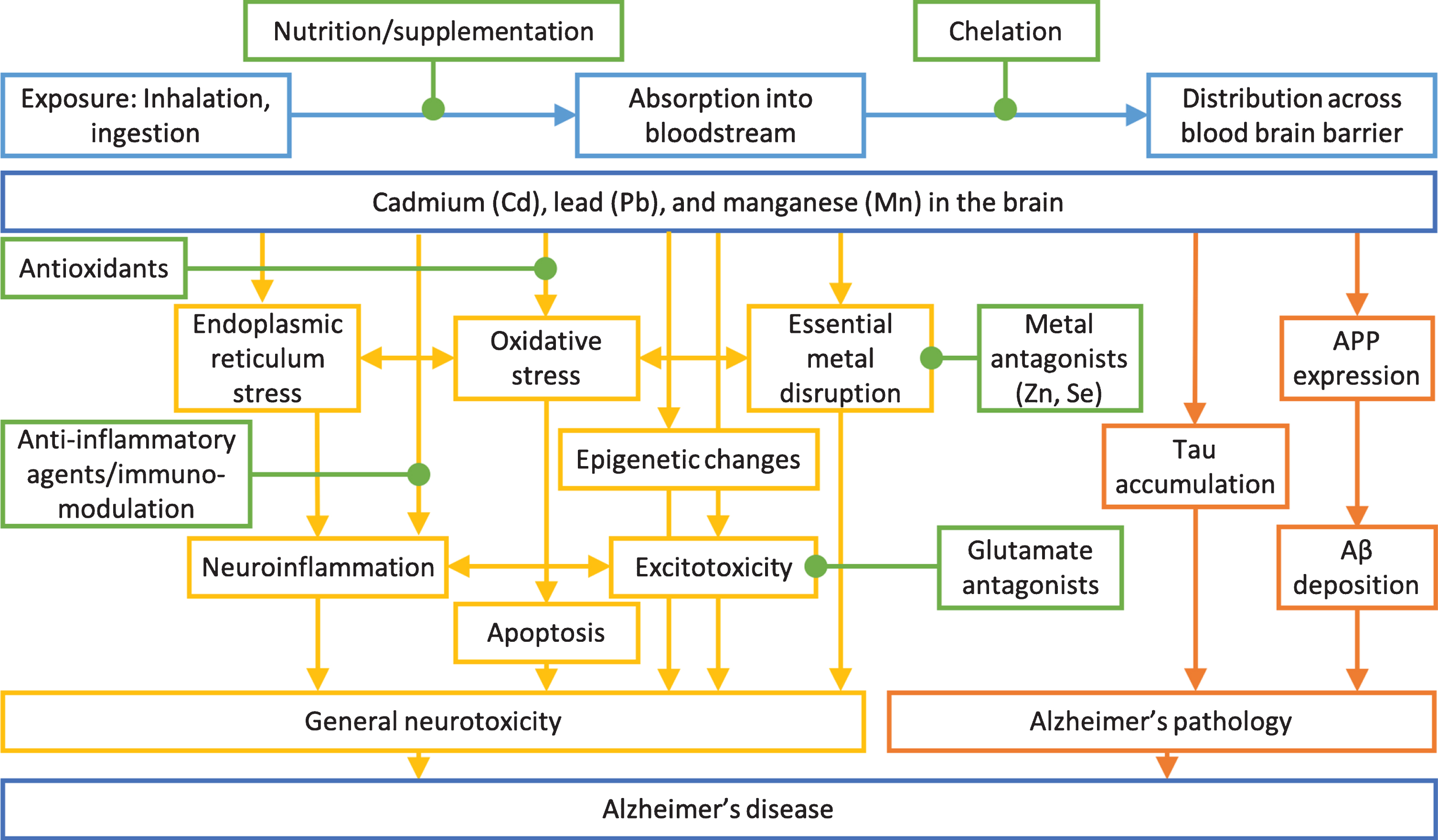
Mechanisms of general neurotoxicity action (yellow) and Alzheimer’s disease specific toxicity (orange) of cadmium, lead, and manganese on Alzheimer’s disease. Possible intervention options (green) and exposure routes and body distribution (light blue) are highlighted. Adapted from [223].
Absorbed lead circulates in the bloodstream. Lead enters cells by hijacking divalent metal transporters, designed to carry essential metals such as iron and copper [34]. Lead crosses the placental barrier and lead can be detected in infant cord blood at similar levels to maternal blood [35]. The blood-brain barrier (BBB) physically separates the brain from water soluble compounds in the bloodstream and transport is tightly regulated. Lead crosses the BBB by substituting for calcium [36] and accumulates in the brain. Lead can also influence the blood cerebrospinal fluid barrier [37]. Lead is distributed in the bloodstream, which is transported to the brain.
Experimental studies linking lead treatment and Alzheimer’s disease
Lead treatment and general neurotoxicity
Lead is a known neurotoxicant causing non-specific brain disruption (Fig. 3). First, lead is a redox-inactive metal that causes oxidative stress by depleting thiols and damaging the antioxidant defense system [38]. Excessive oxidative stress results in endoplasmic reticulum stress, mitochondrial damage, and ultimately apoptosis of neurons [36]. Neurons experience excitotoxic damage from overactivation by calcium associated with lead exposure [36]. Lead disrupts homeostatic levels of essential metals and alters normal metal signaling [34]. These actions together result in neuroinflammation [36]. Similar damage occurs to support cells, such as oligodendrocytes, microglia, astrocytes, and cerebrovascular endothelial cells [36]. Lead exposure causes epigenetic changes and changes in epigenetic regulators in the brain and brain regions [39–41], which may mediate latent effects of early life lead exposure. Lead induces oxidative stress, endoplasmic reticulum stress, neuroinflammation, apoptosis, epigenetic changes, excitoxicity, and essential metal disruption in the brain.
Lead treatment and dysregulation of Alzheimer’s disease pathways
AD mechanisms and symptoms are observed in animal models with lead treatment. Lead effects vary by species, timing, dose, and duration of exposure, though impairments related to AD are consistent. In general, model animals treated with lead have elevated brain levels of amyloid-β protein precursor, amyloid-β (Aβ), and tau as well as altered learning and memory behaviors.
