
Abstract
Background:
A disease-modifying therapy for Alzheimer’s disease (AD) is still an unmet clinical need. The formation of amyloid-β (Aβ) requires the initial cleavage of the amyloid-β protein precursor (AβPP) by BACE1 (beta-site AβPP cleaving enzyme 1), which is a prime therapeutic target for AD.
Objective:
We aimed to design and develop a selective BACE1 inhibitor suitable to AD treatment.
Methods:
The new BACE1 inhibitors consist on a chimeric peptide including a sequence related to the human Swedish mutant form of AβPP (AβPPswe) conjugated with the TAT carrier that facilitates cell membrane permeation and the crossing of the blood-brain barrier. Additionally to the chimeric peptide in the L-form, we developed a D-retroinverso chimeric peptide. The latter strategy, never used with BACE1 inhibitors, is considered to favor a significantly higher half-life and lower immunogenicity.
Results:
We found that both chimeric peptides inhibit recombinant BACE1 activity and decrease Aβ40/42 production in Neuro-2a (N2A) cells expressing AβPPswe without inducing cytotoxicity. The intraperitoneal administration of these peptides to 3xTg-AD mice decreased plasma and brain Aβ40/42 levels, as well as brain soluble AβPPβ production. Also, a reduction of insoluble Aβ was observed in the brain after chronic treatment. Noteworthy, the chimeric peptides selectively inhibited the AβPP-β cleavage relatively to the proteolysis of other BACE1 substrates such as close homologue of L1 (CHL1) and seizure-related gene 6 (SEZ6).
Conclusions:
Overall these new BACE1 chimeric peptideshold promising potential as a selective disease-modifying therapy for AD.
INTRODUCTION
The accumulation of amyloid-β (Aβ) in the brain represents an early event in Alzheimer’s disease (AD) that likely initiates a complex pathogenic cascade [1–4]. The formation of Aβ requires the initial cleavage of amyloid-β protein precursor (AβPP) by beta-site AβPP cleaving enzyme 1 (BACE1) followed by γ-secretase cleavage of the membrane-associated C-terminal fragment. The BACE1 activity is the limiting step during Aβ formation as shown in knockout studies demonstrating that BACE1–/– mice crossed with AβPP transgenic mice do not produce Aβ and do not show Aβ-dependent memory deficits [5]. Therefore, BACE1 is a prime therapeutic target for AD. The likelihood of a therapy modulating Aβ levels as a disease-modifying strategy is highlighted as well by genetic evidence since the carriers of the AβPP Swedish mutation that increases β-site AβPP cleavage exhibit early-onset AD [6]. On the contrary, the AβPP-A673T mutation recently identified in individuals of the Icelandic population protects against AD and age-related cognitive decline possibly due to a decrease in the β-cleavage of AβPP, among other mechanisms [7–9].
Despite the initial findings that BACE1–/– mice did not show perceptible abnormalities, subsequent studies revealed that these mice display complex cognitive and neurochemical phenotypes. Deficient processing of BACE1 substrates associated to synaptic function such as neuregulin 1, close homologue of L1 (CHL1), or seizure-related gene 6 (SEZ6) may contribute to synaptic deficits underlying the BACE1–/– mice phenotypes [3, 10–12]. Nonetheless, several of the phenotypes arise from deficits due to BACE1 inhibition during development and thus may be less relevant to the adult subject. Indeed, mice in which the Bace1 gene was deleted in the whole body of the adult largely lacked the neurological phenotypes observed in germline BACE1 Knockout (KO) mice although the adult conditional BACE1 KO mice still exhibited axonal disorganization in the mossy fiber pathway of the hippocampus [13]. The sequential and increased deletion of BACE1 in adult mice reverses amyloid deposition, reduces gliosis and neuritic dystrophy, and improves synaptic functions [14], which holds BACE1 inhibition has an auspicious therapeutic strategy. On the other hand, many of the phenotypes revealed in mice were found in BACE1 KO animals, which is a more severe interference than pharmacological inhibition. Remarkably, current clinical trials with BACE1 inhibitors have not reported any major safety issues so far [4]. As well, it is worthy to note that a partial inhibition of BACE1 activity might be sufficient to prevent Aβ-driven AD-like pathology. Actually, in PDAPP mice a 50% reduction of BACE1 gene expression is sufficient to protect against Aβ deposition and synaptic dysfunction without compromising brain function [15]. Additionally, healthy middle-aged and elderly heterozygous carriers of the AβPP-A673T protective Icelandic mutation show, on average, a 28% reduction in both Aβ40 and Aβ42 plasma levels [16], suggesting a small decrease in Aβ production might have remarkable protective effects against late-onset AD [7]. Thus, a careful dosage titration of a potential BACE1 inhibitor might help to decrease Aβ production and improve AD patient cognitive functions while minimizing mechanism-based adverse effects.
Over the last years many efforts have been made to discover potent and efficient BACE1 inhibitors. The first generation of BACE1 inhibitors consisted of non-cleavable peptide-based transition state analogues designed after the amino-acid sequence in AβPP at which β-secretase cleaves. Although these large peptidomimetic molecules are very potent BACE1 inhibitors in vitro they did not possess favorable in vivo pharmacological properties, such as oral bioavailability, long serum half-life, or blood-brain barrier (BBB) penetration, hindering their clinical use [17]. More recently, potent third-generation small-molecule BACE1 inhibitors have been developed that achieve satisfactory brain penetration and robust cerebral Aβ reduction in preclinical animal models [17, 18]. Some of these orally bio available BACE1 inhibitor drugs have entered into human clinical trials; however, so far, none of these compounds successfully passed through the phase III [17, 20].
The aim of this work was to develop new BACE1 inhibitors that may exceed some of the limitations of previous compounds [17]. For this purpose we designed several peptide sequences related to the AβPP substrate, which were conjugated to the TAT carrier peptide that enables cell membrane permeation and BBB crossing [21–24]. The endocytic pathways take particular relevance among the translocation mechanisms suggested for carrier peptides and, in many cases, the majority of the imported cargo is trafficked to the endosomal compartments [25, 26], which for our purpose is an advantage since BACE1 localizes preferentially in acidic compartments such as the endosomes. In plus, considering the AβPP cleavage by BACE1 occurs in the endosomes, this feature may allow for the selective inhibition of AβPP-β cleavage while sparing the proteolysis of other BACE1 substrates that are processed in an endocytosis-independent manner [27]. Moreover, in order to increase proteolytic stability, all the chimeric peptides were designed as well with protease-resistant D-amino acids in the retroinverso form (RI) to best mimic the structure of the natural peptide. Besides increasing the stability of biologically active peptides for in vivo application this innovative strategy has the benefit of D-RI-chimeric peptides being less immunogenic. Therefore, we hypothesized the chimeric peptides, related to a BACE1 substrate, would act as selective inhibitors of the AβPP-β cleavage while minimizing the interference with the proteolysis of other endogenous BACE1 substrates beyond endogenous AβPP, and thus enable a decrease in Aβ production and deposition.
The ability of the compounds to inhibit BACE1 was initially evaluated using an in vitro cell-free system. This screening allowed to select the most promising chimeric peptide and its D-RI form, which showed to inhibit BACE1 by decreasing endogenous Aβ40 and Aβ42 production in cellular (N2A-AβPPswe cells) and animal (3xTg-AD mice) AD models without affecting soluble AβPPα (sAβPPα) levels. Accordingly, chronic intraperitoneal (i.p.) administration of the tested chimeric peptides decreased insoluble Aβ in the brain of 3×Tg-AD mice. Remarkably, in N2A cells the chimeric peptides inhibit AβPP-β cleavage but not the processing of other endogenous BACE1 substrates such as CHL1 and SEZ6. This work resulted in the discovery of innovative and selective BACE1 inhibitors for preventing and delaying the progression of AD. Moreover, to the best of our knowledge, this is the first report of peptidic BACE1 inhibitors synthetized with D-amino acids retaining functional activity.
METHODS
Cell-free assay for BACE1 activity
The chimeric peptides were synthesized by Auspep. The compounds ability to inhibit BACE1 was evaluated by an in vitro cell-free assay using the BACE1 Activity Assay kit (CS0010 Sigma). Briefly, recombinant BACE1 was pre-incubated with different concentrations of the chimeric peptides for 15 min at 37°C followed by addition of the substrate 7-Methoxycumarin-4-acetyl-[Asn670, Leu671]-Amyloid β/A4 Precursor Protein 770 Fragment 667–676-(2,4 dinitrophenyl) Lys-Arg-Arg amide trifluoroacetate salt. The experiment was conducted for 2 h at 37°C according to the manufacturer’s protocol. Fluorescence was measured using a 96-well plate for fluorescence assay in a Fluorimeter Spectra Max Gemini EM.
Docking studies
The crystal structure of BACE1 was retrieved from Protein Data Bank (PDB code: 1FKN) and prepared using the Molecular Operating Environment (MOE) 2016.08 software package [28]. The ligand and crystal waters were removed and the receptor structure was protonated (at pH 6.0 and 300 K) using the Protonate 3D tool. OPLS-AA force field [29] was used to assign atom types and charges to each atom in the receptor structure, which was further energy-minimized, using the same force field. The 3D-structure of the chimeric peptides were predicted by using PEP-FOLD3 tool [30], accessible from the online server Mobyle@RPBS (http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/). Their structures were further prepared by using the protocol described for BACE1 3D structure. Afterwards, docking studies with BACE1 and the chimeric peptides were performed by using the Easy interface protocol of HADDOCK server http://haddock.science.uu.nl/services/HADDOCK2.2/ [31]. The active residues were defined by considering the binding site residues, previously identified by the Site Finder tool, in MOE package. The passive residues were automatically defined. Default parameter settings were used for docking calculations.
