
Abstract
Background:
Aging is considered the most important risk factor for Alzheimer’s disease (AD). Recent research supports the theory that immunotherapy targeting the “oligomeric” forms of amyloid-β (Aβ) may halt the progression of AD. However, previous clinical trial of the vaccine against Aβ, called AN1792, was suspended due to cases of meningoencephalitis in patients.
Objective:
To develop a peptide sensitized dendritic cells (DCs) vaccine that would target oligomer Aβ and prevent an autoimmune response.
Methods:
Double transgenic APPswe/PS1 ΔE9 (Tg) and C57BL/6J control mice were used in this study. Cytokine expression profile detection, characterization of antisera, brain GSK-3β, LC3 expression, and spatial working memory testing before and post-vaccination were obtained.
Results:
Epitope prediction indicated that E22W42 could generate 13 new T cell epitopes which can strengthen immunity in aged subjects and silence several T cell epitopes of the wild type Aβ. The silenced T cell epitope could help avoid the autoimmune response that was seen in some patients of the AN-1792 vaccine. The E22W42 not only helped sensitize bone marrow-derived DCs for the development of an oligomeric Aβ-specific antibody, but also delayed memory impairment in the APP/PS1 mouse model. Most importantly, this E22W42 peptide will not alter the DC’s natural immunomodulatory properties.
Conclusion:
The E22W42 vaccine is possibly safer for patients with impaired immune systems. Since there is increasing evidence that oligomeric form of Aβ are the toxic species to neurons, the E22W42 antibody’s specificity for these “oligomeric” Aβ species could provide the opportunity to produce some clinical benefits in AD subjects.
INTRODUCTION
In 2019, an estimated 5.8 million Americans had Alzheimer’s disease (AD) and related dementias [1]. Thus, there is a desperate need to develop and implement an effective strategy for AD treatment and prevention. This is because in the last decade, it has been shown that all FDA approved drugs for AD are only palliative [2]. Several compounds such as Azeliragon [3], Verubecestat [4], and Atabecestat [5] have been tested in clinical trials but have shown no significant therapeutic effects when compared to the placebo. As is well known, the pathological hallmarks of AD are extracellular senile plaques consisting of amyloid-β (Aβ) aggregates and intracellular neurofibrillary tangles consisting of the highly phosphorylated Tau (p-Tau) protein. Thus, immunotherapy against Aβ and p-Tau has emerged in the past 21 years. Vaccines, active immunotherapy, targeting the Aβ protein (i.e., Aβ1–42) and p-Tau have shown some pre-clinical efficacy. This has been demonstrated with studies using an AD transgenic mouse model [6 –11]. However, the transition of these preclinical studies to humans is difficult due to adverse side effects and the inability to specifically target the oligomeric forms of Aβ [12].
The interaction between Aβ in peripheral blood and in the brain has been well studied [13, 14]. Blood Aβ will interact with the immune cells outside of the brain parenchyma and activate the immune system, leading to systemic inflammation. The active immunotherapy using wildtype Aβ will induce a detrimental T cell response and lead to adverse effects. A vaccine targeting aggregated Aβ (particularly soluble Aβ oligomers) will reduce Aβ-related brain pathological functions and inflammation. The clinical trial of the first active immunotherapy (a vaccine against Aβ, AN1792) was suspended due to cases of meningoencephalitis (brain T cell infiltration, auto-T cell response) in patients. Encouragingly, the follow-up to this clinical trial has shown that all responders to the vaccine have slowed down memory decline compared to the control subjects [15 –17]. A next-generation vaccine against Aβ should target specific B epitopes and induce less T cell immune response [18]. However, ACC-001 (vanutide cridificar) is an N-terminal Aβ1–7 amino acid peptide fragment that uses inactivated diphtheria toxin as the carrier. ACC-001 was discontinued during Phase II trials because of a strong autoimmune response [19], another anti-Aβ vaccine developed later was also associated with adverse effects, including the development of cerebral microhemorrhages [10 , 21]. It is important to recognize that the average age of AD patients at diagnosis