Mouse and rat studies identified the timing of susceptibility and molecular targets of lead exposure. C57BL/6J mice exposed to 0.2% lead acetate from post-natal day (PND) 1–20 had altered miRNA expression that targeted epigenetic mediators at 6 months of age [42] and elevated tau protein and mRNA levels at 7 months of age [43]. Similar effects were not observed with adult lead treatment [43]. Mice exposed to lead acetate had variable results on the Morris water maze based on the developmental timing of exposure [44]. Male rats treated with 50mg/kg lead acetate via intraperitoneal injection at 8–9 weeks of age had triple Aβ1–40 levels in the choroid plexus and lower RNA and protein levels of low-density lipoprotein receptor-1 [45]. Rats of both sexes exposed to lead in the maternal drinking water PND1–30 had poorer performance on tests for learning, short term memory, and long term memory, which correlated with reduced number of synapses in the hippocampus and higher tau expression Rats of both sexes exposed to very low levels of lead (0.1%) in the maternal drinking water PND1–21 had increased tau protein in the forebrain and cerebellum and tau hyperphosphorylation, which caused cytoskeleton stability impairment and neuronal dysfunction [46]. Early life lead exposure in mice and rats resulted in impaired memory and AD-relevant pathology.
Transgenic AD susceptibility mice treated with lead are used to test for gene by environment interactions. Amyloid precursor protein (APP) transgenic mice treated with 50 mg/kg lead acetate oral gavages for 6 weeks had elevated Aβ1–40 and Aβ1–42 in the cerebrospinal fluid, cortex, and hippocampus, corresponding with impaired spatial learning on the Morris water maze test [47]. Microtubule associated protein tau (MAPT) transgenic mice treated with 0.2% lead acetate water during PND 1–20 had lead-related altered expression of MAPT and miR-34c, an miRNA that targets MAPT [48]. MAPT transgenic mice similarly treated with early life lead had decreased gene expression of APP at PND 20 and at PND 50 had increased miR-106b, an miRNA targeting APP, and decreased APP protein expression [49]. MAPT transgenic mice given a 10-fold lower dose treatment (0.02% lead acetate) during PND 1–20 were aged, and in midlife (PND 350) no differences were observed with treatment, but APP gene expression, protein expression, and Aβ levels were elevated in late life (PND 600) [50]. With later life lead treatment (exposed from 18 to 20 months of age) no effects on APP gene expression, protein expression, and Aβ levels were observed [50]. Lead treated mice performed poorly on the Morris water maze test at 7 months of age, only when the MAPT gene was knocked out [51]. Mice with both genetic susceptibility and lead treatment had exacerbated AD pathology in early life and latent effects in late life.
A unique long-term exposure model of lead in monkey has provided the strongest evidence for AD related neurodegeneration. Female Macaca fascicularis were exposed from PND1–400 to 1.5 mg/kg/day lead acetate and sacrificed at age 23 [39]. The aging primates exposed to lead exhibited overexpression of APP, Aβ, and enhanced pathologic neurodegeneration [39]. In the same cohort, early life lead exposure was associated with elevated tau mRNA, tau protein, its transcriptional regulators (Sp1 and Sp3), and site-specific tau hyperphosphorylation [52]. Early life lead exposure has a lagged effect on AD related molecular pathways in older life.
Epidemiologic studies of lead exposure and Alzheimer’s disease
Postmortem brain lead concentrations in Alzheimer’s disease
Postmortem brain tissues from AD cases and controls can be compared for overall and region-specific levels of metals. Frontal cortex and ventricular fluid were microwave digested and metal concentrations measured using inductively coupled plasma mass spectrometry (ICP-MS) [53]. No differences in lead concentration were observed between AD cases (n = 14) and controls (n = 14) in either tissue [53], though controls were older than cases and age is well known to be associated with lead levels. By design, postmortem tissues are collected following disease development and any metal differences may be a cause or a consequence of disease.
Epidemiologic studies of lead exposure and cognitive decline, dementia, and Alzheimer’s disease
Epidemiology literature summary for lead exposure and Alzheimer’s disease or cognitive decline
(continued)
AD, Alzheimer’s disease; RR, relative risk; OR, odds ratio; SD, standard deviation; NAS, VA Normative Aging Study; NES2, Neurobehavioral Evaluation System 2; WAIS-R, Weschler Adult Intelligence Scale; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; ALS, amyotrophic lateral sclerosis; SE, standard error; CI, confidence interval; K-XRF, K-Shell X-ray fluorescence; MMSE, Mini-Mental State Examination; PNS, peripheral nervous system; CNS, central nervous system; HR, hazard ratio
Properties of the lead biomarker matrix are important factors for study design and interpretation. Common tissues used for lead measurement and their respective rate of decay in the body are blood (30 day half-life), patella bone (10–15 years half-life), and tibia bone (10–30 years half-life) [54]. Associations may differ based on timing and type of the measurements. In early and mid-life, blood lead is expected to reflect exogenous exposure, while in late-life, blood lead can be attributed to release of sequestered endogenous bone lead. Epidemiologic associations may differ based on timing and type of the exposure biomarker measurements.
Exposure to lead is associated with neurodegeneration in cross-sectional human epidemiology studies [55] (Table 1). In a small matched case-control study of clinically confirmed AD, occupational exposure to lead was associated with a slightly higher, but not statistically significant, odds of AD (odds ratio = 1.12, 95% Cl: 0.63–2.00) [56]. This suggestive observation inspired population-based studies in larger samples to investigate related outcomes. Among men (mean age 66.8 years) in the Veteran’s Affairs Normative Aging Study (NAS), tibia bone lead was associated with poorer cognition, particularly pattern memory and spatial reasoning [57]. The tibia lead association replicated in a larger NAS sample and similar findings were extended to patella lead and blood lead [58]. Soon after, in the Baltimore Memory Study of men and women age 50–70 years, tibia lead was reported to be associated with concurrent lower cognition, while blood lead was not associated with cognition [59]. Lead exposure measured in blood, tibia bone, and patella bone was associated with clinically diagnosed amyotrophic lateral sclerosis in a matched case-control study [60], as well as Parkinson’s disease in a large case-control study [18], suggesting that lead exposure may be associated with multiple neurodegenerative processes and may not be specific to AD or dementia.