Neuroblastoma cell culture
The mouse neuroblastoma cell line stably expressing the human Swedish-mutant AβPP695 (N2A-AβPPswe cells) was provided by Dr. Ciro Isidoro, Università del Piemonte Orientale “A. Avogadro”, Novara, Italy [32]. The N2A-AβPPswe cells were maintained in DMEM (Sigma, D5648) medium supplemented with 10% (v/v) heat inactivated fetal bovine serum (FBS), 0.4 mg/mL geneticin, 3.7 g/L sodium bicarbonate, 1% (w/v) non-essential amino acids and 1 mM sodium pyruvate. The mouse neuroblastoma N2A cell line was cultured in the same medium except for the antibiotic solution (10,000 U/mL penicillin, 10,000μg/mL streptomycin). Cells were cultured in 75 cm2 flasks and maintained in a humidified 5% CO2– 95% air atmosphere at 37°C, and the medium was changed every 3 days. N2A cells were used from passages 22 to 36 and N2A-AβPPswe from passages 13 to 26.
Cell viability assays
The metabolic activity of N2A and N2A-AβPPswe cells was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay as previously described [33]. Cells were plated at a density of 1.3×105 cells/cm2. Twenty-four hours after plating, N2A-AβPPswe cells were incubated in serum-free medium with PEP5 or PEP6 (the two most promising chimeric peptides we developed) and kept at 37°C for 24 h in the incubator. The reduction of MTT was expressed as percentage of the absorbance value obtained in control cells, which was considered to be 100%. Cells not challenged with the chimeric peptides (untreated cells) were used as control.
Cell death was assessed by determining lactate dehydrogenase (LDH) release. For this purpose, N2A and N2A-AβPPswe cells were plated at a density of 1–1.3×105 cells/cm2 and 24 h later were incubated with PEP5 or PEP6 in serum-free medium and kept for 24 h at 37°C in the incubator. The cell culture medium was collected and the LDH release assay (Cytotox 96 Non-Radioactive Cytotoxicity Assay, PROMG17800001) was performed as previously described [33]. The percentage of LDH release was calculated as the ratio of LDH activity in the extracellular medium against the total LDH activity obtained after complete cell lysis with Triton X-100. The Caspase-3/7 activity was determined in N2A-AβPPswe cells by the Caspase-Glo® 3/7 Assay (PROMG8091,) using white-walled 96-well plates (Corning) adequate for cell culture (1×105 cells/cm2) and compatible with the luminometer (Luminometer Lmax II). The assay was performed according to the manufacturer’s protocol. The results were normalized to the control caspase-3/7 activity, which was considered as 1. Untreated cells were used as control.
Monitoring of BACE1 inhibitor cellular uptake by fluorescence microscopy
To analyze the cellular uptake of the BACE1 inhibitor the N2A cells were seeded in μ-Slide 8 Well ibiTreat at a density of 1×105 cells/cm2. Cells were washed twice with PBS, and incubated with 2μg/mL Hoechst 33342 for 10 min. Then, cells were incubated with 20μM PEP6 fluorescently labelled at the N-Terminus with Cy5.5 (Cy5.5-PEP6) (Auspep) for 30 min at 37°C in Phenol Red-free DMEM (Sigma Aldrich, D5030). In order to investigate whether the cellular uptake of PEP6 is carried out by endocytosis, cells were pre-incubated for 30 min at 37°C with 30μM Chlorpromazine (CPZ). The fluorescence was visualized by confocal microscopy (Zeiss LSM710 inverted microscope) using a Plan Apochromat 63×/1.40 Oil objective. The quantitative analysis of microscopy images for PEP6 uptake was performed using Image J software. The threshold levels were visually selected and the integrated fluorescence intensity was obtained by multiplying the area by the mean grey value and then normalized to the total cell number in each image.
In vivo testing of BACE1 inhibitor effects
Experiments were performed using the triple transgenic mouse model of AD (3×Tg-AD mice) [34], a generous gift from Dr. Frank LaFerla, University of California, Irvine, CA, USA. The mice were bred and maintained at the animal house of the Center for Neuroscience and Cell Biology (CNC)-Faculty of Medicine, at the University of Coimbra, under a constant temperature, humidity and a 12 h light/dark cycle. All animals were housed in groups of two to five in enriched cages. Food and water were available ad libitum. In total we used 61 animals in the different assays and mice were grouped as control (n = 21), treated with PEP5 (n = 20), and treated with PEP6 (n = 20). The initial population was 62 mice but one control animal died after intraperitoneally (i.p) injection. Four-month-old mice weighing 20–28 g were injected (i.p) with PEP5 or PEP6 (1.25 mg/Kg), or with saline. Injections were performed in the morning. Twenty-four hours later the animals were sacrificed by decapitation after being anesthetized under halothane atmosphere which is commonly used and has a higher potency and a lower minimum alveolar concentration than isofluorane. Moreover, halothane is less irritant to the respiratory airways and the odor is not intense, which avoids animal distress due to the anesthetic properties [35]. For chronic treatment, 4-month-old mice were daily injected via i.p. for 4 or 6 months and then sacrificed 30 h after the last injection, as described. The brain of each mouse was dissected out in a plate placed on ice and the samples immediately frozen and stored at – 80°C. Also, the blood of animals submitted to the acute and 4-month chronic treatments was collected by exsanguination into EDTA-treated tubes and the plasma obtained after blood centrifugation at 1,000×g for 15 min at 4°C was stored at – 80°C until use. Within each assay control and treated animals were treated and accessed in parallel. The health and clinical status of the animals were monitored daily in order to assess possible adverse effects of the compounds. During the chronic treatment, the animals’ body weight was monitored daily. No animal presented adverse reactions, changes in general health or weight loss. All animal procedures were done in accordance with the European Union Guidelines for Animal Research, Directive 2010/63/EU of September 22, 2010 and approved by the Ethics Committee of the animal house of CNC (ORBEA_39_2013/01032013-2) and the Portuguese Veterinarian Office (Direção Geral de Alimentação e Veterinária – 0421/000/000/2013). The number of animals used was minimized to reduce animal suffering. The in vivo studies were carried out only with the most promising compounds that were initially screened in vitro and in AD cellular models.
Sandwich ELISA
The levels of Aβ40 and Aβ42 in the N2A-AβPPswe cells conditioned medium, in the brain homogenates and in the plasma from 3×Tg-AD mice were determined using a sandwich Enzyme-Linked Immunosorbent Assay (ELISA)(KHB3441, KHB3442, KHB3481, KHB3482, Invitrogen) following the manufacturer’s instructions. For that purpose, N2A-AβPPswe cells were plated at 1×105 cells/cm2 in 6-well plates and 24 h later were incubated in serum-free medium containing the peptides for 24 h at 37°C. After treatment, the conditioned medium was collected and centrifuged at 16,100×g for 10 min at 4°C. Then, the samples were diluted (1 : 15 for Aβ40 and 1 : 5 for Aβ42) and analyzed by ELISA. The results were expressed as percentage of the Aβ levels in the conditioned medium of control cells.
Regarding in vivo studies, to evaluate Aβ soluble levels, the mice brains were homogenized on ice-cold RIPA (Radio immuno precipitation Assay) buffer (250 mM NaCl, 50 mM Tris base, 1% Nonidet P-40, 0.5% Sodium Deoxycholate, 0.1% Sodium Dodecyl Sulfate (SDS), pH 8.0) supplemented with 1% (v/v) of the protease inhibitor cocktail (Sigma), using 2.5 mL of buffer per 0.5 g tissue. After 15 min incubation the lysates were centrifuged at 350,000×g for 20 min at 4°C. The supernatants were collected, diluted (1 : 2) and subjected to ELISA. To determine insoluble Aβ levels, the brain cortex was lysed on ice-cold RIPA buffer (1 mL/150 mg tissue). Lysates were incubated for 15 min and the insoluble material was pelleted by centrifugation for 30 min at 16,100×g at 4°C. The pellets were dissolved in 5 mL of cold 5 M guanidine HCl at room temperature (RT) for 30 min to obtain the Aβ insoluble fraction. Then, samples were diluted in RIPA buffer (1 : 20) and analyzed by ELISA. To measure Aβ peptides in the blood of 3×Tg-AD mice, plasma samples were diluted (1 : 2) and examined by ELISA. The results were normalized to Aβ levels of control animals (injected with vehicle), which were considered as 1.
Western blotting
The supernatant of mice brain lysates was obtained as described above for the ELISA test. The supernatants were stored at – 80°C until used.
The N2A cells were seeded at a density of 1.3×105 cells/cm2. After a 24 h incubation with the chimeric peptides or the β-Secretase Inhibitor IV (Millipore), the conditioned serum-free medium was collected and frozen at – 20°C for protein precipitation and the N2A cells were scrapped and lysed in ice-cold RIPA buffer supplemented with 1% (v/v) of the protease inhibitor cocktail. Cell lysates were maintained on ice for 20 min and then centrifuged at 17,968×g for 10 min at 4°C and the supernatants were collected and stored at – 20°C.
The protein concentration of mice brain and N2A cells lysates was determined by the bicinchoninic acid protein assay (Pierce Biotechnology). The samples were denatured for 5 min at 95°C in 6× concentrated sample buffer [0.5 M Tris, 30% (v/v) glycerol, 10% (w/v) SDS, 0.6 M dithiothreitol, 0.012% bromophenol blue]. To precipitate protein present in the conditioned medium 1.2 mL acetone: water (80 : 20) were added to 800μL of conditioned medium. The samples were maintained at – 20°C for 1 h and then centrifuged at 12,000×g for 10 min at 4°C. The pellet was resuspended in 6× concentrated sample buffer and denatured as described before.