is about 75 years [22], and they have declined immune systems [23, 24]. Regular vaccines may not induce an ideal immune response unless strong adjuvants are introduced into the formula to stimulate the immune system [25]. An alternative approach to active Aβ immunotherapy is anti-Aβ or p-Tau antibody therapy, also known as passive immunotherapy. However, this approach may lead to cerebral microhemorrhages. Passive immunotherapy is expensive and requires frequent subcutaneous administration. In addition, the long-term application of one subtype of IgG will cause an imbalance of immunity in the treated patients [10, 26]. Currently, most of the anti-Aβ antibody-based immunotherapy drugs (e.g., Bapineuzumab [27], GSK933776, and Solanezumab [28]) have been terminated in clinical trials due to the lack of cognitive benefits for AD patients. Importantly, clinical trials with anti-Aβ “isoform-specific” antibodies have also failed [29]. Encouragingly, more recent studies have reported that two other antibodies (Aducanumab at CTAD 2019; BAN2401 at AAIC 2018) have shown substantial reductions in PET amyloid, with many participants becoming amyloid negative by 14 months of high dose treatment, showing that some indication of slowing cognitive decline can be achieved with anti-amyloid immunotherapy [30]. An ideal long-term treatment for AD must simultaneously target the pathological factors (aggregated Aβ and/or p-Tau) and address the impaired immune systems of patients without inducing adverse effects.
There are currently two types of vaccines used to fight diseases. The first type of vaccine is a prophylactic vaccine, which is designed for disease prevention and used for healthy subjects. Their immune systems can typically prevent the overreaction of both B cell and T cell-mediated immune responses. Therefore, adjuvants are allowed in the formula to provide a cost-effective and efficient vaccine [31]. The second type of vaccine is a therapeutic vaccine, which is designed to be used for disease treatment. This type of vaccine should avoid using a strong adjuvant to prevent over-stimulating the immune system [32]. Also, the therapeutic vaccine should produce a long-lasting and low or moderate antibody titer response without yielding any obvious adverse effects. Therefore, the therapeutic vaccine should be more suitable for patients with impaired immune systems, such as those with AD or PD.
We have developed a therapeutic vaccine by using mutated Aβ-sensitized mouse bone marrow-derived dendritic cells [33 –35]. The major advantages of such a vaccine is the use of a natural adjuvant—the dendritic cells (DCs). Our research data demonstrate that a mutation in a T cell epitope can break tolerance in APP mice and aged mice [35 –37]. This new DC vaccine was developed by sensitizing mouse DCs with a mutated Aβ (E22W42, also named as CAO22W: Aβ1–42 No. 22 amino acid of Aβ1–42 mutated to W). E22 is an important location for FAD/CAA mutations such as Osaka (delE22), Dutch (E22Q), Arctic (E22G), and Italian (E22K) mutations. E22W42 could potentially stabilize a specific oligomeric form of Aβ. The oligomeric form of Aβ plays a primary pathogenic role in the pathological cascade of AD development, so the best approach to stop the cascade response is to reduce the oligomeric Aβ load. Because we use DCs as a carrier, this vaccine can coordinate both innate and acquired immunity to overcome age-related impairments of the immune system [38 –40]. Thus, this DC-based vaccine has immunomodulatory effects and can potentially be used as a therapeutic vaccine for AD.
MATERIALS AND METHODS
Materials
All peptides used in the study were purchased from Biomer Tech Inc. (CA, USA).
Human wild type Aβ1–42 (WT42), Aβ1–35 (WT35), and Aβ1–25 (WT25) sequences are as follows (the length of each amino acid type is delineated by the subscript numbers): D1AEFRHDSGYEVHHQKLVFFAEDVG25SNKGAIIGLM35VGGVVIA42.
GenBank access number: Q29149
Mutant Aβ1–42 (E22W42), E22W1-25 (E22W25), and E22W1-35 (E22W35) sequences are as follows (the length of each amino acid type is delineated by the subscript numbers): D1AEFRHDSGYEVHHQKLVFFAWDVG25SNKGAIIGLM35VGGVVIA42.
Primary Aβ antibody A8 was obtained from Dr. Ying Zhang’s laboratory at Beijing Jiaotong University (Beijing China); A8 targets oligomer Aβ. The 6E10 antibody was purchased from Bio Legend (CA, USA, Cat# 803001).