Epidemiology evidence is strengthened by the use of longitudinal studies to assess temporal relationships between exposure and disease. In the NAS when at least two Mini-Mental State Exam (MMSE) scores were available, one interquartile range (IQR) (20μg/g of bone mineral) higher patella bone lead concentration was associated with 0.24 points lower MMSE scores (95% CI: –0.44, –0.05) [61]. In a follow-up NAS analysis of up to 5 repeated cognitive measures over 18 years, an IQR higher level of patella lead was associated with 0.016 points lower MMSE score per year (95% CI: –0.032, –0.0004) [62]. Importantly, these differences in MMSE reflect cognitive performance and do not indicate clinical significance. Clinical cases of AD in Qu
Current epidemiologic studies of lead exposure are limited in the reach of their exposure measures [64]. Adult bone lead estimates of cumulative lead stretch into mid-life. The brain has periods of particular vulnerability to toxicants and exposure during vulnerable periods may increase risk of AD. Newer exposure methods include tooth lead, where through targeted laser ablation, timing of metal exposure can be pinpointed [65], including exposures that occur in early life [66]. Future clinical studies of AD may incorporate lead exposure measures. AD is the most common form of dementia in late life, representing 70% of dementia cases [4]; however, diagnosis requires specific clinical or pathological characteristics. Many lead exposure studies were conducted in population-based samples and a large study sample would be required to observe enough cases to rigorously test AD’s association with lead exposure.
Lead summary
Lead exposure is widespread due to current and previous industrial uses. Lead is ingested, inhaled, or dermally absorbed and then it travels in the bloodstream and can cross the BBB. Lead is a potent neurotoxicant causing non-specific brain disruption, resulting in oxidative stress, endoplasmic reticulum stress, mitochondrial damage, excitotoxicity, altered homeostatic metal signaling, inflammation, and ultimately neuronal apoptosis. In animal models, lead treatment causes AD-related pathology including changes in AβPP, Aβ, and tau, as well as memory deficits. In older adults, lead exposure is associated with lower cognitive status and longitudinal declines in cognition. To assess lead exposure risk on AD specifically, prospective evidence in human clinical samples is needed.
CADMIUM (Cd) AND ALZHEIMER’S DISEASE
Cadmium introduction
Cadmium overall health effects
Cadmium has no essential physiologic function in humans and is classified as a Group-I carcinogen by the International Agency for Research on Cancer [67]. Long-term exposure to low-level cadmium increases risks for kidney damage, osteoporosis, hypertension, lower lung function, and diabetes [68]. Recently, cadmium has emerged as a neurotoxicant, although evidence in humans is still limited.
Current cadmium exposure levels
Most people are exposed to cadmium, and exposure is most commonly measured in blood and urine biosamples. Blood cadmium levels represent current exposure (approximately 75 days [69–72]), while urine cadmium levels represent cumulative exposure (10–15 years [73]) due to long-term retention in the kidneys [74, 75]. In the general population (≥1 year of age), the geometric mean blood level of cadmium is 0.32μg/L and the geometric mean urine level (≥6 years of age) is 0.19μg/g creatinine (0.19μg/L) [76]. Cadmium levels are generally higher in women than men as low iron increases cadmium absorption, and cadmium levels are higher in smokers than non-smokers [74, 75].
Cadmium exposure sources
Cadmium is a bluish-white metal naturally found in the earth’s crust and cadmium is environmentally persistent. Anthropogenic sources of cadmium include mining and refining, combustion of fossil fuels, waste incineration and disposal, and the manufacture and application of phosphate fertilizers [75]. Diet is the primary cadmium exposure source [68] and cigarette smoking is another important source for non-smokers and smokers. Ingestion of contaminated foods and inhalation of air cadmium are major routes of exposure.
Cadmium transport to the brain and into neuronal cells
Cadmium absorption into bloodstream and travel to the brain
Cadmium exposure from inhalation and ingestion sources interfaces with gastrointestinal tract and lung (Fig. 2). Cadmium is taken up by these tissues and enters the bloodstream. Under normal conditions, only small amounts of cadmium can cross the BBB in adults [77]. The choroid plexus, a component of the blood-cerebrospinal fluid barrier, restricts blood toxicant access to the cerebrospinal fluid and maintains internal central nervous system homeostatic environment [78]. The choroid plexus is the main site of cadmium accumulation in the brain [78].
The olfactory nervous system may be a direct transport pathway of cadmium to the brain and therefore, bypassing the BBB. Cadmium concentrations in the olfactory mucosa and olfactory bulbs increased with intranasal instillation of cadmium in mice [79]. Increased concentrations of cadmium in the olfactory bulbs led to reductions in odorant-evoked neurotransmitter release from the olfactory nerve and axonal projections from the olfactory epithelium to olfactory bulbs [80]. Cadmium-treated mice showed lower performance in hippocampus-dependent spatial learning and memory and olfactory memory [81]. Cadmium directly passes into the central nervous system through the olfactory system causing persistent, irreversible damage by inhibiting adult neurogenesis in the hippocampus and olfactory bulb.
Cadmium transport into cells
The transport systems for divalent essential elements play a role in the cellular uptake of cadmium. Calcium, iron, and zinc transport systems (e.g., divalent metal transporter-1 (DMT1), calcium transporter-1, and calcium channels), transport cadmium [82]. Intestinal absorption of cadmium primarily occurs through DMT1 and depends on the body stores of other metals, especially iron. Iron deficiency increases intestinal absorption of cadmium through DMT1 [83]. DMT1, calcium transporters, and zinc transporters are expressed in neurons and vascular endothelial cells of the brain [84, 85].
Experimental studies linking cadmium treatment and Alzheimer’s disease
Cadmium treatment and general neurotoxicity
Toxicological studies support underlying biological mechanisms by which cadmium exerts neurotoxic effects. Direct effects through oxidative stress, neuroinflammation, and apoptosis in neuronal cells are well defined. Cadmium may also induce neurotoxicity by changing permeability of the BBB and interacting with other neurotoxicants, leading to Aβ aggregation and tau neurofibrillary tangle production. Pathogenic processes following cadmium exposure result in cognitive impairment and AD pathology (Fig. 3).
