
Abstract
Background:
Small vessel disease (SVD) and Alzheimer’s disease (AD) frequently coexist; however, it remains unclear how they collectively affect cognition.
Objective:
We investigated associations between SVD and AD biomarkers, namely amyloid, tau, and neurodegeneration (ATN) in young onset dementia (YOD) and explored how SVD and ATN interact to affect cognition.
Methods:
80 YOD individuals were recruited from a memory clinic. SVD burden (SVD+) was operationalized as a score >1 on the Staals scale and ATN was measured using cerebrospinal fluid (CSF).
Results:
SVD+ was associated with lower CSF Aβ1–42 (B = –0.20, 95% CI: –0.32 to –0.08) and greater neurodegeneration, indexed as hippocampal atrophy (B = –0.24, 95% CI: –0.40 to –0.04). SVD+ was not associated with tau. Cognitive impairment was associated with CSF Aβ1–42 (B = –0.35, 95% CI: –0.55 to –0.18) but not SVD. Rather, SVD was indirectly associated with cognition via reduced CSF Aβ1–42, specifically with global cognition (B = –0.03, 95% CI: –0.09 to –0.01) and memory (B = 0.08, 95% CI: –.01 to .21). SVD was indirectly associated with cognition via increased neurodegeneration in grey matter (Global cognition: B = –0.06, 95% CI: –0.17 to –0.03; Memory: B = 0.05, 95% CI: 0.01 to 0.18) and the hippocampus (Global cognition: B = –0.05, 95% CI: –0.11 to –0.01; Memory: B = 0.06, 95% CI: 0.01 to 0.17).
Conclusion:
In YOD, SVD burden was associated with AD pathology, namely CSF Aβ1–42. SVD indirectly contributed to cognitive impairment via reducing CSF Aβ1–42 and increasing neurodegeneration.
INTRODUCTION
Young-onset dementia (YOD) represents individuals with dementia onset before the age of 65 years. YOD is associated with greater economic burden [1], faster cognitive deterioration, and shorter survival compared to late-onset dementia (LOD) [2], although shorter survival has been debated in more recent studies [3, 4]. In addition, patients with YOD are likely to be in paid employment with significant financial commitments and with young children and elders to support. Therefore, identifying disease models of YOD constitutes an important step to reduce disease burden and improve clinical outcomes for this vulnerable group.
The most common etiology of YOD is Alzheimer’s disease (AD), followed by small vessel disease (SVD) [5]. Both AD and SVD frequently co-exist and are strong predictors of cognitive impairment and dementia [6, 7]. Imaging biomarkers of SVD include white matter hyperintensities (WMH), lacunes, microbleeds, and perivascular spaces [8]. Each of these biomarkers increase the risk of cognitive impairment [9–12], with executive dysfunction being the most vulnerable [13]. Biomarkers of AD pathophysiology involve amyloid-β (Aβ1–42), phosphorylated tau protein (p-tau), and neurodegeneration, measured as total tau (t-tau) or grey matter atrophy [14, 15]. Collectively these three biomarkers are referred to as the ATN pathway, which each have an effect on cognitive decline [16].
The association between SVD and AD pathology remains controversial. Studies have shown that SVD risk factors, such as pulse pressure, hypertension, and diabetes, are associated with abnormal CSF Aβ1–42 levels [17, 18], while other studies show that SVD is associated with CSF tau, independent of Aβ1–42 [19]. On the other hand, several studies show that SVD is not associated with CSF Aβ1–42 [7, 19–23]. The interaction between Aβ1–42 and SVD in the process of cognitive impairment also remains unclear. Some suggest cognitive impairment includes the synergistic effect of both Aβ1–42 and SVD [21], while others suggest both pathologies have independent effects on cognition [7, 22]. Most of these studies have focused on sporadic LOD. As such, little is known about the associations of SVD with AD pathology in patients with YOD. Given young age is a strong moderator of vascular injury and AD pathology [21], it is imperative to determine the associations between SVD and AD pathophysiology, and their effect on cognition in patients with YOD.