The mice brain lysates and N2A cells lysates containing 75μg of protein and the total content of precipitated protein from the conditioned medium were separated by electrophoresis in 10% (w/v) SDS polyacrylamide gels (SDS/PAGE). Proteins were then transferred to Polyvinylidenedifluoridemembranes (Millipore) in a suitable buffer for 2 h at 750 mA. The membranes were blocked with 5% (w/v) BSA in Tris-buffered saline (TBS) (150 mM NaCl, 25 mM Tris-HCl, pH 7.6) containing 0.1% Tween 20 (TBS-T) for 1 h at RT and incubated overnight at 4°C with the primary antibodies, which were as follows: rabbit anti-AβPP (1 : 5,000; # A8717 Sigma), monoclonal mouse Anti-BACE (2μg/mL; # MAB5308 Millipore), mouse anti-human sAβPPβ-swe (5μg/mL; #18957 IBL), mouse anti-human sAβPPα (5μg/mL; #11088 IBL), rabbit anti-sAβPPβ-wt (1 : 1,000; #813401 BioLegend), goat anti-CHL1 (1 : 200; #AF2147 R&D Systems, 2018) and sheep anti-SEZ6 (1 : 200; #BAF4989 R&D Systems), a kind gift from Dr Moechars, Janssen Pharmaceutica, Belgium. After six washes with TBS-T, the membranes were incubated for 1 h, at RT, with an alkaline phosphatase-linked secondary antibody, specific to goat (1 : 5,000; #sc-2771 Santa Cruz Biotechnology), mouse (1 : 20,000; GE Healthcare), rabbit (1 : 20,000; #NIF1317 GE Healthcare) or sheep (1 : 10,000; #713-055-147 Jackson Immuno Research). The protein immunoreactive bands were visualized by chemifluorescence with the Enhanced Chemifluorescence substrate (GE Healthcare) in a Versa Doc Imaging System (Bio-Rad). After stripping, the membranes of the lysates were reprobed with a monoclonal anti-β-actin antibody (1 : 10,000; #A5441 Sigma) for protein loading control. In the case of the conditioned medium, the membranes were stained with Coomassie Blue (Phastgel blue R – Sigma) to visualize total protein. The optical density of the bands was quantified with the Quantity One Software (Bio-Rad). The results were normalized to β-actin or total protein and were expressed as the relative amount compared to the control.
Plasma cytokine levels
Mice chronically treated with the chimeric peptides or saline for 4 months were sacrificed and the blood was collected by exsanguination. The plasma levels of ten cytokines interleukin (IL)-1β, IL-6, Tumor Necrosis Factor Alpha (TNF-α), IL-2, IL-12, IL-4, IL-5, IL-10, Granulocyte-macrophage Colony-stimulating Factor (GM-CSF), Interferon Gamma (IFN-γ) were determined through a multiplex array using the Bio-Plex™ system (a service provided at Biocant Park, Cantanhede, Portugal).
Statistical analysis
To test if the values come from a Gaussian distribution we used the D’Agostino-Pearson Omnibus normality test. When normality was not followed the results were analyzed by using the Mann-Whitney non-parametric test, or the Kruskal-Wallis non-parametric test followed by Dunn’s post hoc test for multiple comparisons. Otherwise, results were analyzed using the one-way analysis of variance followed by Dunnett’s post hoc test. A value of p < 0.05 was considered significant. No sample calculation was performed. Prism 6.0 (GraphPad Software) was used for all statistical analysis and the results were expressed as mean±SEM. We did not use any test to identify outliers and any data point was excluded.
RESULTS
The chimeric peptides inhibit BACE1 activity in vitro
The development of BACE1 inhibitors faces many challenges and at present there is no drug approved for clinical use. Thus, our goal was to conceive a new inhibitor of BACE1 that could overcome some of the limitations of the molecules previously developed. For this purpose we designed several AβPP-based peptide sequences (11 amino acids) that were conjugated to the TAT carrier peptide (Table 1), which permeate the cell membrane and cross the BBB [21–24]. The chimeric peptides contain the TAT (48–57) peptide covalently linked to the sequence flanking the Met-Asp672 β-cleavage site present on human AβPP or to the sequence flanking the Leu-Asp672 β-cleavage site on the human Swedish mutant AβPP (AβPPswe) that has an increased affinity for BACE1. Also, we designed another peptide based on the AβPP sequence flanking the β-cleavage site on AβPPswe but with a variation of the amino acids sequence (hereafter designed by Artificial Variant). Of note, the chimeric peptides do not include the full Aβ sequence in order to avoid amyloid formation and/or accumulation.
BACE1 activity in the presence of the chimeric peptides
The amino acid sequences of the chimeric peptides (synthesized by Auspep, Australia) including a peptide related to AβPP-or AβPPswe-β-cleavage site conjugated to the TAT (48-57) carrier peptide (GRKKRRQRRR) are shown. A screen for the BACE1 inhibitory activity of the chimeric peptides (100μM) was performed in cell-free system using a fluorescence resonance energy transfer-based assay kit. The results are expressed as the percentage of BACE1 activity inhibition and represent the mean±SEM of 3–7 independent experiments.
The number of amino acids in the AβPP-related sequences was chosen bearing in mind that the BACE1 active site pocket accommodates eight side chains and considering three more residues to work as a spacer between the cargo and the TAT carrier. Noteworthy, the chimeric peptides should be selectively recognized by BACE1 without interfering with AβPP cleavage mediated by α-secretase and BACE2. Therefore, we selected the AβPP sequence flanking Asp672 instead of the sequence flanking the Tyr-Glu682 cleavage site that is also recognized by BACE1, to use an AβPP sequence distant from the cleavage site recognized by α-secretase and from the in vivo preferential cleavage sites of BACE2.
Additionally, the chimeric peptides were designed as well with protease-resistant D-amino acids in the retroinverso form (RI) to increase proteolytic stability. The D-RI form leaves the surface topology of the amino acids side chains intact, mimicking the structure of the L-peptide. This double inversion of the peptide was shown to allow, in many cases, an in vivo application without loss of function [23, 37]. Moreover, the C-terminal of all peptides was modified by amidation.
The efficacy of the chimeric peptides to inhibit BACE1 was analyzed by an in vitro cell-free assay as described in the Methods section. As shown in Table 1, for the same concentration (100μM), the chimeric peptide based on AβPPswe (PEP3) had a higher inhibitory effect than the chimeric peptide based on AβPP wild type (PEP1), as expected due to the higher affinity of AβPPswe for BACE1. The most effective peptides as BACE1 inhibitors were PEP3 (% of inhibition 79.86±3.09) and PEP5 (% of inhibition 88.38±3.67) that is the chimeric peptide including the Artificial Variant of AβPPswe. Thus, we selected PEP5 as one of the most promising peptides to continue our study. Also, since we were interested in investigating the biological activity of a D-RI-chimeric peptide we selected PEP6 (% of inhibition 60.45±4.12) that is the D-RI form of PEP5. Figure 1 shows the percentage of BACE1 activity inhibition at different concentrations of PEP5 and PEP6.

Inhibition of BACE1 activity versus the PEP5 and PEP6 concentration. Recombinant BACE1 was incubated with different concentrations (0–100μM) of the PEP5 or PEP6 chimeric peptides as indicated in the Methods section. The results are expressed as the percentage of BACE1 activity inhibition and represent the mean±SEM of 3–7 independent in vitro experiments (n = 3, 0.01–100μM PEP6 and 10, 50μM PEP5; n = 6, 0.010 and 0.1μM PEP5; n = 5, 1μM PEP5; n = 7, 100μM PEP5) carried out in triplicate.
Binding mode of the two most potent chimeric peptides to BACE1
In order to predict the binding mode of the most promising peptides, PEP5 and PEP6, into BACE1, docking studies were carried out using HADDOCK 2.2. The docking results revealed that both chimeric peptides can enter through the active site cleft via the PEP5/PEP6 cargo sequence (Fig. 2A), establishing different types of interactions with active site residues, namely the catalytic aspartate dyad (Asp32 and Asp228), the flap region (Tyr71, Thr72, Gln73, and Gly74), and the 10 s loop (Ser10 and Gln12). Figures 2B and 2C show all protein-peptide interactions between BACE1 and PEP5, and between BACE1 and PEP6, respectively. Hydrogen bond interactions involving Gly74 on BACE1 were established with both PEP5 (Ser16) and PEP6 (Glu4). Moreover, Asp32 plays an important role in the interaction profile of both peptides, by establishing a hydrogen bond interaction with PEP5 (Asn17) and a salt bridge with PEP6 (Asp1).
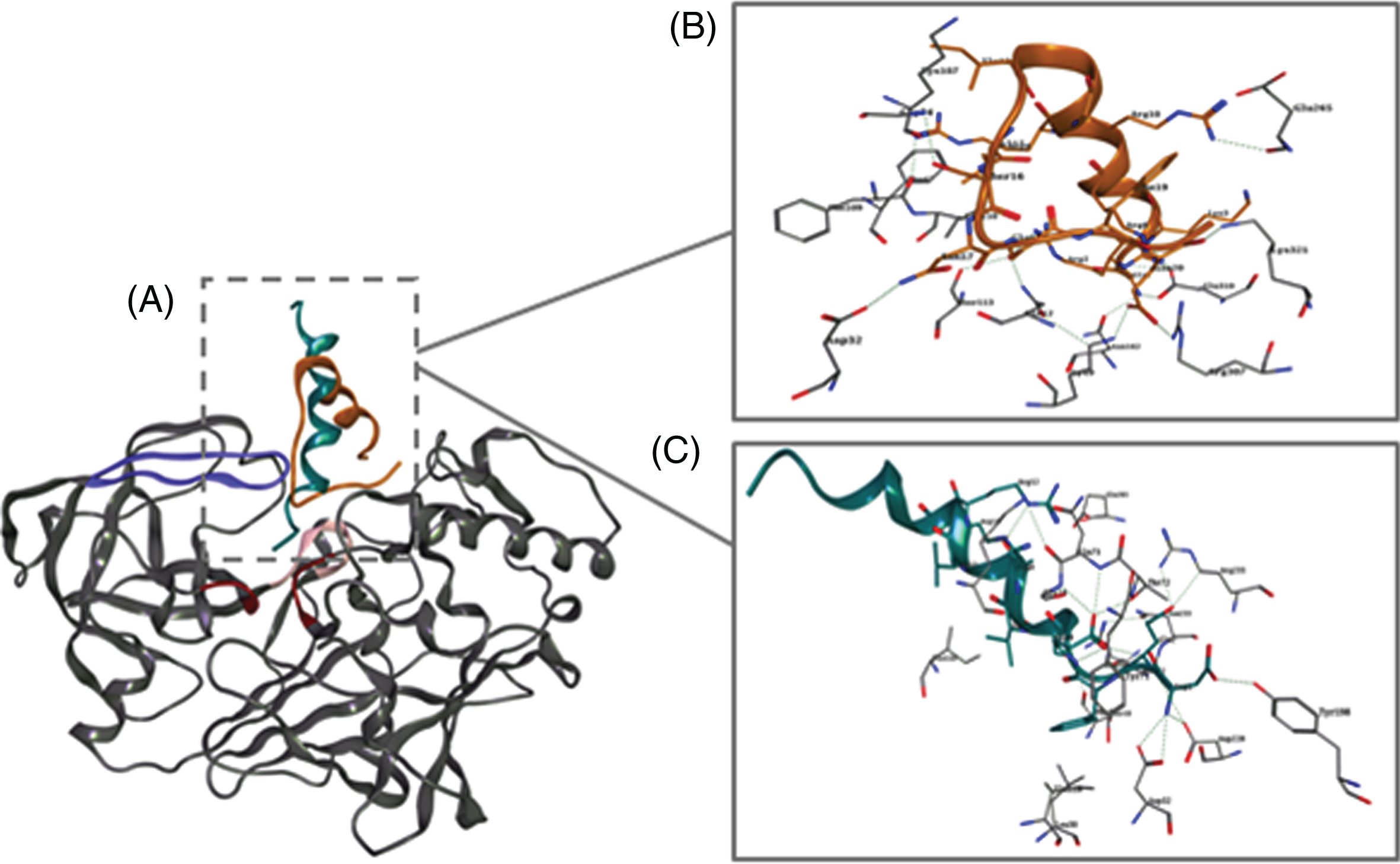
Docking simulations of PEP5 and PEP6 with human BACE1 (PDB code 1FKN) using software HADDOCK 2.2. A) Predicted binding mode of PEP5 (orange) and PEP6 (cyan) structures with BACE1 (grey). Molecular interactions between PEP5 (B) and PEP6 (C) with the active site residues of BACE1. Both PEP5 and PEP6 establish important interactions with key active site residues (e.g., Asp32, Asp228, Gly74, Tyr71). Red, purple, and light pink represent the catalytic motif, the flap region, and the 10s loop, respectively.