Cadmium is a redox-inactive metal that indirectly induces oxidative stress [38]. Cadmium has a high affinity for sulfhydryl group of thiols, such as glutathione and metallothionine [86]. Acute high-level exposure or long-term persistent low-level exposure interferes with the antioxidant defense system [87]. Cadmium induces oxidative stress in neuronal cells [86] and brain endothelial cells [88]. At low cadmium doses, glutathione detoxification is activated. At higher doses, with continued oxidative stress glutathione depletion occurs. Cadmium causes oxidative stress-dependent neuroinflammation and impaired neurodevelopment in young rats, enhanced with exposure to mixtures of lead, cadmium, and arsenic [89]. Rats treated with N-acetyl cysteine, a medication typically used to increase glutathione levels following acetaminophen overdose, had toxic effects of cadmium reversed, including memory deficits, increased thiobarbituric acid reactive substances (a marker of lipid peroxidation), and decreased hippocampus, cerebellum, and hypothalamus acetylcholine esterase activity [90]. Cadmium treatment induced brain oxidative stress and treatment with an antioxidant ameliorated cadmium neurotoxicity.
Cadmium-induced oxidative stress initiates neurodegeneration signaling pathways, such as mitogen-activated protein kinase (MAPK), protein kinase B (Akt), mammalian target of rapamycin (mTOR), and CD95/APO-1 (Fas)/Fas Ligand (FasL)-mediated mitochondrial apoptotic pathways, leading to neuronal apoptosis [91–93]. These signaling pathways are essential for growth, proliferation, and survival of neurons and are central in synaptic plasticity and learning and memory formation in the brain [94].
Metallothionein and trace metals also play a role in cadmium neurotoxicity via signaling pathways. Metallothionein, a low-molecular-weight sulfhydryl-rich metal-binding protein, can protect against cadmium toxicity by binding free cadmium ions within cells [95]. Metallothionein-III is downregulated in the brain of AD patients [96]. Insufficient production of metallothionine-III by prolonged exposure to cadmium causes neuronal apoptosis [97]. Cadmium exposure disrupts intracellular calcium homeostasis and increases extracellular calcium influx, triggering neuronal apoptosis via activation of MAPK and mTOR signaling pathways [98]. Cadmium also impairs the cerebral microvascular endothelium and increases permeability of the BBB, disrupting brain ion balance and nutrient uptake [99]. Cadmium treatment induces oxidative stress, neuroinflammation, and apoptosis in neuronal cells.
Cadmium treatment and dysregulation of Alzheimer’s disease pathways
Animal studies support biological links between cadmium exposure and Aβ aggregation and tau neurofibrillary tangle accumulation [100]. Treatment with 2.5 mg Cd/kg/day in drinking water in APP/presenilin-1 (PS1) transgenic mice increased Aβ1–42, reduced α-secretase protein expression, and reduced soluble AβPPα (sAβPPα) [101]. Cadmium-treated mice showed deteriorated learning and memory abilities and senile plaque depositions in the brain. Cadmium-related learning and memory deficits may be attributed by inhibition of α-secretase and promotion of the amyloidogenic AβPP processing (AβPP metabolism through the β-secretase pathway), which in turn leads to Aβ1–42 accumulation and senile plaque deposition [101, 102].
Cadmium treatment in vitro induced aggregation of the third repeat (R3) fragment of the microtubule-binding domain of tau [103]. R3 is critical in the nucleation of the tau filament formation process [104]. Cadmium forms Cd-tau dimers by binding to the nitrogen atoms of imidazole groups of histidine residues, affecting the nucleation step on tau aggregation [103]. The static electric strike of cadmium ion to the surrounding R3 peptide chains can prompt conformation conversion and enhance interactions with the R3 dimers, leading to enhanced aggregation through the elongation step [103]. Cadmium treatment increases Aβ production and tau tangles.
Cholinergic neuron toxicity is another potential cadmium-AD pathway. Cadmium exposure increases cell death on cholinergic neurons, leading to alterations in acetylcholinesterase and degeneration of basal forebrain cholinergic neurons [105]. Memory deficits seen in AD is associated with the loss of cholinergic neurotransmission due to degeneration of cholinergic neurons in the basal forebrain [106]. In SN56 cholinergic murine neuroblastoma cell line model of the basal forebrain, cadmium treatment induced apoptosis, mediated by blockade of muscarinic M1 receptors (related to memory loss in rats and humans), overexpression of neurotoxic acetylcholinesterase-S, downregulation of neuroprotective acetylcholinesterase-R, and increased Aβ and tau protein levels.
Epidemiologic studies of cadmium exposure and Alzheimer’s disease
Postmortem brain cadmium concentrations in Alzheimer’s disease
There are limited studies that examined the associations between cadmium exposure and AD in human populations. A study using postmortem brain tissues found that AD brain tissues had higher concentrations of cadmium (hippocampus: 0.547 g/g dry weight (d.w); cerebral cortex: 0.518 g/g d.w.) compared with age-matched control brain samples (hippocampus: 0.472 g/g d.w; cerebral cortex: 0.496 g/g d.w.) in an Eastern Canada sample but not in a United Kingdom sample [107]. In a recent study using postmortem brain samples from AD patients and nondemented elderly controls, cadmium concentrations in the frontal cortex were lower in AD cases (20 ng/g) than in controls (30 ng/g) [53]. This finding should be interpreted with caution because AD patients (mean age = 78 years) were younger than nondemented controls (mean age = 88 years). A meta-analysis including 8 studies covering 405 AD patients and 424 control subjects found that circulating concentrations (either whole blood, serum, or plasma) of cadmium were significantly higher in AD (standardized mean difference = 0.62 (95% CI, 0.12, 1.11) versus controls) [108]. This same meta-analysis reported that circulating lead concentrations were lower in AD patients. Again, it should be noted that the findings from postmortem brain tissues are subject to confounding by AD risk factors, especially age.