Animals
Double transgenic APPswe/PS1 ΔE9 (Tg) and C57BL/6J control mice were used in this study, and the number of mice in each group is no fewer than nine. APPswe/PS1 ΔE9 mice with C57BL/6J background were originally purchased from JAX MMRRC (Stock# 034829) and bred in our facility. All APP/PS1 mice were initially genotyped by PCR at the time of grouping. The PCR result was further confirmed by using blood Aβ1–40 measurements. Only the mice expressing Aβ1–40 were used as transgenic mice. Pre-behavior results of RAWM (error and latency) and the plasma Aβ1–40 levels were balanced among Tg groups. Mice were caged individually and maintained on a 12/12 h light/dark cycle and in a temperature-controlled room. Food and water were available ad libitum. Mice were 10 months of age when treatment began.
All described procedures were conducted in accordance with the United States Public Health Service’s Policy on Humane Care and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of South Florida. All animals were housed in the vivarium at the USF College of Medicine.
B and T cell epitope prediction
The Immune Epitope Database (IEDB, https://tools.iedb.org/) was used to analyze the T cell and B cell epitope of wild type and mutated Aβ peptides. After the performance of B cell epitope prediction using DNAstar and IEDB, there were no differences among the wild type and mutant peptides [37]. Peptides were then subjected to MHC binding prediction. Binding affinity was predicted for a set of random natural peptides of 8-14 amino acid lengths for the 27 most frequent HLA class I alleles. This set of alleles is considered to cover >97% of the global population. The binding prediction was made using the NetMHCpan-4.0 method as implemented in the IEDB MHC binding prediction tool TepiTool [41 –43]. The prediction tool provides the predicted binding affinity in terms of percentile ranks ranging from 0 to 100; a peptide that has a lower percentile ranking is a better binder.
Monomer Aβ preparation
The purchased Aβ peptides (E22W42, WT42) were dissolved in HFIP at 1 mg/ml and incubated at 37°C for 3 days on a rotator then sonicated in a water bath for 1 h to disrupt any pre-existing aggregates and then aliquoted and lyophilized. Samples were then stored at –80°C for future applications.
Oligomer Aβ preparation
HFIP pretreated Aβ (E22W42, WT42) film was diluted at 5 mg/ml with DMSO and further diluted to 20μM with Ham’s F12 medium for aggregation, which was achieved by 72 h of incubation at 37°C. The oligomer formation was detected and confirmed with western blot.
Bone marrow and immature DC preparation
Mouse bone marrow dendritic cells (mDCs) were harvested from the femur and tibia of 10-week-old non-transgenic mice (C57BL/6J). On day 0, after removing all muscle tissue with gauze from the femurs and tibias, the bones were washed with 1XPBS and soaked in 70% ethanol for 30 s. The bones were then washed three times with 1XPBS, and both ends were cut in a dish. The marrow was flushed with RPMI 1640 with a syringe, then gently resuspended and passed through a 70μm strainer into a centrifuge tube. Following centrifugation at 300×g for 10 min, the cell pellets were loosened by gentle vortex. Then red blood cells were lysed by incubation with 5 ml ACK buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM EDTA, pH 7.3) for 30 s while shaken at room temperature, and 45 ml 1XHBSS was added to stop lysis. After centrifugation, cells were resuspended with RPMI 1640 (10% FBS, and 1% penicillin-streptomycin, 50μM of 2-mercaptoethanol, 10 ng/ml of recombinant mouse GM-CSF (BD, Cat: 554586), and 10 ng/ml of recombinant mouse IL-4 (BD Cat:550067)), then brought to 1×106 cells/ml and seeded in 6-well plates with 3 ml/well. The medium was replaced on day 1, and cell treatment was initiated on day 4 by aspirating off 1ml of medium and then adding back 1 ml/well fresh medium containing 60μg/ml WT42 or E22W42. The cells were harvested on day 8, and the concentration was adjusted to 5×106 cells/ml for injection.