Here, in a cross-sectional study of YOD patients with mild AD, we tested the association between SVD and ATN biomarkers. We further investigated how SVD and ATN biomarkers interact to affect cognition using moderation and mediation models. We hypothesized that the prevalence of SVD would be associated with a greater burden of ATN biomarkers. We further hypothesized that SVD would moderate the effect of ATN biomarkers on cognition, that is strengthen the associations between ATN and cognition. We also hypothesized a mediation effect where SVD would indirectly be related to cognitive impairment via increasing burden of ATN biomarkers. This direction of effect proposed for the mediation, where SVD affects ATN biomarkers, is in line with pathological studies showing that SVD promotes amyloid aggregation [24], restricts the clearance of amyloid [25] and causes vascular-related amyloid [26].
METHODS
Participants
Study participants were selected from the Singapore Young-Onset Dementia Cohort (SYNC), which is a cohort of patients with mild AD experiencing onset of symptoms below the age of 65 years; recruited from a tertiary neurology center (National Neuroscience Institute, Singapore) between 2015–2018. From SYNC, we selected patients with CSF, MRI, and neuropsychological assessments (consort diagram in the Supplementary Material). A diagnosis of mild dementia of the Alzheimer’s type was made based on DSM-V [27] criteria and supported by a Clinical Dementia Rating Scale (CDR) [28] of 0.5 to 1. Exclusion criteria for the SYNC study included significant neurological or psychiatric comorbidities, a history of alcohol or drug abuse and presence of other neurodegenerative conditions.
Ethics approvals and patient consents
The SYNC study was approved by the Singhealth Centralized Review Board. Informed written consent was obtained from all participants according to Declaration of Helsinki and local clinical research regulations.
Measures
Demographic characteristics, including the participants’ age, gender, race, and years of education were collected using a structured interview with the patient or next of kin.
Cardiovascular risk factors, including blood pressure, diabetes, hyperlipidemia, and history of stroke were noted from medical records. Total cardiovascular risk was indexed using the Framingham office-based cardiovascular disease risk prediction model, which took into account age, gender, BMI, systolic blood pressure, smoking, and diabetes [29].
Cognitive functioning was indexed using global and domain assessments. Global cognition was assessed using the Montreal Cognitive Assessment (MoCA) [30]; memory was assessed using the Wechsler Memory Scale version 4 (WMS-IV) Story Recall delayed and immediate tests [31] and the Alzheimer’s Disease Assessment Scale- cognitive subscale (ADAS-Cog) delayed and immediate recall tasks [32]; executive function was assessed using the Frontal Assessment Battery [33] and Color Trails 1 and 2 [34]; and visuospatial skill was assessed using the WAIS-IV Block Design test [35] and the Rey Complex Figure test [36].
CSF
CSF was collected using a lumbar puncture. The analysis of Aβ1–42, p-tau, and t-tau were performed using ELISA immunoassays in accordance with prescribed protocol requirements (INNOTEST tTau Ag, INNOTEST PHOSPHO-TAU(181) and INNOTEST β-AMYLOID(1–42); Innogenetics Inc., Alpharetta, GA). CSF cut offs were based on laboratory parameters: Aβ1–42 < 480 pg/mL, p-tau >61 pg/mL, t-tau >425 pg/m (Fujirebio, Ghent, Belgium).
MRI protocol
Patients underwent a 3T MRI scan (Achieva 3.0; Philips Medical Systems, Best, Netherlands) within six months of clinical and neuropsychological evaluation. Scan specifications include, (a) T1-weighted MPRAGE (axial acquisition, 176 slices, matrix size = 256×256, voxel size = 1.0×1.0×1.0 mm3, echo time (TE) = 3.2 ms, repetition time (TR) = 7 ms, inversion time (TI) = 850 ms, flip angle = 8° field of view (FOV) = 256×256 mm2), and (b) T2-weighted FLAIR imaging (170 slices, matrix size = 256×256, voxel size = 1.0×1.0×1.0 mm3, TE = 340 ms, TR = 8000 ms, TI = 2400 ms, FOV =240×240 mm2). Scans were visually-rated for WMH using the 0–3 Fazekas Scale [37] on axial FLAIR sequences for deep and periventricular WMH in the left and right hemispheres; lacunes and microbleeds using STandards for Reporting Vascular changes on nEuroimaging (STRIVE) [38]; and global cortical atrophy using the Pasquier scale [39]. SVD burden was calculated using the Staals score [8] which combines WMH, lacunes, microbleeds, and perivascular spaces into a total SVD-related brain damage score, ranging from 0–4. Visual-ratings were performed by two independent trained raters and any differences in ratings that differed by a large margin was resolved by discussion and ensuring that the appropriate image slices were used for rating.