Moreover, for PEP5, an additional hydrogen bond was established between the chimeric peptide Glu18 and Gln12 on BACE1, as well as a salt bridge between the chimeric peptide Glu20 and Arg307 on BACE1. Among others, these interactions seem to facilitate the peptide-binding and its stability into BACE1 catalytic domain. On the other hand, the N-terminal region of PEP6 (Asp1) binds into a negative electrostatic region, establishing different interactions with BACE1 binding site residues, such as Asp32 and Asp228 (salt bridge), as well as with Thr72 and Tyr198 (hydrogen bond). Additionally, several interactions between Glu2 of PEP6 and BACE1 residues were found, such as a hydrogen bond with Asp228, two hydrogen bond with Thr232 and with Asn233, and a salt bridge with Arg235. Also, Phe3 of PEP6 establishes a π-interaction with Tyr71 of BACE1. A hydrogen bond network is observed as well between Glu4 of PEP6 and Thr72, Gln73, and Gly74 of BACE1. Furthermore, the presence of hydrophobic residues in both peptide structures, namely Ile13 and Val15 on PEP5, and Val7 and Ala8 on PEP6, seems to contribute to the establishment of alkyl interactions with Lys107 and Ile110 on BACE1, which can ultimately contribute to a better accommodation of both peptides into the cleft. These results might support the observed BACE1 enzymatic inhibition in presence of the chimeric peptides PEP5 and PEP6.
The new BACE1 inhibitors enter N2A cells by endocytosis
One of the limitations of previously developed BACE1 inhibitors has been its low cellular permeability [38]. Since we hypothesized that the new BACE1 inhibitors are internalized by endocytosis, we assessed the cellular uptake pattern of fluorescently labelled PEP6 (CY5.5-PEP6) in the N2A cell line in the presence or in the absence of CPZ, a cationic amphipathic drug that, in a micromolar range, inhibits clathrin-mediated endocytosis [39].
Preincubation of the cells with CPZ strongly inhibited the cellular uptake of 20μM CY5.5-PEP6 (Fig. 3A), suggesting that the new BACE1 inhibitor PEP6 enters the cells mainly through clathrin-mediated endocytosis, as expected.
As demonstrated by the MTT reduction assay (Fig. 3B) and the LDH release assay (Fig. 3C) the new BACE1 inhibitors PEP5 and PEP6 did not induce toxicity in N2A cells after an incubation period of 24 h.

New BACE1 inhibitors enter N2A cells by endocytosis and do not alter cell viability. N2A cells were treated with CPZ for 30 min, or remained untreated, followed by incubation for a further 30 min in the presence of PEP6 labelled with the fluorescent dye Cy5.5. The nuclei were labelled with Hoechst 33342. Live cells were visualized by confocal microscopy and the fluorescence intensity quantification was represented graphically (A). The fluorescence of N2A cells incubated with PEP6 in the absence of CPZ was considered as 100%. The results represent the mean±SEM of 5 independent experiments performed in duplicate. The viability of N2A cells treated with 50μM and 75μM PEP5 and 20μM and 50μM PEP6 was assessed by the MTT reduction assay (B) and cell death was assessed the LDH release assay (C) after 24 h incubation at 37°C. Untreated cells were used as control. The MTT results expressed as percentage of control represent the mean±SEM of at least 6 independent cell culture preparations performed in triplicate. The LDH results expressed as the percentage of total LDH represent the mean±SEM of at least 8 independent experiments performed in triplicate. Statistical analysis was made by non-parametric Mann-Whitney test for confocal microscopy and using the Kruskal Wallis test followed by Dunn’s multiple comparison post hoc test for cell viability assays. **p < 0.01 significantly different compared to control group.
BACE1 inhibitors decrease Aβ40 and Aβ42 generation in N2A-AβPPswe cells
The ability of PEP5 and PEP6 to inhibit Aβ production through the amyloidogenic AβPP processing pathway, as a consequence of BACE1 inhibition, was assessed in N2A-AβPPswe cells. After 24 h incubation with increasing concentrations (12.5–200μM) of PEP5 or PEP6 we analyzed the levels of Aβ40 (Fig. 4A) and Aβ42 (Fig. 4B) in the culture medium using a sandwich ELISA specific for each Aβ peptide. As expected, both chimeric peptides inhibited endogenous BACE1 activity. It was observed that 50μM PEP5 decreased the levels of secreted Aβ40 and Aβ42 to 59.33±8.74% and 38.68±5.45%, of control, respectively, whereas 75μM PEP6 decreased the levels of Aβ40 and Aβ42 to 54.67±8.34% and 37.38±7.62% of control, respectively. The IC50 values (Table 2) indicate that PEP5 inhibits more effectively Aβ production than PEP6 in N2A-AβPPswe cells, which is in agreement with the results regarding the inhibition of BACE1 activity obtained using a cell-free system.
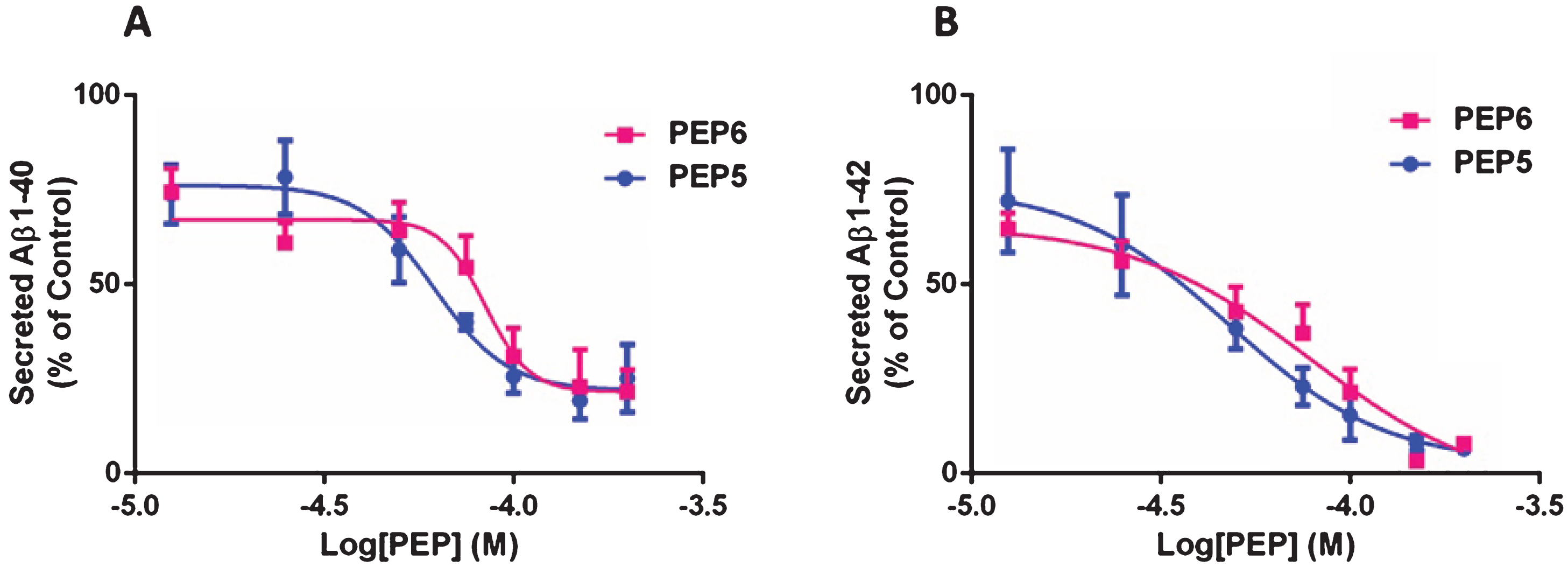
The new BACE1 inhibitors decrease Aβ40 and Aβ42 levels in Neuroblastoma-2A cells constitutively expressing AβPPswe. N2A-AβPswe cells were incubated in FBS-free medium with 12.5 to 200μM PEP5 or PEP6 for 24 h, at 37°C, in a humidified incubator with 5% CO2. Cells not challenged with the chimeric peptides (untreated) were used as control. The levels of secreted Aβ40 (A) and Aβ42 (B) were determined by ELISA. The results are expressed as percentage of control and represent the mean±SEM of 3–7 independent cell culture preparations.
Peptides IC50 determined in a cellular assay
The IC50 refers to the peptide concentration that inhibits endogenous Aβ40 and Aβ42 production in N2A-AβPPswe cells by 50%.