Immunization procedure
10-month-old APPswe/PS1 ΔE9 mice received a dose of 1.5×106 DCs per mouse (0.3 ml in 1×PBS) administered by intraperitoneal (IP) injection bi-weekly for the first three injections. For the following two injections, DCs dosage decreased to 1×106 cells in 0.2 ml 1×PBS administered monthly by IP injection. A total of five injections were administered. Blood was collected 10 days after each vaccination [36].
Radial arm water maze (RAWM)
The radial arm water maze test was used to monitor the cognitive function of the mice before and after vaccinations. It contained 6 swim paths (arms) radiating from an open central area with a hidden escape platform located at the end of one of the arms. The pool was surrounded by several extra maze cues to allow spatial navigation. On each trial, the mouse was allowed to swim for up to 60 s to find the escape platform. The platform was located in the same arm on each trial. On day one, mice were given 15 trials alternating between a visible platform (above the water) and a hidden platform (below the water). On day two, mice were given 15 additional trials, with all the trials using a hidden platform. The start arm varied for each trial so that mice relied upon spatial cues to solve the task instead of learning motor rules. The goal arm for each mouse was different to avoid odor cues from revealing the goal arm. Entry into an incorrect arm (all four limbs within the arm) was scored as an error. Failure to make an arm entry within 15 s was also scored as an error. The errors for blocks of three consecutive trials were averaged for data analysis. Mice averaging 1 error or less by the end of day two are considered to have reached the learning criterion. On the third day, a reversal trial was performed, with the goal platform placed in the arm 180° from the original location. Mice were given 15 trials, all with a hidden platform. Latency to find and ascend the platform was recorded (60 s maximum) [44].
Brain tissue preparation
After the RAWM test, mice were anesthetized with SomnaSol (Henry Schein Animal Health, Cat#024352) and intracardially perfused with 50 ml of saline. The brains were carefully removed, and the right hemisphere was frozen at –80°C. On the day of final brain tissue preparation, frozen tissue was thawed and homogenized in the RIPA buffer containing proteinase inhibitor (100 mM Tris [45], 150 mMNaCl, 0.5% DOC, 1% NP-40, 0.2% SDS,1 mM Na3VO4, 10 mM NaF, 1 mM PMSF, 20μM Leupeptin) with a pellet pestle motor and 10 s sonication, then centrifuged for 20 min at 21,000 g at 4°C. Crude protein concentrations were determined by Bio-rad DC protein assay (Bio-Rad Cat:5000112) and adjusted to the same level for all the samples. The supernatants obtained from this protocol were stored at –80°C. The left hemispheres were transferred into a 4% paraformaldehyde solution for future immunohistochemistry tests.
Blood sample collection and detection
Blood samples were collected in EDTA tubes by submandibular phlebotomy bleeding before the vaccination and 10 days after every injection. On the euthanasia day, mice were anesthetized and blood was drawn from each mouse via cardiac puncture. All plasma samples were analyzed for plasma Aβ level, plasma anti-Aβ antibody purifications, plasma antibody titer, plasma cytokine, and plasma antibody Ig isotyping detection.
Purification of plasma antibodies
Mouse plasma antibodies were purified by SulfoLink Coupling Resin (Thermo Scientific Prod # 20402) and conjugated with synthesized cysteine residue Aβ peptide according to the manufacturer’s instructions. Briefly, after equilibrating the resin with binding buffer, the diluted plasma was passed through the resin five times. The antibody was then washed with binding buffer and eluted with the Elution Buffer. The eluted antibodies were immediately adjusted to physiologic pH by adding Neutralization Buffer, and the concentration was determined by Bio-rad DC protein assay (Bio-Rad Cat:5000112).