Statistical analysis
Data preparation involved imputing variables with less than 30% missing data, based on recommendations by the American Psychological Association Task Force on Statistical Inference [40]. Multiple imputation using the five chained equations procedure was used to perform logistic regression with original weights to estimate grey matter volumes (19% missing) using the predictors age, gender, education, MoCa, CSF Aβ1–42, Fazekas score, lacunes, microbleeds, and global cortical atrophy. Patients were grouped as having SVD burden (SVD+) based on a Staals score [8] ≥1 or no SVD (SVD–) based on a Staals score of 0. Skewed variables included Staals score, p-tau, and t-tau, and were log transformed. For variables containing 0 (Staals score), a 1 was added prior to transformation.
Main statistical analysis
Group differences
Differences between patients with SVD+ and SVD–were determined for demographics, cardiovascular disease risk factors, cognition, CSF biomarkers, and grey and white matter volume. A t-test was used for continuous variables, with Welsh adjustment for unequal variances, and χ2 test used for categorical variables.
Mediation analysis
A mediation analysis quantifies whether a predictor is indirectly related to an outcome via a third variable. Our a priori mediation hypothesis predicted that SVD (predictor) would indirectly affect cognition (outcome) via the ATN pathway (mediating variable) (Fig. 1a). ATN was indexed as CSF Aβ1–42, CSF p-tau, and neurodegeneration represented as CSF t-tau, total grey matter volume (as a measure of general neurodegeneration), and hippocampal volume (as a measure of AD-specific neurodegeneration). Cognition was indexed as global cognition and domain-specific cognition, namely memory, executive function and visuospatial skills. Cognitive domains were created by z-scoring each test and averaging the total z-score over the number of tests for each domain.
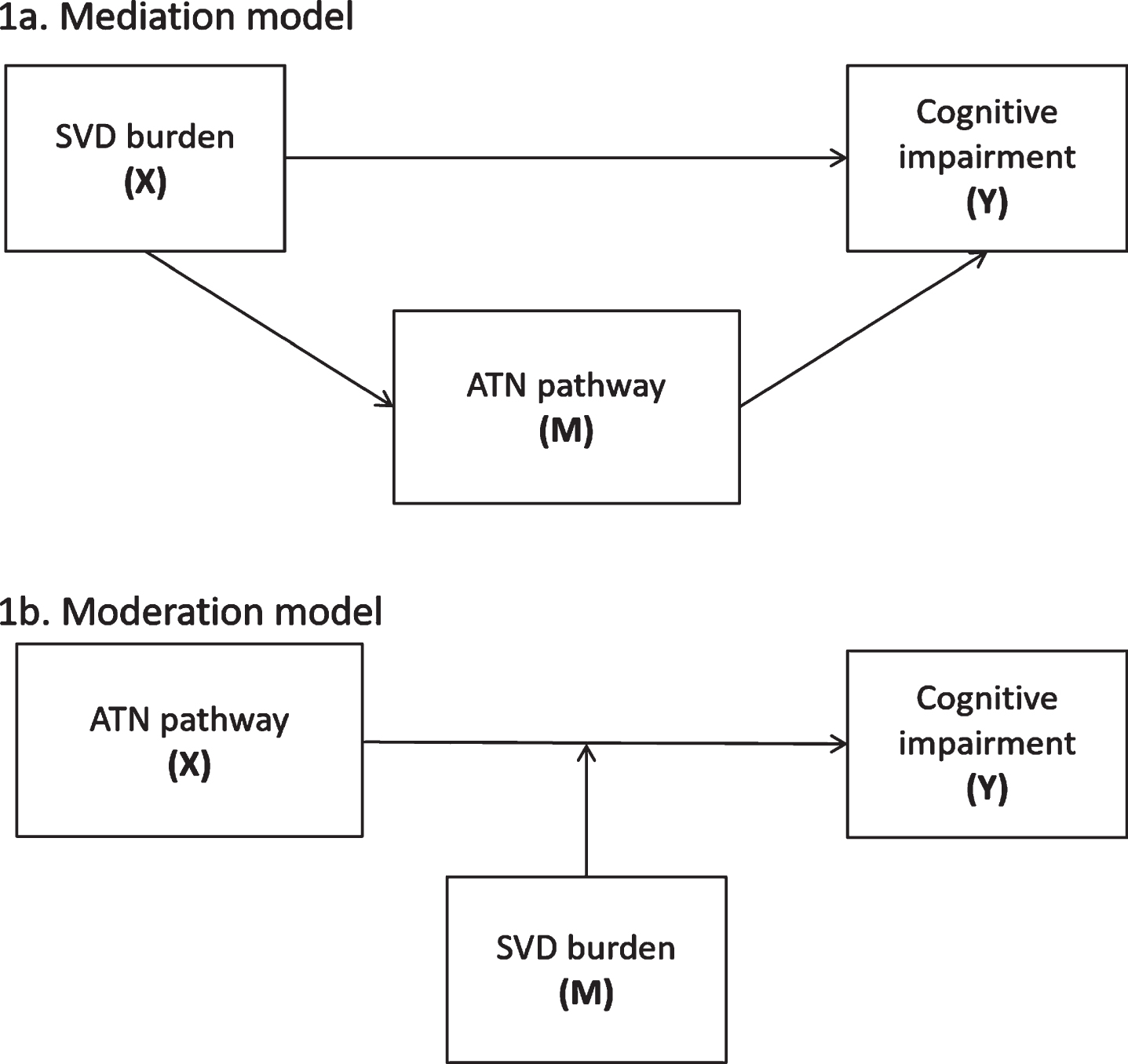
a) The mediation model illustrates that SVD (X) indirectly affects cognition (Y) via the ATN pathway (M) (X ⟶ M ⟶ Y). b) The moderation model illustrates that the relationship between ATN components (X) and cognition (Y) may be strengthened in the presence of SVD (M) (XM ⟶ Y).