Epidemiologic studies of cadmium exposure and cognitive decline, dementia, and Alzheimer’s disease
Epidemiologic studies linking cadmium exposure to AD risks (prevalence or incidence) have rarely been conducted due to methodologic challenges such as lack of relevant exposure data, low incident rate or prevalence, and late onset (Table 2). Instead, a few studies have examined cognition as an early indicator of future AD risks and they consistently report an association between cadmium exposure and decreased cognitive function in older adults [109–111]. A cross-sectional study with 2,068 older adults from the US National Health and Nutrition Examination Survey (NHANES) 2011-2014 showed a significant association between cadmium exposure measured in whole blood (median = 0.35μg/L) and lower cognitive function [111]. An earlier NHANES study (NHANES-3) reported an association between urinary cadmium, a longer-term biomarker of cadmium exposure, and a measure of attention and perception (Symbol Digit Substitution Test) only among never smokers, but not in the entire population [109].
Epidemiology literature summary for cadmium exposure and Alzheimer’s disease or cognitive decline
(continued)
AD, Alzheimer’s disease; MMSE, Mini-Mental State Examination; CI, confidence interval; NHANES, National Health and Nutrition Examination Survey; HR, hazard ratio; CSID, Community Screening Instrument for Dementia; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; IU, Indiana University; NES2, Neurobehavioral Evaluation System 2; SRTT, Simple Reaction Time Test; SDST, Symbol-Digit Substitution Test; SDLT, Serial Digit Learning Test; DDST, Digit-Symbol Substitution Test.
A possible link of cadmium and AD in human populations has been supported by two studies examining incident AD mortality. In NHANES 1999–2004 cycles, participants in the top quartile of blood cadmium (>0.6μg/L) had an adjusted hazard ratio of 3.83 (95% CI, 1.38, 10.6) compared with those in the lowest quartile (≤0.3μg/L) [112]. Urinary cadmium, a longer-term biomarker of cadmium exposure, was associated with a 58% higher rate of AD mortality per 0.51μg/L increase in urinary cadmium [113]. Both studies were limited by underestimation of AD cases and possibly low power (i.e., high false positive) due to low mortality rate (1.1–1.3% AD risk over mean 7.5 follow-up years [113]). Competing risk is another challenge in mortality studies, where highly exposed individuals are more likely to die from other causes before having a chance to die of AD.
Cadmium summary
Cadmium exposure primarily occurs through dietary and cigarette sources. Inhaled cadmium can enter the brain through the olfactory bulb. Cadmium can also enter the brain through the blood cerebrospinal fluid barrier. In animal models in the brain, cadmium causes oxidative stress, neuroinflammation, and neuronal apoptosis. Cadmium also induces neurotoxicity by changing permeability of the BBB, causing Aβ aggregation and producing tau neurofibrillary tangles. In human aging studies, cadmium may be associated with decreased cognitive function and clinical AD specifically. However, the pathophysiologic link between environmental cadmium exposure and AD is limited given the uncertainty in cadmium transport to the brain.
MANGANESE (Mn) AND ALZHEIMER’S DISEASE
Manganese introduction
Manganese overall health effects
Manganese is the fifth most abundant metal in the environment and the twelfth most abundant element overall on earth [114]. It is an essential trace metal required for proper growth and physiological processes, such as bone growth, blood clotting, immune response, carbohydrate metabolism, and brain function [115]. Manganese is a cofactor for normal cell function enzymes, including arginase, pyruvate carboxylase, glutamine synthetase, and manganese superoxide dismutase (MnSOD; SOD2). Despite the importance of manganese in human health, excessive manganese is neurotoxic, and exposure to high levels of manganese may accumulate in the brain, causing an irreversible parkinsonian syndrome known as manganism [116–118]. Exposure to high levels of manganese results in impaired cognitive function and contributes to the pathogenesis of AD [119].
Current manganese exposure levels
Adequate adult intake of manganese is 1.8 mg per day for women, and 2.3 mg for men [120]. Manganese exposure can be measured in several different specimen types. Normal range for manganese levels in the general population is 4–15μg/L in blood, 1–8μg/L in urine, and 0.4–0.85μg/L in serum, though the usefulness of urine and serum manganese as biomarkers for exposure is limited [121, 122]. Blood manganese levels have a half-life of approximately 40 days [123] and are higher with female sex, younger ages, and Asian origins [124, 125].
Manganese exposure sources
Diet is the primary source of manganese in the general population. Manganese is abundant in plant-based foods, including whole grains, rice, nuts, and leafy vegetables. Animal foods, including meat, fish, poultry, eggs, and dairy, lack this nutrient [126]. Daily intakes of manganese typically range from 2 to 6 mg, of which ∼1–5% is normally absorbed [127]. Due to the dual role of manganese as an essential nutrient and a potent toxicant, whole-body manganese levels are tightly controlled by regulating intestinal absorption and excretion of the metal through homeostatic mechanisms [128]. Thus far, manganese toxicity from high dietary manganese intakes in humans has not been reported [129].
Manganese toxicity classically results from elevated exposure levels in drinking water or air. Manganese is widely used in industrial processes and commercial products. Excessive occupational exposure to manganese is most common in mining, welding, ore processing, dry battery manufacture, and organochemical fungicide use [130–133]. Manganese toxicity can also arise from an impaired or under-developed excretion system, including in patients receiving total parenteral nutrition therapy [134, 135], patients with hepatic encephalopathy [136], and abusers of ephedrone (methcathinone) [137].
Manganese transport to the brain and into neuronal cells
Manganese absorption into the bloodstream and travel to the brain
Manganese can be absorbed and transported into various body tissues, including brain. Dietary manganese is absorbed from the intestine, and it can cross the BBB. The blood-cerebrospinal fluid barrier may also be a major interface for brain manganese uptake [138]. Airborne manganese can be absorbed through the pulmonary system into the systemic circulation or through the olfactory nervous system into the brain. The nasal-brain pathway circumvents the BBB and allows for direct contact with the brain; thus airborne manganese exposure has been a major concern for neurotoxicity [139]. While three major routes, through the BBB, cerebrospinal fluid, or nasal-brain pathways, transport manganese to the brain, the mechanisms by which manganese is absorbed and distributed in the brain are not well understood.