Antibody titer determination
Plasma anti-Aβ antibody levels were determined by ELISA analysis. A 96-well plate was coated with 50μL of Aβ1–42 peptides at 10μg/mL in carbonate bicarbonate (CBC) buffer. A CBC buffer plate was also used in order to measure control background binding. The Aβ1–42 peptide experimental plate and CBC-bound control plate were incubated overnight at 4°C. The plates were then washed with 1XPBST and blocked with 1.5% BSA in PBST for 1 h at room temperature, followed by extensive washing. Plasma samples were then diluted serially in the blocking buffer and loaded to both the Aβ1–42 peptide plate and CBC coated plates. Plates were then incubated for 1 h at 37°C and washed, then HRP-conjugated anti-mouse IgG (Sigma Cat: A9044) at 1 : 5000 dilutions with blocking buffer was added into each well and incubated for 1 h at 37°C. After washing, TMB peroxidase substrate (Surmodics Cat: TMBS-1000) was added to each well and incubated at room temperature for 10 min. The reaction was stopped by adding 100μL/well of 0.4 M H2SO4. Absorbance at 450 nm was read with a BioTek Synergy H4 microplate reader. Samples with OD450 nm absorbance 3 times higher than controls were considered as positive, the highest dilution being designated as the endpoint titer.
Antibody Ig isotyping
Isotyping of immunoglobulin in plasma was performed using magnetic bead-based multiplex assay (Millipore Cat: MGAMMAG-300K). The assay was performed according to the manufacturer’s instructions. The results were obtained by reading the plates on the Bioplex MAGPIX Multiplex Reader, and the concentration was calculated by using Bio-plex manager 6.1 software. A total of 6 markers (IgA, IgG1, IgG2a, IgG2b, IgG3, and IgM) were tested and quantified upon each standard.
Plasma cytokine expression profile detection
Plasma cytokine expression was performed using a magnetic bead panel 96-well plate assay (Millipore Cat: MCYTOMAG-70K) according to the manufacturer’s instructions. The results were obtained and analyzed by Bio-plex MAGPIX Multiplex Reader and Bio-plex manager 6.1 software, and each cytokine was calculated upon each standard.
Plasma Aβ1–40 and Aβ1–42 level detection
The concentrations of Aβ40/42 were measured by the Aβ1–40 and Aβ1–42-specific sandwich ELISA kit (Mega Nano Biotech, FL, USA). Each well of a 96-well plate was coated with 50μl G1–42 (goat anti-human Aβ1–42) antibody diluted to 10μg/mL in 1XPBS and incubated overnight at 4°C. The plate was washed 5 times and blocked by adding a 200μl blocking buffer at 37°C for 1 h. After washing and drying the plate, the plate was kept at room temperature ready for assay.
In a 96-well sample preparation plate, 50μl diluted detection antibody was added into each well and 50μl diluted peptide standard (Aβ1–40 or Aβ1–42) or 50μl diluted plasma samples were added at different dilutions to each designated well. Then the solutions were immediately transferred into the antibody-coated plates. The plates were incubated at 4°C for overnight. After washing, 100μl diluted secondary antibody was added to each well and incubated for 45 min on an orbital shaker at room temperature. The plates were washed, and TMB peroxidase substrate (Surmodics Cat: TMBS-1000) was added to each well and incubated at room temperature for 10 min. The reaction was stopped by adding 100μl/well of 0.4 M H2SO4. Absorbance at 450 nm was read with a BioTek Synergy H4 microplate reader. The concentration was calculated upon the peptide standard.
Western blot detection for protein expression
Equal amounts of mouse brain protein samples were denatured with a loading buffer (Invitrogen Cat: NP0007) containing 6% β-mercaptoethanol and heated at 70°C for 10 min. Protein samples were then loaded to each well and separated using a 10% Bis-Tris gel. Precision plus protein dual color standards (Bio-rad, #1610374) were used as molecular standards. The separated samples were transferred with semi-dry assay to the PVDF membrane (Millipore Cat: IPFL00010). Membranes were first blocked with 0.2% Iblock buffer for 1 h at room temperature, and then incubated with the primary antibody at designated dilutions in blocking buffer on a shaker overnight at 4°C. After washing with 1XPBST three times for 5 min, blots were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody in blocking buffer for 1 h. The enhanced chemiluminescence substrate (Thermo Scientific Prod # 34078) was used to develop the blots. Image J software was used for gel quantification.
The primary antibodies used were phospho-GSK-3β (Cell signaling Cat: 9336S), GSK-3β (Cell signaling Cat: 9315S), phospho-Erk (Cell signaling Cat:4370S), Erk (Cell signaling Cat: 4695S), LC3I/II (Cell signaling Cat: 4108S), A8 [46] (Beijing Jiaotong University), and β-Actin (Sigma Cat: A5441).