Mediation was assessed using path analysis, which is a form of multiple regression. Path analysis tests for both the direct associations between all variables in the model (X⟶Y; X⟶M; M⟶Y in Fig. 1a) and the indirect associations (X⟶M⟶Y in Fig. 1a). An independent path analysis model was conducted for each mediator (CSF Aβ1–42, CSF p-tau, CSF t- tau, total grey matter volume, and hippocampal volume) and each cognitive outcome (global, memory, executive functions, and visuospatial skills). The mediating variables that were not the primary target in the analysis were included as covariates, in addition to the Framingham cardiovascular risk score which accounted for demographics and cardiovascular risk factors.
All moderation and mediation analyses were conducted using SPSS Amos version 20 (SPSS, Inc, Chicago, IL). As a non-parametric estimation of effects, SEs and biases for all moderation and mediation analyses, bias-corrected (BC) bootstrapping was applied with 1000 resamples [41]. Bootstrapping measures the variability of the linear approximation of each path in the model and estimates the bias of this linear approximation to the population [42]. BC bootstrapping has been empirically validated as a tool for multiple comparison correction, deriving robust parameter estimates based on maximized power and limited type 1 error rates [43]. The significance of the BC bootstrap estimate was indicated by confidence intervals that did not contain 0. Path analysis model fit was assessed using published recommended criteria [44]: a) χ2 p-value: >0.05, b) Bentler comparative fit index (CFI: >0.95), and c) root mean error of approximation (RMSEA: <0.04). Effect sizes for the direct effects were indexed using the standardized coefficient of the slope (B), which is identical to the benchmark set for Pearsons r; a coefficient of 0.10, 0.30, and 0.50 indicated small, moderate and large effects respectively [45]. Effect size for indirect effects were indexed by squaring Cohen’s [45] estimations because indirect effects represent a product of two effects [46]; a coefficient of 0.01, 0.09, and 0.25 indicated small, moderate, and large effects, respectively.
Moderation analysis
A moderation analysis quantifies whether the relationship between a predictor variable and an outcome is strengthened or weakened in the presence of a third variable. Our a priori moderation hypothesis predicted that the relationship between ATN components (predictors) and cognition (outcome) would be strengthened in the presence of SVD (moderating variable) (Fig. 1b).