In order to exclude that the reduction in Aβ production was due to N2A-AβPPswe cell death we investigated cell viability by the MTT reduction assay, the LDH release assay, as well as by determining caspase-3/7 activity. A 24 h exposure to the new BACE1 inhibitors (PEP5 and PEP6), at concentrations close to the IC50 concerning Aβ42 production, did not affect the MTT reduction capacity of N2A-AβPPswe cells (Fig. 5A), indicating the compounds did not alter the general metabolic status of the cells. Also, 50μM PEP5 or 75μM PEP6 did not significantly change the release of LDH (Fig. 5B), nor altered caspase-3/7 activity (Fig. 5C). A positive control for the caspase-3/7 activity was performed with 10 nM staurosporine, (data not shown). These results rule out the possibility that the decrease in Aβ40 and Aβ42 production by the BACE1 inhibitor is a consequence of a decrease in cell viability, and support the contention that it is instead due to the inhibition of the AβPP amyloidogenic processing.

The new BACE1 inhibitors PEP5 and PEP6 at concentration near the IC50 do not induce cytotoxicity. N2A-AβPPswe cells were incubated with 50μM PEP5 or 75μM PEP6, in FBS-free culture medium for 24 h, at 37°C, in a humidified incubator with 5% CO2. Untreated cells were used as control. General metabolic activity was evaluated by the MTT reduction assay (A), necrotic cell death was assessed by evaluating the release of LDH (B), and apoptotic cell death was evaluated by measuring the activity of caspases 3/7 (C) at the end of the incubation period. The MTT results expressed as percentage of control represent the mean±SEM of 6 (PEP6) or 9 (PEP5) independent cell culture preparations. The LDH results expressed as the percentage of total LDH represent the mean±SEM of 7 independent cell culture preparations. The results of the caspase3/7 assay were normalized to control cells caspase activity and represent the mean±SEM of 2–4 independent cell culture preparations (n = 2 for 15 min and n = 4 for the 30–180 min time points). Ns, not statistically different from control cells as determined by Kruskal Wallis non parametric test followed by Dunn’s test for multiple comparisons.
The new BACE1 inhibitors decrease brain and plasma Aβ levels in 3×Tg-AD mice
After establishing that the chimeric peptides PEP5 and PEP6 inhibit Aβ40 and Aβ42 production in a cellular model of AD we next evaluated their efficacy in vivo using the 3×Tg-AD mouse model [34]. These mice progressively develop plaques and tangles, the neuropathological correlates of AD. Oddo et al. [34] described that Aβ oligomers begin to accumulate between 2 and 6 months of age, with continued age-dependent increase observed between 12 and 20 months. The extracellular Aβ deposits become apparent in 6-month-old mice in the cerebral cortex. Thus, 4-month-old mice were submitted to a single intraperitoneal administration of PEP5 or PEP6 (1.25 mg/Kg), or injected with saline, and sacrificed 24 h later. The dose was selected based on preliminary studies showing that 1.25 mg/Kg was the minimal dose tested that caused a significant decrease in Aβ levels 24 h after treatment (data not shown). Of note, our aim was to inhibit only a small percentage of Aβ production since it is known from the literature that this translates into a considerably greater effect on insoluble Aβ levels upon chronic administration [15]. As shown, both compounds decreased soluble brain and plasma Aβ levels. The chimeric peptides significantly decreased brain soluble Aβ40 (Fig. 6A, PEP5 : 0.730±0.064 and PEP6 : 0.783±0.036 versus control 1.000±0.035) and Aβ42 (Fig. 6B, PEP5 : 0.807±0.048 and PEP6 : 0.823±0.044 versus control 1.000±0.027) levels. Similar results were obtained in 3×Tg-AD mice blood plasma. We observed a significant decrease in Aβ40 levels (Fig. 6C) in mice treated with PEP5 (0.685±0.016) or PEP6 (0.655±0.031) when compared with control animals injected with saline (1.000±0.055). Both peptides were shown to decrease Aβ42 plasma levels as well (Fig. 6D, PEP5 : 0.876 767±0.065 and PEP6 : 0.737±0.069 versus control: 1.003±0.126) although the result is not statistically significant.
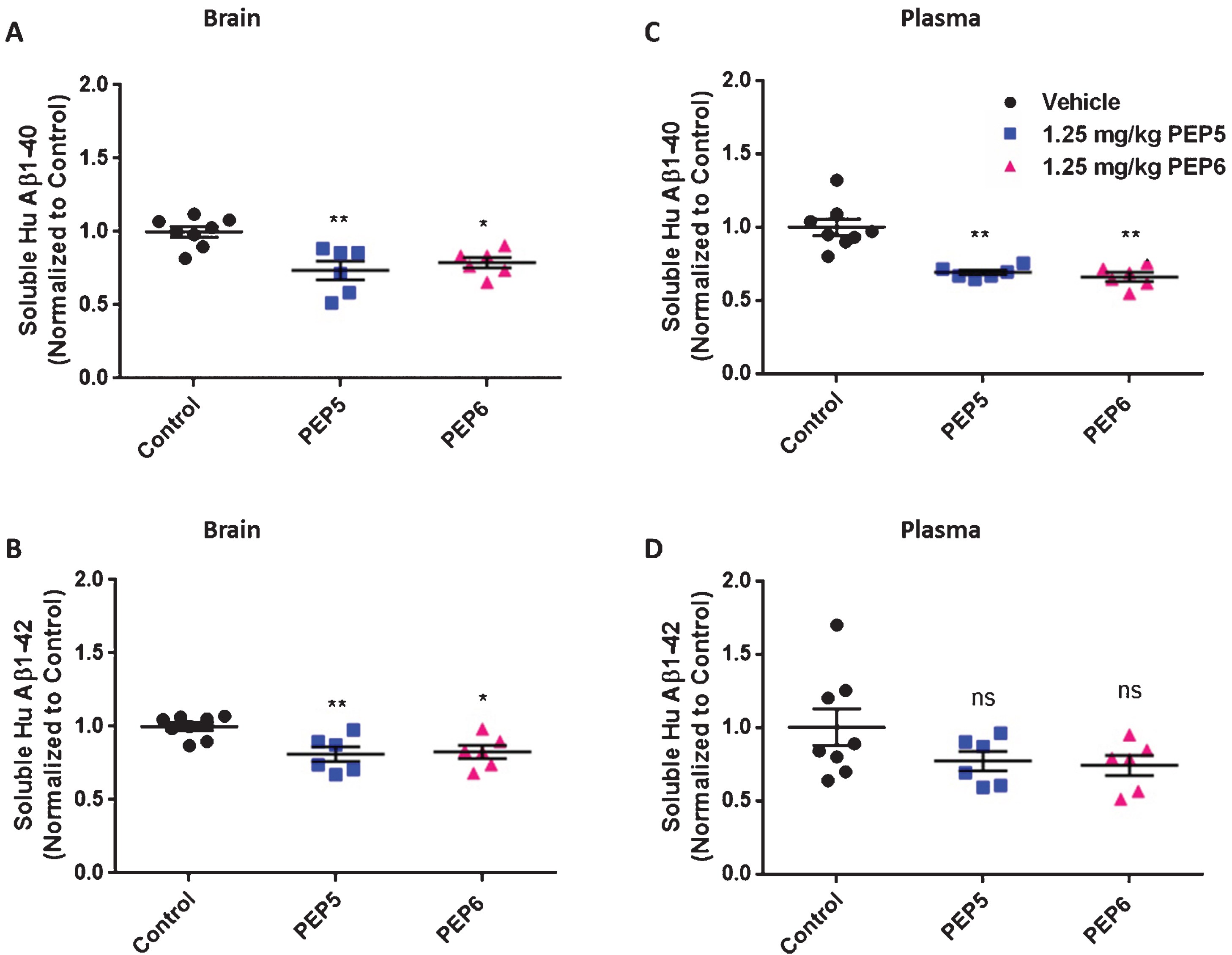
The new BACE1 inhibitors PEP5 and PEP6 decrease brain and plasma Aβ levels in 3×Tg-AD mice. Twenty-four hours after a single i.p. administration of the new BACE1 inhibitors PEP5 and PEP6 (1.25 mg/Kg) mice were sacrificed and brain (A and B) and plasma (C and D) soluble Aβ40 and Aβ42 levels were evaluated by ELISA. Control mice were injected with the vehicle (saline) in the absence of chimeric peptides. The results were normalized to control and represent the mean±SEM of n = 6–8 male mice per group. Statistical significance was determined by Kruskal Wallis non-parametric test followed by Dunn’s post hoc for multiple comparisons. *p < 0.05, **p < 0.01, significantly different compared to control group. ns: not statistically different from control group.
The new chimeric peptides selectively inhibit AβPP-β cleavage in vivo
The sAβPPβ fragment is the direct product of BACE1-mediated AβPP cleavage. Therefore, in addition to Aβ evaluation, we analyzed by Western blot the protein levels of sAβPPβswe in the brain of 3×Tg-AD submitted to treatment with the chimeric peptides. The administration of PEP5 and PEP6 at 1.25 mg/Kg decreased the levels of sAβPPβswe (Fig. 7A, B, PEP5 : 0.724±0.066 and PEP6 : 0.791±0.069 normalized to control considered as 1) but did not decrease either AβPP (Fig. 8A, B) or BACE1 protein levels (Fig. 8C, D), which demonstrates that the reduction in Aβ and sAβPPβswe levels is due to the inhibition of BACE1 activity and not because of a drop on the enzyme or its substrate amount. Moreover, we investigated whether the chimeric peptides could interfere with the non-amyloidogenic processing of AβPP by analyzing the protein level of the sAβPPα fragment, which results from AβPP cleavage by α-secretase. The results show that PEP5 and PEP6 did not alter brain sAβPPα levels (Fig. 7C, D, PEP5 : 1.063±0.105 and PEP6 : 1.001±0.037 normalized to control considered as 1), indicating that peptides 5 and 6 selectively inhibit BACE1 activity in 3×Tg-AD mice.