Statistical analysis
The data were analyzed by using GraphPad Prism 6 software. Statistical evaluation of the results was initially performed using a one-way ANOVA involving all groups. This was then followed by a post-hoc pairwise analysis of group differences, and then Fisher LSD test was used. Pearson correlation was used to analyze the correlation. The level of statistical significance was set at α= 0.05. Results were presented as mean±SEM.
RESULTS
Antigenicity prediction and immune response of the DC preparation in vivo
For optimal vaccine function, both B cell and T cell responses must be induced. The B cell response results in antibody production. In addition, the T cell response results in cytokine production and T cell surface marker expression of both CD4 and CD8. These T cells are activated through peptides presented by MHC-II and MHC-I, respectively. The TepiTool tools predict peptide binding to MHC class I and class II molecules. When compared to WT42, 13 novel T cell epitopes were discovered when analyzing E22W42 (Table 1). These 13 new peptides were all from mutations from E to W at amino acid number 22. The results demonstrate that these new peptides have a strong affinity to MHC class I and class II molecules. Moreover, the point mutation can help break Aβ-related immune-tolerance.
The HLA-I Affinity Analysis of E22W42 Against Human HLA Alleles
Antigenicity prediction results of novel HLA-I T cell epitope and antibody response of E22W42 peptide fragments sensitized DCs as a vaccine. Antigenicity results of the novel HLA-I T cell epitope of E22W42. Only the new T cell epitopes against human HLA-I were listed; no sequences in the WT42 were detected. The prediction is a combination of rank and IC50. Ranks below 1 are considered as high affinity. IC50 < 50 is high binding to MHC-I, <500 is moderate binding to MHC-I, and <5000 is low binding to MHC-I. Mouse binding in silico was also checked. However, no major differences among all MHC alleles were discovered (data not shown). Note: In addition, the Aβ1–42 peptides were predicted against mouse MHC I alleles. Only E22W42 peptide was identified to have a strong affinity based on the recommended threshold (percentile rank 1.0). The peptide is from AA20 to AA 31 FAWDVGSNKGAI against H-2-Db.