The moderation effect was calculated by centering the ATN and SVD variables, then multiplying SVD with each component of ATN to create an interaction variable. This interaction variable was the predictor in the regression analysis and cognition was the outcome, with Framingham cardiovascular risk score as the covariate. An independent analysis was run for each SVD and ATN interaction variable, and for each cognitive outcome (global cognition and cognitive domains). A post-hoc exploratory analysis investigated the interaction between SVD and Aβ1–42 with hippocampal volume as the outcome.
Data availability
Deidentified participant data will be made available upon reasonable request from the corresponding author.
RESULTS
Group differences
SVD– and SVD+ patients showed no differences with demographics, APOE ɛ4 status, global cognition, cardiovascular risk factors, Framingham cardiovascular risk score, p-tau, t-tau, total white and grey matter (Table 1). SVD+patients were trending on having lower CSF Aβ1–42 (t [78] = –1.97, p < 0.10) and lower hippocampal volume (t [78] = –1.91, p < 0.10) compared to SVD–patients.
Participant characteristics
1Total available genetics data N = 31 SVD+ (18 missing) and N = 20 SVD- (13 missing). SVD+, positive for cerebrovascular burden; SVD-, no cerebrovascular burden; FAB, Frontal assessment battery; ADAS, Alzheimer’s Disease Assessment Scale; RCFT, Rey Complex figure test; SD, standard deviation; IQR, interquartile range. at-test df = 1,78; bχ2 df = 1; cWelsh adjusted t-test; dFazekas rated for deep and periventricular WMH in the left and right hemispheres, with total score out of 12. ∗p < 0.05, ∗∗p < 0.01
Mediation analysis
The path analysis regression models with SVD as the predictor, ATN components as the mediator and cognition as the outcome had excellent model fit according to recommended criteria [44].
Direct associations
X (SVD) ⟶Y (Cognition): SVD was trending on being associated with memory, while controlling for cardiovascular risk factors, CSF Aβ1–42, CSF p-tau, and CSF t-tau (B = 0.18, SE = 0.05, bootstrapped p < 0.10, BC95% CI: –0.00 to 0.33). SVD was not directly associated with any other cognitive outcomes, including global cognition, executive functions or visuospatial skills.
X (SVD) ⟶ M (ATN components): SVD was associated with CSF Aβ1–42, while controlling for cardiovascular risk factors, CSF p-tau, and CSF t-tau (B = –0.20, SE = 0.07, bootstrapped p < 0.01, BC 95% CI: –0.32 to –0.08). SVD was also associated with neurodegeneration, indexed as hippocampal volume, while controlling for cardiovascular risk factors, CSF Aβ1–42 and CSF p-tau (B = –0.24, SE = 0.10, bootstrapped p < 0.05, BC 95% CI: –0.40 to –0.04). SVD was not associated with CSF t-tau, total grey matter volume or CSF p-tau.
As a post-hoc analysis we investigated the association between CSF Aβ1–42 and neurodegeneration, specifically hippocampal atrophy given it was the only neurodegeneration marker associated with SVD. CSF Aβ1–42 was associated with hippocampal volume while controlling for cardiovascular risk factors, SVD and p-tau (B = 0.01, SE = 0.01, bootstrapped p < 0.05, BC 95% CI: 0.00 to 0.01).
M (ATN components) ⟶ Y (cognition): CSF Aβ1–42 was significantly associated with memory, while controlling for cardiovascular risk factors, CSF p-tau, CSF t-tau and SVD (B = –0.35, SE = 0.01, bootstrapped p < 0.01, BC95% CI: –0.55 to –0.18). CSF Aβ1–42 was not associated with any other cognitive outcomes. Neurodegeneration indexed as CSF t-tau was significantly associated with visuospatial skills, while controlling for cardiovascular risk factors, CSF Aβ1–42, CSF p-tau, and SVD (B = –0.56, SE = 0.27, bootstrapped p < 0.05, BC95% CI: –1.07 to –0.16). Neurodegeneration indexed as total grey matter was significantly associated with global cognition (B = .04, SE = 0.01, bootstrapped p < 0.01, BC 95% CI: 0.02 to 0.06), memory (B = –.02, SE = 0.01, bootstrapped p < 0.05, BC95% CI: –0.02 to –0.00), and visuospatial skills (B = 0.00, SE = 0.01, bootstrapped p < 0.01, BC95% CI: 0.00 to 0.01), while controlling for cardiovascular risk factors, CSF Aβ1–42, CSF p-tau, and SVD. Neurodegeneration indexed as hippocampal volume was significantly associated with global cognition (B = 1.34, SE = 0.71, bootstrapped p < 0.01, BC 95% CI: 1.25 to 3.91), memory (B = –0.02, SE = 0.05, bootstrapped p < 0.05, BC95% CI: –0.21 to –0.01), and visuospatial spatial skills (B = 0.42, SE = 0.10, bootstrapped p < 0.01, BC95% CI: 0.15 to 0.51), while controlling for cardiovascular risk factors, CSF Aβ1–42, CSF p-tau, and SVD. CSF p-tau was not directly associated with any cognitive outcomes. Executive functions were not directly associated with any of the ATN components.