Manganese transport into cells
The essential, yet toxic, nature of manganese necessitates precise homeostatic mechanisms to maintain appropriate manganese body levels. While several transporters are involved in the transport of manganese into or within the brain, most of them also transport other essential metals, such as iron and zinc, and have not been rigorously tested in physiological contexts.
Recent genetic studies revealed that three metal-ion transporters are essential in maintaining manganese homeostasis: solute carrier family 30, member 10 (SLC30A10), SLC39A14, and SLC39A8. Loss-of-function mutations in SLC30A10 were reported in patients with elevated manganese levels in blood, manganese accumulation in the liver and brain, and parkinsonism [140–142]. Slc30a10-deficient mice hyperaccumulate manganese in the blood, liver, and brain [143]. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity [144, 145]. Similarly, mutations in SLC39A14 were reported in patients with high manganese levels in blood and accumulations of manganese in the brain, but not in the liver, and with juvenile-onset dystonia-parkinsonism [146]. Slc39a14-deficient mice [147–149] recapitulate these human phenotypes, including manganese accumulation in the brain, but not in the liver [147–149]. On the other hand, loss-of-function mutations in SLC39A8 were reported in patients with severe manganese deficiency in the blood [150–152]. Slc39a8-inducible global knockout and liver-specific knockout mice also showed abnormally reduced manganese levels in the blood and in multiple tissues [153]. SLC39A8 is a cell-surface manganese import transporter, and that disease-associated mutations abrogated its uptake activity [154]. Taken together, SLC30A10, SLC39A8, and SLC39A14 are required for maintaining manganese levels, but their roles in brain manganese homeostasis and transport remain largely unknown.
Manganese and iron are similar in atomic masses, radii, and electron structures, and they share transport mechanisms. DMT1 is the primary manganese importer [155–157]. Belgrade rats deficient in DMT1, however, showed the same concentration of manganese in the brain as wild-type rats [158], suggesting that DMT1 may not be the major brain transporter of manganese. Dietary iron deficiency increases the expression of DMT1 in rat olfactory epithelium, resulting in elevated blood manganese after a single dose of intranasal instillation of radio labeled 54Mn [159]. Dietary iron deficiency increases manganese uptake through upregulation of DMT1 and potentiates apoptosis in the olfactory bulb in rats and human neuronal cell line [160]. The iron exporter ferroportin can also export both iron and manganese from the cell [161, 162]. Flatiron mice deficient in ferroportin have impaired manganese metabolism [163] and accumulate manganese and other metals, including iron in the brain [164]. Ferroportin exports manganese, in addition to iron, and is protective against manganese-induced toxicity and oxidative stress in dopaminergic SH-SY5Y cells [165]. Neurons may acquire manganese through transferrin uptake mechanisms [166]. DMT1 transports divalent metals such as Fe2 + and Mn2 +, but the transferrin-transferrin receptor (Tf-TfR) system is involved in the uptake of trivalent metals such as Fe3 + and Mn3 + [167, 168].
Experimental studies linking manganese and Alzheimer’s disease
Manganese exposure and general neurotoxicity
Manganese-induced neurotoxicity has been well studied. Underlying mechanisms include oxidative stress, mitochondrial dysfunction, autophagy dysregulation, accumulation of intracellular toxic metabolites, and apoptosis [169–171]. Mitochondria play critical roles in aging-related neurodegenerative diseases, including AD [172]. Mitochondrial dysfunction is involved in the pathogenesis of AD via mitochondrial reactive oxygen species production [173, 174]. Manganese accumulates in the brain mitochondria, although the efflux is very slow [175, 176]. MnSOD is a potent antioxidant enzyme located in the mitochondria. MnSOD activity declines during the aging process [177]. Excess manganese can impair MnSOD activity, thus increasing reactive oxygen species production and eventually leading to mitochondrial dysfunction [178].
Manganese and dysregulation of Alzheimer’s disease pathways
In addition to general neurotoxicity from manganese exposure, manganese is involved with AD pathology. To examine a causal relationship between oxidative stress and Aβ pathology, partially MnSOD deficient mice (one allele of MnSOD knockout) were crossed mice overexpressing a doubly mutated human APP [179]. Partial deficiency of MnSOD induced oxidative stress, and increased brain Aβ levels and Aβ plaques [179]. In contrast, MnSOD overexpression improved resistance to Aβ, slowed plaque formation or increased plaque degradation, and markedly attenuated the AD phenotype [180]. Furthermore, the APP/PS1 mouse model has an age-dependent accumulation of Aβ in the brain and an accelerated decline in mitochondrial function associated with a decrease in MnSOD activity [181]. These studies suggest close relationships between manganese induced mitochondrial dysfunction and oxidative stress in AD pathophysiology.
Manganese specifically binds to ligands in the N-terminus of Aβ1–40, as demonstrated in Aβ/micelle studies using Mn2 + ions as paramagnetic probes [182], similar to Cu2 + and Zn2 + ions [183, 184]. A weak binding affinity between Mn2 + ions bind to the N-terminus of Aβ1–40 in the millimolar to micromolar range was confirmed using nuclear magnetic resonance spectroscopy [185]. The discovery of additional metal Mn2 + ion binding to Aβ revealed more complex AD metal chemistry than the previously well-defined role for Cu2 + and Zn2 + ions in AD. High levels of manganese induce Aβ-related neurotoxicity in both cultured neurons and rodent brains [186]. Mouse N2a neuroblastoma cells stably expressing both wild-type PS1 and Swedish mutant APP (APPsw) treated with manganese led to dose-dependent neurotoxicity and increased Aβ levels [186]. Moreover, high levels of manganese induced Aβ-related cognitive impairment in the APP/PS1 mouse model of AD [186]. This study further demonstrated the possible mechanisms related to impaired Aβ degradation; high manganese reduces expression of two major enzymes involved in Aβ degradation, neprilysin and insulin degrading enzyme, without altering AβPP expression [186]. Furthermore, manganese chelator reduced the concentration of manganese in the brain, and it restored the cognitive function of the AD model along with decreased Aβ peptides in the AD model, suggesting manganese chelation therapy as a possible strategy for the intervention of AD pathogenesis [186]. In addition, exposure to manganese can cause tau hyperphosphorylation [187], which may lead to the formation of neurofibrillary tangles, one of the key clinical structure changes in AD. Manganese has an affinity for Aβ and exposure to high levels of manganese may accelerate the accumulation of Aβ in the brain, thereby increasing Aβ neurotoxicity and accelerating the disease’s progression.