The new BACE1 inhibitors decrease sAβPPβswe but do not alter sAβPPα levels in 3×Tg-AD mice. Twenty-four hours after a single i.p. administration of PEP5 or PEP6, mice were sacrificed and brain lysates were used to determine sAβPPβswe (A and B) and sAβPPα (C and D) levels through Western blot analysis. Actin was used as loading control. The control mice were injected with the vehicle (saline) in the absence of chimeric peptides. Representative images for each protein are presented above the graph (V: Vehicle and T: Treated). In each case (A–D), the representative image was obtained from the same membrane. The results normalized to β-actin and expressed as the relative amount of control represent the mean±SEM of 4–6 male mice per group. Statistical analysis was performed by non-parametric Mann-Whitney test. *p < 0.05, significantly different compared to control group. ns: not statistically different from control group.
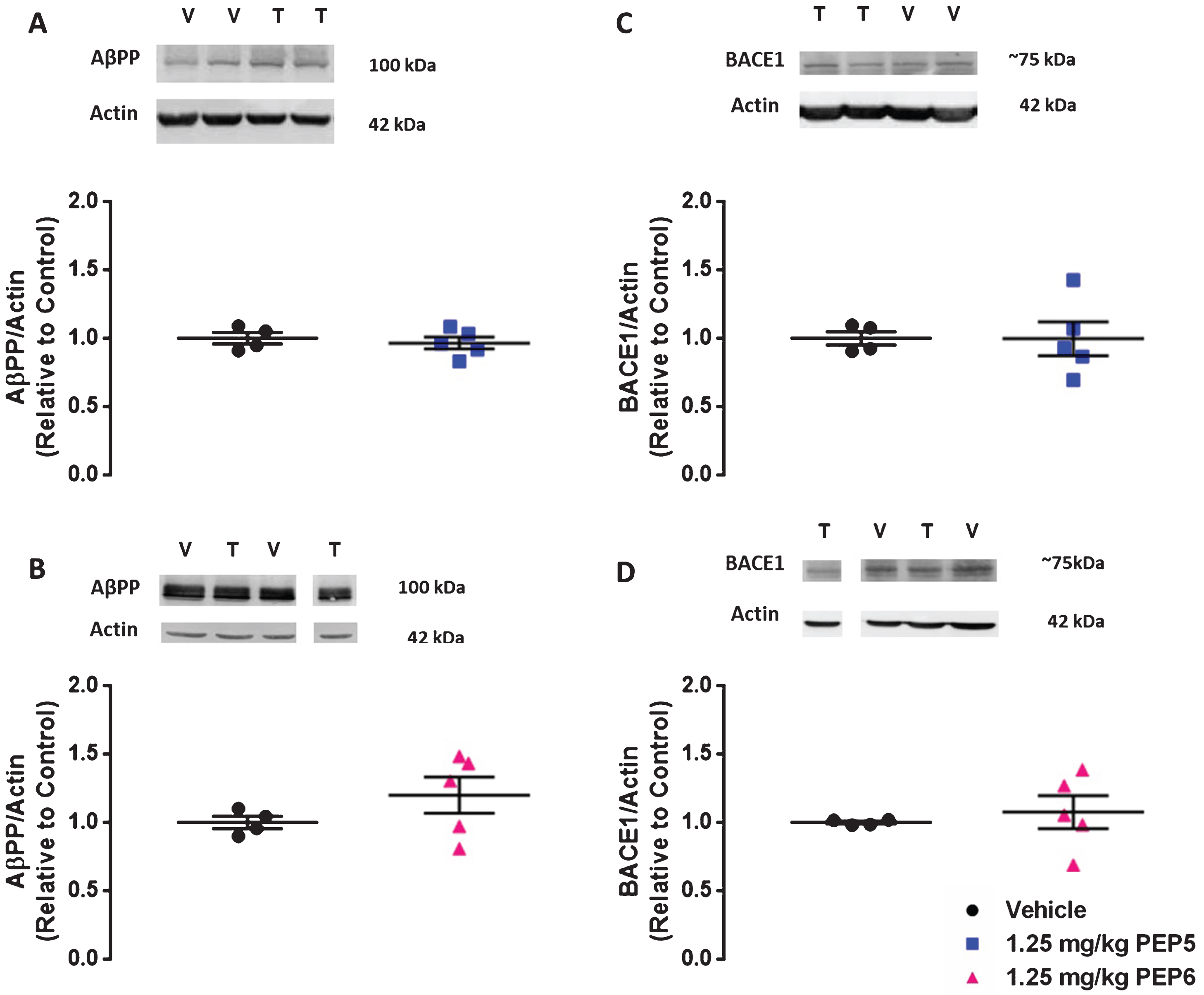
The new BACE1 inhibitors do not alter AβPP nor BACE1 protein levels in 3×Tg-AD mice. Twenty-four hours after a single i.p. administration of PEP5 or PEP6, mice were sacrificed and brain lysates were used to determine AβPP (A and B) and BACE1 (C and D) protein levels through Western blot analysis. Actin was used as loading control. The 3×Tg-AD mice injected with the vehicle (saline) in the absence of chimeric peptides was used as control. Representative images for each protein are presented above the graph (V: Vehicle and T: Treated). In each case (A–D), the representative image was obtained from the same membrane. The results normalized to β-actin and expressed as the relative amount of control represent the mean±SEM of 4–5 male mice per group. Statistical analysis was performed by non-parametric Mann-Whitney test. *p < 0.05, significantly different compared to control group. ns: not statistically different from control group.
Chronic treatment with the new BACE1 inhibitors decreases brain insoluble Aβ levels
We next evaluated the effect of a chronic treatment with the chimeric peptides on Aβ pathology using the same mouse model. The 4-month-old 3×Tg-AD mice were injected intraperitoneally (six in seven days) with PEP5 or PEP6 (1.25 mg/Kg), or with saline, during 4 or 6 months according to the animal studies flow-chart. After treatment, the mice were sacrificed and the insoluble Aβ40 and Aβ42 brain levels determined by ELISA. As shown in Fig. 9, both peptides induced a statistically significant decrease in brain insoluble Aβ42 (Fig. 9A, PEP5 : 0.470±0.030; PEP6 : 0.448±0.027 versus control: 0.998±0.206), which is the Aβ species more prone to aggregate and therefore more toxic than Aβ40. Likewise, the chronic treatment with PEP6 decreased insoluble Aβ40 and PEP5 treatment showed a trend to decrease insoluble Aβ40 (Fig. 9B, PEP5 : 0.597±0.022; PEP6 : 0.452±0.037 versus control: 0.998±0.216). Taken together these results suggest that chronic treatment with the new BACE1 inhibitors decrease amyloid deposition in the brain of 3×Tg-AD mice.

Insoluble Aβ levels decrease in 3×Tg-AD mice submitted to a chronic treatment. The levels of insoluble Aβ42 (A) and Aβ40 (B) were determined by ELISA. The results, normalized to control mice, represent the mean±SEM of 5–6 mice per group (control: 3 males and 2 females, PEP5 and PEP6 : 3 males and 3 females). Statistical significance was determined by Kruskal-Wallis non-parametric test followed by Dunn’s post hoc for multiple comparisons. *p < 0.05, **p < 0.01 significantly different compared to control group, ns: not statistically different from control group.
Chronic treatment with the new BACE1 inhibitors does not induce an immunological reaction in 3×Tg-AD mice
Although cell permeable peptides usually display low toxicity [40], at present there are limited studies concerning the possible toxic effects and immunogenicity of TAT and TAT chimeric peptides in vivo. Therefore, we evaluated the immunological status of 3×Tg-AD mice submitted to a chronic treatment (4 months) with PEP5 or PEP6 by determining the plasma levels of several cytokines, namely IL-1β, TNF-α, IL-6, IL-2, Il-4, IL-5, IFN-γ, IL-10, IL-12, and GM-CSF, through a multiplex array system. Our results did not show any change in the levels of the cytokines under study, which include inflammatory cytokines and cytokines that are involved in the stimulation of T and B lymphocytes and in allergic responses (Fig. 10). On the other hand, the 3×Tg-AD mice submitted to the chronic treatment with the new BACE1 inhibitors for the period of 4 months did not present differences in body weight, food consumption, and survival rate between cohorts (data not shown).