Indirect associations (X ⟶ M ⟶ Y)
SVD was indirectly associated with global cognition, as mediated by CSF Aβ1–42, while controlling for cardiovascular risk factors, CSF p-tau and t-tau (Table 2). SVD was also indirectly related to global cognition as mediated by total grey matter volume and hippocampal atrophy, while controlling for cardiovascular risk factors, CSF Aβ1–42 and CSF p-tau.
Regression weights for the indirect associations between SVD and cognition as mediated by ATN
B, standardized beta; SE, standard error; CI, confidence interval. ∗bootstrapped p < 0.05, ∗∗bootstrapped p < 0.01.
SVD was indirectly associated with memory, as mediated by CSF Aβ1–42, while controlling for cardiovascular risk factors, CSF p-tau and hippocampal atrophy (Table 2). SVD was also indirectly associated with memory as mediated by total grey matter volume and hippocampal atrophy, while controlling for cardiovascular risk factors, CSF Aβ1–42 and CSF p-tau.
SVD was not indirectly related to executive function or visuospatial function via the ATN pathway.
Moderation analysis
The path analysis regression models with SVD interacting with ATN components as the predictors and cognition or hippocampal volume as the outcome had excellent model fit according to recommended criteria [44].
SVD did not interact with CSF Aβ1–42, CSF p-tau or CSF t-tau to affect global cognition or cognitive domains. A post-hoc analysis indicated that SVD did not interact with CSF Aβ1–42 to affect hippocampal volumes.
DISCUSSION
Main findings
In a cohort of patients with YOD, we showed that the prevalence of SVD burden was highly concomitant with the prevalence of low CSF Aβ1–42, even when SVD+and SVD- participants were matched on age, gender, education, APOE ɛ4 status, global cognition, cardiovascular risk factors, and grey and white matter volume. SVD was associated with lower CSF Aβ1–42 and greater neurodegeneration in an AD-specific region, the hippocampus. Meanwhile no associations were observed between SVD and CSF p-tau or CSF t-tau. Cognitive impairment was directly associated with low CSF Aβ1–42, but not with SVD. Rather, SVD was indirectly associated with global and memory impairment via reduced CSF Aβ1–42 levels and increased neurodegeneration in total grey matter and in the hippocampus. SVD was not found to moderate the strength or direction of the relationship between ATN and cognition. Our findings suggest that in YOD, SVD is linked to the ATN pathway and indirectly drives cognitive impairment via this pathway.
Associations between SVD, amyloid, and neurodegeneration
Aβ1–42 deposition is believed to be first in a line of upstream events that cause AD-related dementia [14]; however, opinions building on this theory suggest that vascular dysregulation may be the earliest pathological event contributing to the development of AD [47]. Interestingly, the role of vascular dysregulation on the cascade of ATN events appear to vary between LOD and YOD. Previous studies in LOD identified that SVD had a more delayed effect on the ATN sequelae by affecting tau aggregation [19]. While our findings in YOD suggested that SVD was associated with earlier mechanisms such as Aβ1–42 deposition, but not tau aggregation. Thus while vascular dysregulation may be one of the earliest pathological events contributing to AD [47], the mechanism by which it contributes to Aβ1–42 and tau pathology may vary between LOD and YOD. How SVD affects Aβ1–42 in YOD remains unclear, although insights from in vitro studies suggest that the vascular system may be involved in promoting Aβ1–42 aggregation [24], restricting clearance of Aβ1–42 [25] and causing vascular-related Aβ1–42 [48].