WT42 has a major T-cell epitope located at the central regions encompassing residues 16–24 [33]. Mice administered E22W35 peptide-sensitized DC vaccine can induce a much higher antibody response compared to E22W25, WT25, and WT35 (Fig. 1). Therefore, the optimized minimum length for the DC vaccine activation should be 1–35 or longer.
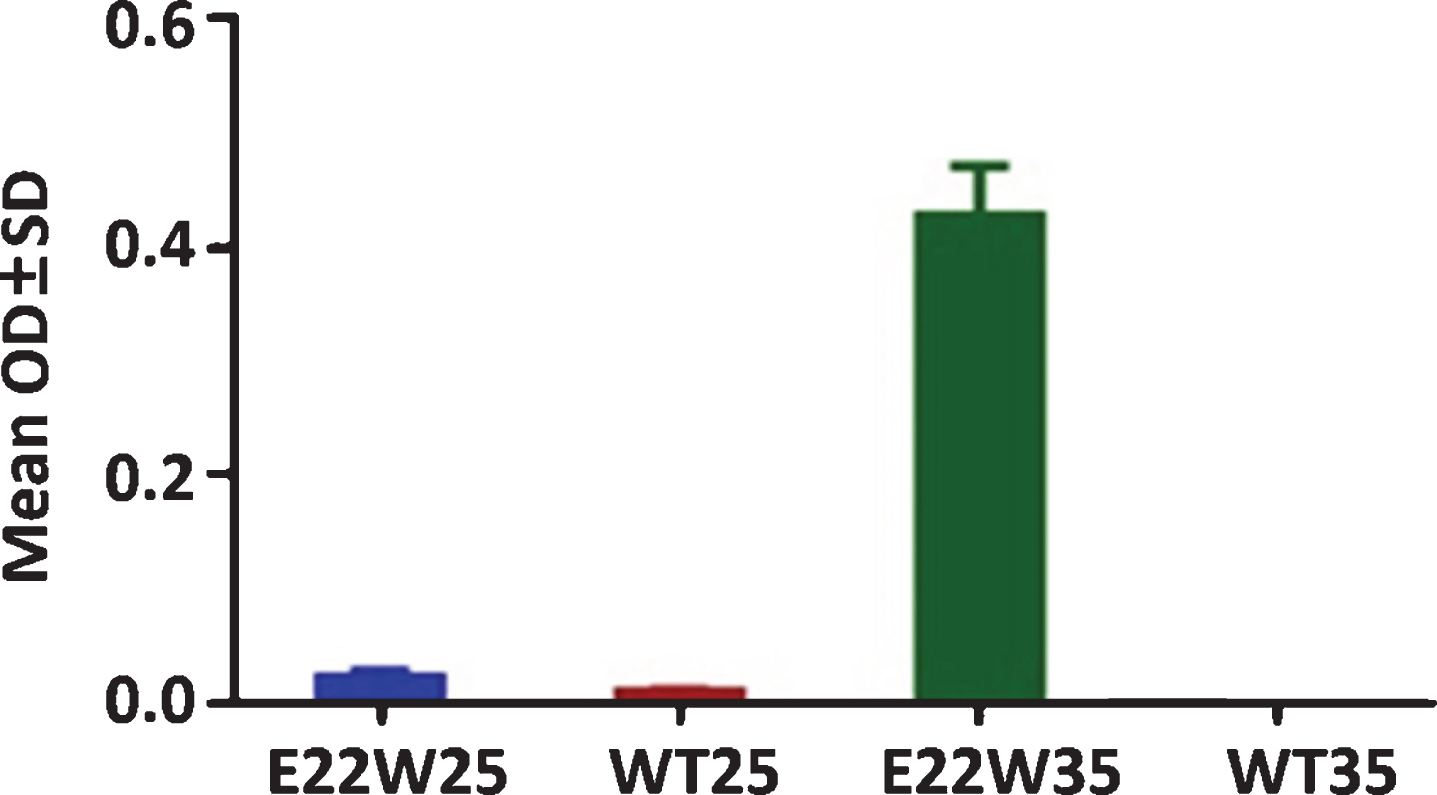
E22W35 is the minimum peptide length to sensitize DCs to induce the immune response. Antibody titers of the C57/BL6 mice after two injections by using E22W25, E22W35, WT25, and WT35 sensitized DCs vaccine at 6-month-old. Only E22W35 can induce an antibody response (n = 4 per group).
E22W42-sensitized DC vaccine targeting oligomeric Aβ
Aβ42 plays a key role in the pathogenesis of AD and is a core biomarker for the diagnosis of AD. In addition, oligomeric Aβ is well accepted as the major toxic isoform to neurons and may play a role in AD development [40]. Therefore, it is essential to determine whether E22W42 can generate an oligomer-specific antibody compared to other groups. As demonstrated in Fig. 2a, E22W42 group mice showed a good antibody response and these antibodies bound non-aggregated Aβ42 peptide even at 1 : 800 dilutions. Moreover, our data did not show a linear decrease in absorbance when the antibody was diluted from 1 : 200 to 1 : 800 (1 : 4 dilution). Furthermore, the data in Fig. 2a also demonstrates that there was a high level of anti-oligomer Aβ antibody in plasma (1 : 800 dilution) because there was an excellent response to dilution from 1 : 200 to 1 : 800 (coated antigen is pre-aggregated WT42). The antibody isolated from the E22W42 DCs vaccinated mice plasma was then compared with the other anti-Aβ antibodies by western blot assay. In Fig. 2c, the blot was probed with the A8, an oligomer-specific antibody [46]. In Fig. 2d, the blot was probed with a purified antibody from plasma of the E22W42 DCs vaccinated mice. In Figure 2e, the blot was probed with 6E10 antibodies. Monoclonal antibody 6E10 is among the first anti-Aβ monoclonal antibodies. The antibody 6E10 targets both monomeric and oligomeric Aβ [47]. The significance is that the E22W42 DCs vaccine produced an oligomer-specific antibody. Both ELISA assay and western blot show this vaccine targets the toxic isoform of Aβ (Fig. 2). Figure 2f shows the antibody production and time duration for the immunization. The results show that the antibody level can be continuously boosted up until the 5th immunization injection, and no significant changes in level occurred, even after 2 months.