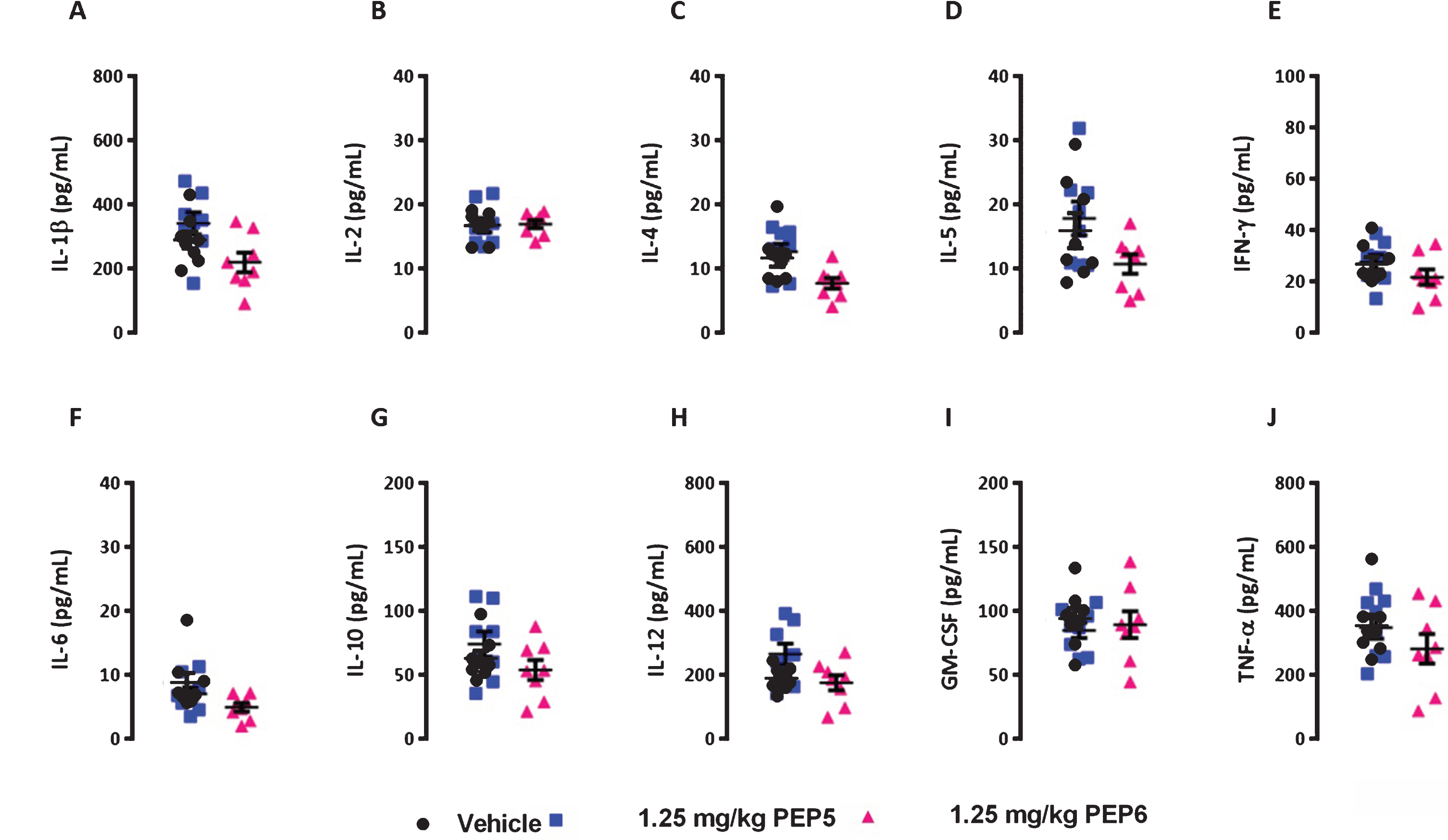
The new BACE1 inhibitors PEP5 and PEP6 do not induce an immune response in 3×Tg-AD mice. The 4-months-old 3×Tg-AD mice were injected (i.p.) six in seven days for a period of 4 months with PEP5 or PEP6. After mice sacrifice the blood plasma was obtained and the levels of IL-1β (A), IL-2 (B), IL-4 (C), IL-5 (D), IFN-γ (E), IL-6 (F), IL-10 (G), IL-12 (H), GM-CSF (I), and TNF-α (J) were measured by multiplex cytokine array using the Bio-Plex™ system, according to manufacturer’s protocol. Control mice were injected with the vehicle (saline) in the absence of chimeric peptides. The results (pg per mL) represent the mean±SEM of 8 mice per group (Control and PEP5 : 4 female and 4 male mice; PEP6 : 3 female and 5 male mice). No statistically significant differences between vehicle and treated animals were observed, (p > 0.05), as determined byone-way analysis of variance, followed by Dunnett’s post hoc test.
The new BACE1 chimeric peptide inhibitors do not affect the processing of the physiological BACE1 substrates SEZ6 and CHL1
To investigate whether the new BACE1 inhibitors PEP5 and PEP6 are selective toward non-amyloidogenic BACE1 substrates, we evaluated by Western blot the levels of CHL1 and SEZ6 proteins, as well as the levels of sAβPPβ, in the N2A cell lysates and in the conditioned medium of cultures incubated with the chimeric peptides for 24 h. In accordance with the previous results, both chimeric peptides decreased the levels of the sAβPPβ fragment in the conditioned medium of N2A cells, which is due to the inhibition of AβPP-β cleavage (Fig. 11A). It was observed that 50μM and 75μM PEP5 decreased the levels of sAβPPβ to 0.80±0.06 and 0.61±0.03 of control, respectively, whereas 20μM and 50μM PEP6 decreased the levels of sAβPPβ to 0.61±0.03 and 0.46±0.04 of control, respectively. Interestingly, as shown in Fig. 11B and 11C, the chimeric peptides at concentrations that inhibit the AβPP cleavage by about 39% (75μM PEP5) and 54% (50μM PEP6) did not affect the protein levels of either CHL1 or SEZ6 in N2A cell lysates or in the conditioned medium. These findings show that both PEP5 and PEP6 selectively inhibit BACE1 activity, decreasing the levels of the sAβPPβ fragment, the direct product of BACE1-mediated AβPP cleavage, without affecting the proteolysis of the BACE1 physiologic substrates CHL1 and SEZ6. This selectivity toward the inhibition of AβPP-β cleavage is a ground-breaking feature of the BACE1 chimeric peptide inhibitors that is not seen in previously developed BACE1 inhibitors. Noteworthy, we used the commercially available β-Secretase Inhibitor IV (1μM and 5μM) for comparison and we observed a significant reduction not only in the levels of sAβPPβ, that decreased by about 55% (1μM) and 63% (5μM), but also in the levels of CHL1 and SEZ6. Indeed, in the conditioned medium of N2A cells the CHL1 levels were reduced by about 24% and 41% for 1μM and 5μM β-Secretase Inhibitor IV, respectively, whereas SEZ6 levels decreased by about 52% and 73% for 1μM and 5μM, respectively, showing that this BACE1 inhibitor affects the processing of both amyloidogenic and non-amyloidogenic BACE1 substrates (Fig. 11A, B).
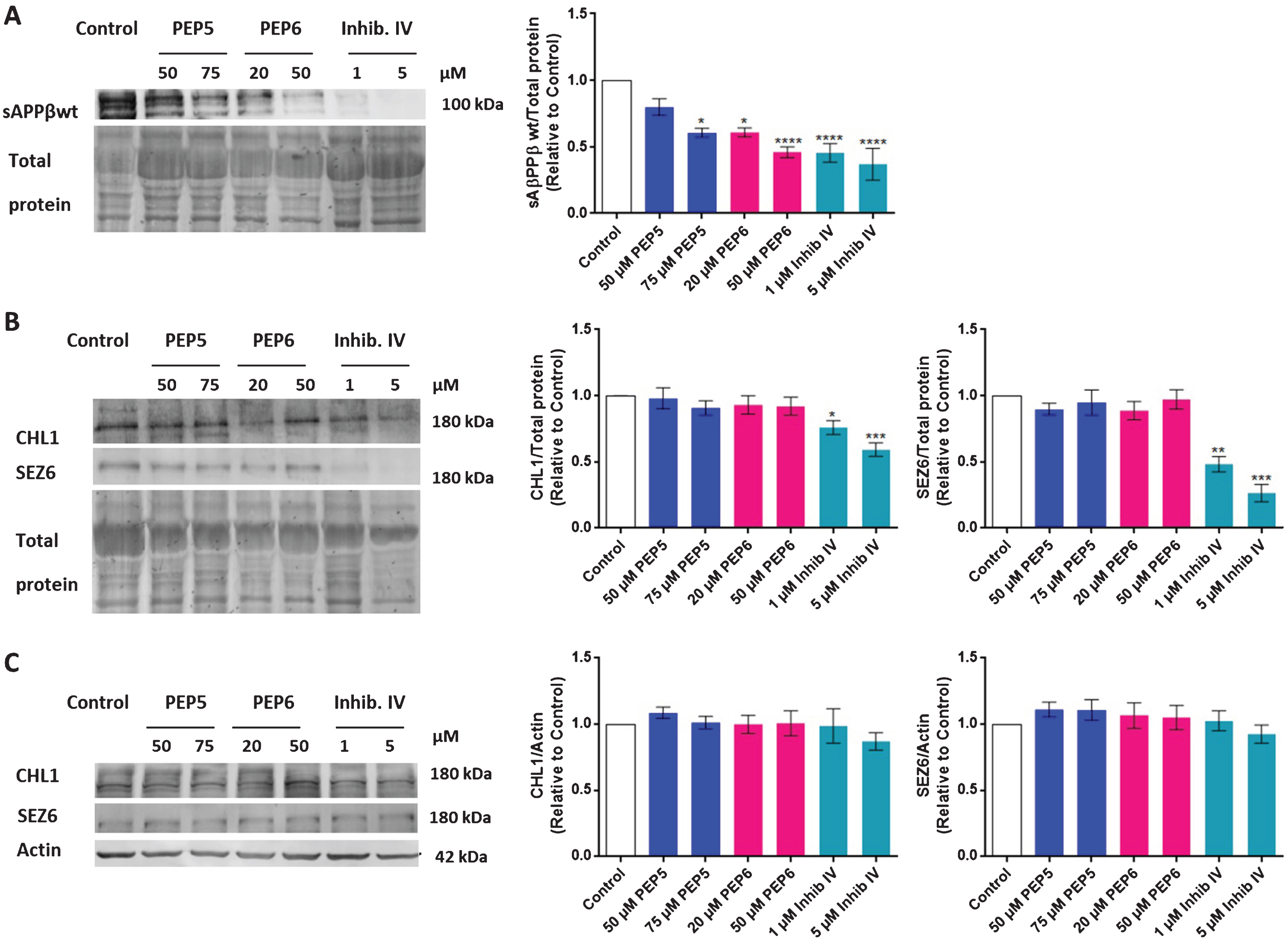
The new BACE1 inhibitors selectively inhibit the AβPP-β cleavage in N2A cells. After 24 h incubation with PEP5 or PEP6, the sAβPPβ levels in N2A cells conditioned medium (A), and the SEZ6 and CHL1 levels in conditioned medium (B) and in N2A cell lysates (C) were evaluated by Western blot. The β-Secretase Inhibitor IV (Inhibit IV, Calbiochem) was used as control. The results, normalized to total protein (conditioned medium) or to β-actin (cell lysates), and expressed relatively to control, represent the mean±SEM of at least 5 independent cell culture preparations. Statistical significance was determined by Kruskal Wallis non-parametric test followed by Dunn’s post hoc for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, **** p < 0.0001 significantly different compared to control.
DISCUSSION
Here we provide evidence that the innovative chimeric peptides we designed, including a D-RI peptide, selectively inhibit BACE1 activity in vitro leading to a decrease in Aβ40 and Aβ42 production without cytotoxic effects. Moreover, using the 3×Tg-AD mice we demonstrate that the compounds decrease plasma and brain Aβ levels and that chronic treatment leads to a reduction of insoluble Aβ in the mice brain. The partial reduction in the levels of brain soluble Aβ triggered by a 24 h treatment is ultimately implicated in a significant depletion of Aβ insoluble levels observed after a 6-month chronic treatment. Importantly, the BACE1 chimeric peptide inhibitors selectively inhibited the AβPP-β cleavage relatively to the proteolysis of the BACE1 substrates CHL1 and SEZ6, a pioneering feature that contributes to minimize possible mechanism-derived side effects of a therapeutic with a BACE1 inhibitor.