A frontal cortex gene expression profiling experiment was performed in Cynomolgus macaques who received 3.3–5.0 mg/kg of manganese weekly for 10 months [188]. Manganese treatment upregulated 61 genes compared to controls, from a total of 6,766 genes. The most highly upregulated gene was Amyloid Beta Precursor Like Protein 1 (APLP1), a member of the APP family associated with AD [188]. Immunohistochemistry confirmed increased APLP1 expression and revealed Aβ diffuse plaques in manganese-treated frontal cortex [188]. Neurological function mediated by the frontal cortex is affected in manganese-exposed animals and provides further explanation of visuospatial associative learning deficits in these same animals [189]. Manganese-induced neurotoxicity is likely attributable to the translational inhibition of AβPP and heavy chain ferritin resulting in excessive iron accumulation and exacerbated neurotoxic oxidative stress [190].
Epidemiologic studies of manganese exposure and Alzheimer’s disease
Postmortem brain manganese concentrations in Alzheimer’s disease
The physiological concentration of manganese in the normal human brain is estimated to be from 5.32 to 14.03 ng Mn/mg protein, which corresponds to 20.0–52.8μM Mn [191]. In mammalians, manganese-induced neurotoxicity occurs when manganese concentrations in the brain are elevated by ∼3-fold, which corresponds to 15.96–42.09 ng Mn/mg protein or 60.1–158.4μM Mn [191]. These calculations suggest that Mn levels in the brain must be tightly controlled within a narrow physiological range.
Few studies have measured manganese concentrations in the brain of AD patients and normal controls, reporting mixed results. One earlier study measured manganese concentrations in the human brain of AD and aging participants using instrumental neutron activation analysis [192], a nuclear process for determining the metal concentrations in a vast range of materials [193]. Manganese levels in all brain regions were not different between controls and AD subjects (0.261μg/g versus 0.245μg/g) with the highest manganese levels detected in the basal ganglia in both groups [192]. In contrast, in two brain regions, the parietal cortex and the cerebellum [194], metal concentrations were measured by ICP-MS, a well-established method for quantifying various trace elements [195]. Higher levels of manganese were observed in the parietal cortex of the AD brain compared to controls [194].
Abnormal manganese concentrations are noted in AD and may play a role in its pathogenesis. The AD brain is under intensive oxidative stress [196] and MnSOD plays a role in AD progression. MnSOD is localized in the cerebral cortex and hippocampus of patients with AD [197], suggesting MnSOD is associated with the formation of Aβ plaques. Brains of AD patients have increased expression but reduced enzyme activity of MnSOD [198]. In summary, these studies suggest that brain manganese and MnSOD alterations may contribute to AD pathology.
Epidemiologic studies of manganese exposure and cognitive decline, dementia, and Alzheimer’s disease
Studies have examined associations between manganese exposure and cognitive functions (Table 3). Manganese was usually measured in blood, hair, drinking water, or air. Studies using hair [199], blood [200–202], or both hair and blood [203] biomarkers reported significant associations between adult manganese levels and impaired cognitive function.
Epidemiology literature summary for manganese exposure and Alzheimer’s disease or cognitive decline
Epidemiology literature summary for manganese exposure and Alzheimer’s disease or cognitive decline
Mn, manganese; SD, standard deviation; CDR, Clinical Dementia Rating Scale (0.5 = MCI, 1 = Mild Dementia, ≥2 = Dementia); MMSE, Mini-Mental State Examination; CI, confidence interval; SMD, standardized mean difference; OR, odds ratio; WAIS-III, Wechsler Adult Intelligence Scale; WMS, Wechsler Memory Scale; RAVLT, Rey Auditory Verbal Learning Test; MCI, mild cognitive impairment.
In occupational manganese settings, exposure is typically higher than in environmental settings. Those with occupational exposure have reported deficits in attention and concentration, memory, visuospatial function, verbal learning, and executive and other cognitive functions, and manganese blood levels have a dose-effect relationship with cognitive function [202]. Following environmental manganese exposures in Quebec, women with higher manganese blood levels had lower visual memory scores, while men with higher manganese blood levels had poor initial learning and recall, or both, on visual and verbal test scores [200]. In two rural Mexico communities living within a manganese mining district, higher blood manganese levels were associated with low-level cognitive function on the MMSE [201]. Also in a Mexican mining district, air manganese concentrations were associated with adult attention impairment [204]. In the environment from a ferromanganese alloy plant in Brazil, hair manganese in mothers was negatively associated with nonverbal cognitive ability, measured on Raven’s Progressive Matrices [203]. In two communities near a ferromanganese refinery in Brazil, hair and fingernail manganese levels were inversely associated with visual working memory and intelligence [199]. In a cross-sectional study of adults residing in Marietta and East Liverpool, Ohio, USA who were exposed to high levels of environmental airborne manganese from industrial sources, manganese exposure was associated with lower working memory, visuospatial memory, and verbal skills [205]. Together, these studies suggest exposure to high levels of manganese results in decreased cognition in adults.