We found SVD and CSF Aβ1–42 were each associated with AD-pattern neurodegeneration, namely hippocampal atrophy. SVD did not interact with Aβ1–42 to affect hippocampal atrophy, suggesting each mechanism had an independent effect on neurodegeneration. Previously, SVD has been shown to have a stronger effect on temporal lobe atrophy than Aβ deposition in healthy elderly, and this association was strongest in individuals with amyloid burden [49]. An in vivo study demonstrated that while hippocampal atrophy was similar between patients with AD and SVD, the cause of neural loss differed; neural loss in AD was caused by amyloid deposition and neural loss in SVD was caused by microvasculature pyramidal cell loss [50]. Consistent with these in vivo studies, our findings suggest that SVD and CSF Aβ1–42 each have additive effects on AD-pattern neurodegeneration, likely resulting from different pathogenic mechanisms.
Associations between SVD and amyloid with cognition
Low CSF Aβ1–42 was associated with memory impairment. A moderate effect size was observed, suggesting that YOD patients with low Aβ1–42 may exhibit observable memory deficits. CSF Aβ1–42 was not associated with any other cognitive domains, which is consistent with repeated findings that amyloid pathology has a greater influence on memory-related systems than other cognitive domains [51]. In comparison to Aβ1–42, SVD was not directly associated with cognitive impairment nor did it interact with Aβ1–42 to affect cognition. While SVD was associated with hippocampal atrophy, it did not interact with Aβ1–42 to affect hippocampal atrophy. Thus despite being associated, SVD and Aβ1–42 work independently to effect both cognitive impairment and neurodegeneration. Comparatively, studies with LOD cohorts have demonstrated SVD predicts cognitive decline alone [19] and in some cases synergistically with Aβ1–42 [21]. Thus, it is likely that young age may protect against the effects of SVD on cognition; however, may not protect against the effects of AD pathology on cognition.
One mechanism by which SVD was related to cognitive impairment was indirectly via increasing neurodegeneration and lowering CSF Aβ1–42. This was observed for both global cognition and memory. The former indirect effect suggests that neural loss may be critical for SVD to manifest clinically in YOD patients. The later indirect effect suggests that while SVD is not related to clinical outcomes in YOD, it is related to other disease mechanisms such as Aβ1–42 deposition. We further note that the size of the mediation effect was small when the outcome was global cognitive impairment, while the effect size was moderate when the outcome was memory; suggesting that the effect of SVD lowering CSF Aβ1–42 may be most detrimental for memory functions. These findings support previous perspectives [47] that improving the integrity of small blood vessels in the brain may be a means to prevent AD before it begins. We build on this past research by demonstrating that in YOD, improving the integrity of small blood vessels may potentially reduce the accumulation of AD pathology such as Aβ1–42 and neurodegeneration, which may have consequences on reducing cognitive impairment. We note this perspective needs to be interpreted with caution as our study was cross-sectional and further longitudinal research is required to investigate causal links between SVD, ATN, and cognition.
Limitations and future research
One limitation is that our findings were based on a modest sized sample with cross-sectional data, thus causal relationships cannot be inferred. We note that the clinical diagnosis of AD was not biomarker supported. Clinical criteria were used to diagnose patients given the AD biomarkers were used as predictors in the analysis. We further note the mean CSF Aβ1–42 level in our cohort was in the normal range, while the tau levels were in the abnormal range, suggesting AD in our cohort could have been predominantly tau driven. We further note we did not have a comparison group, such as healthy controls or an LOD cohort. Future research would benefit from comparing YOD with LOD from a single cohort to ensure consistency in methodology.
Conclusion
In young patients with dementia, the prevalence of SVD is highly concomitant with the prevalence of low CSF Aβ1–42. Cognitive impairment in this young population was directly associated with low CSF Aβ1–42, but not with SVD. Rather, SVD was related to AD disease mechanisms, namely Aβ1–42 deposition and hippocampal atrophy. By affecting upstream and downstream phases of the ATN pathway, SVD was indirectly associated with cognitive impairment. These findings support further research into evaluating whether management of SVD will impact the clinical course of YOD.
Availability of data and materials
The dataset analyzed during the current study are not publicly available but is available upon reasonable request from the corresponding author.