Although many factors contribute to AD pathogenesis, Aβ dyshomeostasis is still the most extensively validated and compelling therapeutic target [1, 4]. BACE1 is a crucial enzyme for Aβ generation [5, 41] and several BACE1 inhibitors have been developed aiming to act as a disease-modifying drug [17–20]. Despite the numerous efforts, it remains a daunting task to find a drug suitable for clinical use. Besides the pharmacological properties and the pharmacokinetic requirements that should be met, the development of BACE1 inhibitors faces the challenge of possible mechanism-based side effects due to the inhibition of the proteolysis of other BACE1 substrates that are relevant for neuronal physiological functions, namely for synaptic transmission [10–12]. Also, physiological concentrations of Aβ have been shown to facilitate synaptic plasticity [42]. Consequently, novel approaches in the design of drugs avoiding complete inhibition of BACE1 or enabling a substrate-selective inhibition are needed.
Therefore, instead of a small-molecule BACE1 inhibitor we developed chimeric peptides including a peptide sequence related to a BACE1 substrate conjugated to a cell-penetrating peptide (CPP), the TAT carrier peptide, which facilitates cell membrane permeation and the crossing of the BBB [21–24]. Due to the carrier, the new BACE1 inhibitors have the potential to be internalized by the cells mainly through endocytosis [25, 26], promoting the co-localization of the drug with BACE1 in the endosomes, which is the preferential subcellular compartment for the BACE1-AβPP interaction and subsequent AβPP-β cleavage in the neurites [9]. Moreover, the activation of the neuronal endocytic pathway is an early response in AD [43], which adds to a possible enhancement of the chimeric peptide cellular uptake and efficacy. Our results using N2A cells suggest that PEP6 is internalized by a clathrin-mediated endocytosis since CPZ strongly decreased the PEP6 uptake (Fig. 3A). Remarkably, the endocytic uptake of the new BACE1 inhibitors we developed might favor the selective inhibition of AβPP-β cleavage without interfering with the proteolysis of other BACE1 substrates that are cleaved in different subcellular compartments [27]. Indeed, our results demonstrate that the new BACE1 inhibitors we developed have the ability to selectively inhibit the AβPP-β cleavage without interfering with the proteolysis of CHL1 and SEZ6 (Fig. 11). As described before, the development of BACE1 inhibitors may have possible mechanism-based side effects due to the inhibition of the proteolysis of other physiological BACE1 substrates. These include, but are not limited to, CHL1 and SEZ6 which have important physiological roles in the central nervous system [10–12]. A recent study using long term in vivo two photon imaging and ex-vivo electrophysiological recordings demonstrated that SEZ6 is important for maintaining normal dendritic spine dynamics and synaptic functions and that SEZ6 is critically involved in BACE1 inhibition-induced synaptic deficits in adult mice [44]. Moreover, adult conditional BACE1 KO mice exhibit axonal disorganization in the mossy fiber pathway of the hippocampus, a phenotype correlated with a deficit in BACE1-mediated CHL1 processing, a protein involved in axonal guidance [13].
The in vitro studies we performed using recombinant BACE1 showed that PEP5 and PEP6 inhibit the enzyme activity (Fig. 1, Table 1). Consistently, docking studies revealed that both chimeric peptides can enter the enzyme active site cleft via the cargo sequence and can establish several interactions with active site residues, namely with the catalytic aspartate dyad (Fig. 2). Together, these results support an interaction between the chimeric peptides and BACE1 leading to enzyme inhibition. The ability of both chimeric peptides to inhibit BACE1 activity was further confirmed in a cellular model of AD through a reduction in Aβ40 and Aβ42production (Fig. 4).
Beyond chimeric peptides in the L-form, we developed molecules with a structural modification, specifically D-retroinverso chimeric peptides, backing a significant half-life and lesser immunogenicity of the peptides [23, 45]. To the best of our knowledge this innovative approach with D-RI-peptides was never used in the design of a BACE1 inhibitor. Thus, we further advanced the understanding on this matter by proving that D-RI-chimeric peptides may not lose biological function, retaining the ability to inhibit BACE1 in vitro and in vivo. Among the compounds we developed, the chimeric peptide PEP6, which is the D-RI form of PEP5, achieved in vivo a similar effect to PEP5 in the reduction of Aβ production with the possible benefit of allowing a reduction in the frequency of administration, an advantage to the clinical use.
Several studies report the beneficial effect of BACE1 inhibitors in AD mouse models reversing cognitive deficits and reducing Aβ levels, plaque burden and associated pathology, even in aged animals [14, 46–49]. The pharmacological inhibition of BACE1 activity rescues neuronal hyperactivity and repairs local neuronal circuits and long-range network connectivity whose dysfunction underlies cognitive impairment [47]. Also, the clinical trials with BACE1 inhibitors showed the ability of these drugs to reduce Aβ levels in the cerebrospinal fluid of human subjects, supporting the view that BACE1 is the primary β-secretase responsible for Aβ production in humans as well [17, 50–52]. Still, the randomized trial with the BACE1 inhibitor Verubecestat (MK-8931) failed to improve the cognitive or functional decline in mild-to-moderate AD patients suggesting that once dementia is present it may be too late to carry out treatments targeting Aβ production [53]. Because the amyloid pathology initiates about 22 years before clinical symptoms become apparent [54], it has been proposed that BACE1 inhibition should be implemented as early as possible, before the onset of clinical symptoms. Indeed, using transgenic mouse models of AD, it was recently demonstrated that the initial process of plaque formation, rather than the subsequent phase of gradual plaque growth, is the most sensitive to BACE1 inhibition [55]. This observation supports previous studies indicating that therapeutic BACE1 inhibition efficacy in ameliorating AD-like pathology and memory deficits is hindered if initiated when the transgenic mice have already a massive Aβ deposition [56]. Nevertheless, emerging data showed that combining passive anti-Aβ immunization with chemogenetic arrest of new Aβ production restored the cognitive performance of mice with severe amyloid pathology [57], stressing the value of the combination of anti-Aβ therapies including BACE1 inhibitors in more advanced stages of AD.
In our study we have used 4 month-old 3xTg-AD mice as the starting point to initiate the treatments with the chimeric peptides since at this age they do not present Aβ plaques despite they show increased intraneuronal soluble Aβ levels that correlate with the earliest cognitive impairment [34, 58]. Our results demonstrate that both PEP5 and PEP6 selectively inhibit BACE1 activity decreasing the brain levels of sAβPPβ and of soluble Aβ40 and Aβ42without interfering with AβPP cleavage by α-secretase (Figs. 6 and 7). The partial reduction in the levels of brain soluble Aβ, of about 18–20% for Aβ42, sustained a reduction of about 53–55% in the insoluble levels of Aβ42 after a 6-month chronic treatment (Figs. 6B and 9). This finding, similar to the effect obtained with the BACE1 inhibitor TAK-070 [49], is in agreement with observations in PDAβPP transgenic mice heterozygous for BACE1 in which soluble Aβ levels were lowered by only 12% at a young age but insoluble Aβ was reduced by 50% in elderly animals [15].
Though reportedly cell permeable peptides display low toxicity [26, 40], at present there are limited studies concerning the toxic and immunogenic effects of the TAT peptide in vivo in the long term. In our study PEP5 and PEP6 did not altered N2A-AβPPswe cell viability at the concentration near the IC50 for Aβ42 production (Table 2 and Fig. 5). Also, chronic treatment with the chimeric peptides did not induce any change in the blood levels of the cytokines here tested, which include inflammatory cytokines and cytokines involved in the stimulation of T and B lymphocytes and in allergic responses (Fig. 10) in agreement with a previous study regarding in vivo TAT immunogenicity after a single administration of the peptide [59]. Of note, we assessed cytokine levels after a daily administration of 1.25 mg/Kg of the new BACE1 inhibitors for the period of 4 months which, as far as we know, is the most extended period of an in vivo TAT-peptide administration leading to the evaluation of the immune response. Significantly, several clinical trials with CPP-derived therapeutics are now under development, namely XG-102 and AM-111 that include a D-RI peptide conjugated to TAT entered phase III clinical trials [26]. Moreover, the 3×Tg-AD mice submitted to the chronic treatment with the new BACE1 inhibitors did not present differences in body weight, food consumption, and survival rate between cohorts, neither any overt signs of toxicity (data not shown). These profiles should be a merit of these compounds, considering the long treatment period.
Our current findings provide evidence that PEP5 and PEP6 appear to be pharmacologically effective and safe, supporting the validity of partial BACE1 inhibition as a disease-modifying therapy for AD, especially if initiated in an early phase of the disease. Notably, these new BACE1 chimeric peptide inhibitors selectively inhibit the AβPP-β cleavage, an innovative feature that contributes to minimize possible mechanism-derived side effects of a BACE1 inhibitor therapeutic.
Footnotes
ACKNOWLEDGMENTS
We thank to Dr. Diederik Moechars, Neuroscience Department, Johnson & Johnson Pharmaceutical Research and Development, Janssen Pharmaceutica, Belgium, for the kind gift of the anti-SEZ6 antibody (#BAF4989 R&D Systems). This work was supported by FCT (PTDC/NEU-SCC/1351/2012), Pest-C/SAU/LA0001/2013-2014, CENTRO-07-ST-24-FEDER-002002, UIDB/04539/2020, INOV.C INC-2014-09-002-5645, FEDER, and COMPETE HealthyAging2020:CENTRO-01-0145-FEDER-00-0012, CENTRO-01-0247-FEDER-003269 (Drugs-2CAD) and Bolsa de Investigação Edgar Cruz e Silva (granted by Grupo de Estudos de Envelhecimento Cerebral e Demência and sponsored by Santa Casa da Misericórdia de Lisboa). The Post-Doctoral Researcher Contract SFRH/BPD/101028/2014 to Rosa Resende was funded by the European Social Fund.