Few studies have specifically tested the relationship between manganese exposure and AD, and the results have been inconsistent. In a retrospective ecological study, clinical cases of AD in Qu
Potential link between manganese and overlapping cases of AD-Parkinson’s disease
Manganese impacts AD pathology [119, 186] and manganese has a well-established connection with Parkinson’s disease [207, 208]. In cases where both AD and Parkinson’s disease overlap, little is known of manganese’s contribution. AD and Parkinson’s disease are the two most common neurodegenerative diseases with substantial overlap in pathological and clinical features. Mild cognitive impairment is associated with a risk of progression to AD and risk of progression to Parkinson’s disease [209, 210]. Clinically, approximately 30% of patients with AD develop parkinsonism [211], and a high percentage of these patients have Lewy bodies [212]. Over 50% of patients with Parkinson’s disease eventually develop dementia [210]. Up to 50% of AD cases display α-synuclein aggregation into Lewy bodies [213–215], and cerebrospinal fluid from patients with either AD or Parkinson’s disease has similar α-synuclein levels [216]. Patients with dementia with Lewy bodies-AD typically exhibit more accelerated cognitive dysfunction than is seen in patients with AD alone [217, 218]. Transgenic mice that develop dementia with Lewy bodies and AD pathologies display cognitive decline associated with a dramatic enhancement of Aβ, tau, and α-synuclein pathologies [219].
The ATP13A2 (PARK9) gene is an interesting factor that may link manganese to mixed cases of Parkinson’s disease and AD. Mutations in ATP13A2 were identified in patients with Kufor-Rakeb syndrome, an autosomal recessive juvenile-onset Parkinson’s disease that is characterized by supranuclear upgaze paresis and dementia [220]. ATP13A2 is suggested to be involved in the transport of metals, including manganese, into cells [221]. Overexpression of ATP13A2 reduces intracellular manganese concentration and protects cells against manganese-induced neurotoxicity and cell death [221]. Patients with Lewy body disease had reduced ATP13A2 protein levels correlated with increased α-synuclein and Aβ in all Lewy body disease cases [222]. Given the role of manganese in the etiology of Parkinson’s disease and AD, further studies to determine potential mechanisms linking manganese to overlapping cases of Parkinson’s disease and AD are warranted.
Manganese summary
Manganese is an essential metal that is primarily received through dietary sources. However, exposure to high levels of the metal via inhalation can lead to brain manganese accumulation and a parkinsonian-like disorder known as manganism. Dietary manganese crosses the BBB, whereas inhaled manganese is absorbed through the olfactory transport pathway, thus resulting accumulation of the metal in the brain. In animal models, excessive manganese causes oxidative stress through impaired MnSOD and causes AD pathology including Aβ accumulation and tau phosphorylation. In human epidemiologic studies, manganese binds Aβ and elevated manganese exposure is associated with cognitive declines, though the prospective association with clinical AD has not yet been demonstrated. Extremely high exposure to manganese is associated with manganism, a specific neurodegenerative disease. The neurological effects of manganese exposure depend on levels of additional essential metals.
CONCLUSION
AD and related dementias are presently incurable and represent major public health challenges. There are 50 million people currently estimated to have dementia around the globe, and this number is expected to reach 152 million in 2050 [3]. AD and related dementias are most likely to occur in aging populations. As the aging population grows, the burden of disease, especially in developing countries, is tremendous. Randomized pharmacological trials for AD and related dementias have been largely unsuccessful [5], and trial investigators emphasize the importance of identifying risk factors for disease [7]. Identification of modifiable risk factors is critical to prevention of AD and related dementias and would have a significant public health impact. Likely risk factors for AD and related dementias include exposure to heavy metals, such as lead, cadmium, and manganese.
The metals lead, cadmium, and manganese occur naturally and persist in the environment. Lead and cadmium are non-essential metals that serve no required biological purpose in the human body and serious adverse effects are observed with increasing exposure levels. The general population is primarily exposed to lead via house dust from lead-based paint and drinking water from lead pipes, while the general population is primarily exposed to cadmium through cigarette smoking and diet. Manganese is an essential metal for normal physiological processes and the general population is primarily exposed to manganese through diet. Adverse effects can occur with manganese exposure that is either too low or too high.
Exposures to lead, cadmium, and manganese are ubiquitous in our environments and stored in our bodies. Lead, cadmium, and manganese are divalent metals that can be transported into cells and around the body using endogenous divalent metal transporter systems, similar to normal transport for calcium and iron. Older adults carry historic lead exposure in bones from periods when lead was more commonly used in commercial products. Older adults also retain high body burdens of cadmium due to cadmium’s long half-life in the kidney. Manganese similarly has a fairly long half-life in tissues, especially in the bones, and accumulates in the brain.
Lead, cadmium, and manganese are well-documented neurotoxicants acting through multiple pathways to contribute to AD pathology. In cell and animal model systems, lead, cadmium, or manganese treatment induces oxidative stress, neuroinflammation, and apoptosis in neurons. In addition, animal models treated with these metals observe AD-related pathological features in the brain (Aβ and tau tangles) as well as memory deficits. Human epidemiologic studies have consistently shown lead, cadmium, and manganese are associated with impaired cognitive function and cognitive decline in adults. No longitudinal human epidemiology study has assessed lead or manganese exposure on AD specifically. Two human studies using data from the US NHANES reported a possible link between cadmium exposure and AD mortality. Though lead, cadmium, and manganese have characterized neuronal toxicological pathways, cause AD-related pathology and memory deficits in model systems, and are associated with declines in cognition in older adults, evidence in humans linking these to AD is very limited. More longitudinal epidemiologic studies with high-quality time course exposure data and incident cases of AD are warranted to confirm and estimate the proportion of risk attributable to these exposures.
Older adults are poised to experience lead-, cadmium-, and manganese-related accelerated declines in cognition as they age. Given the widespread and global exposure to lead, cadmium, and manganese, even small increases in the risks of AD and related dementias would have a major population impact on the burden on disease. Exposure management should be considered to reduce the risks of AD and related dementias that may be attributable to exposure to lead, cadmium, or manganese. Modifying exposure levels to the known neurotoxicants and suspected AD and related dementia risk factors, lead, cadmium and manganese, should be a public health priority to prevent disease.
Footnotes
ACKNOWLEDGMENTS
Dr. Bakulski was supported by National Institutes of Health grants (R01 ES025531, R01 ES025574, R01 AG055406, R01 MD013299, P30 AG053760) and the ALS Association. Dr. Park was supported by National Institutes of Health grants (R01 ES026578, R01 ES026964). Dr. Seo was supported by National Institutes of Health grants (R21NS112974 and R00ES024340). This work was supported by National Institutes of Health grants (UG3 OD023285, UH3 OD023285, P30 ES017885).