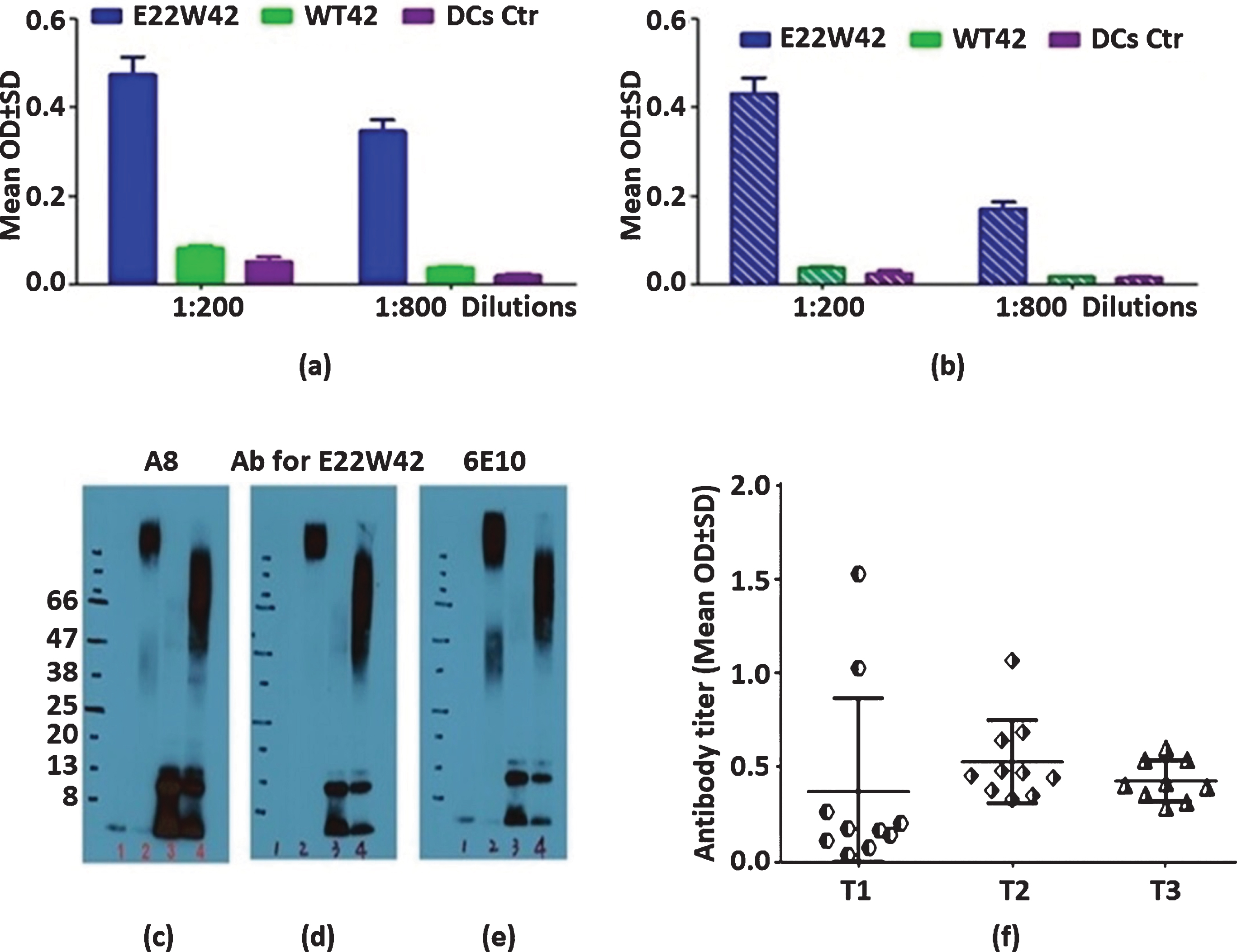
E22W42 DCs vaccines can successfully induce high level antibody binding to monomeric and oligomeric Aβ. Characterization of antisera from E22W42 DCs vaccine. a) The binding ability of plasma collected from mice treated by different peptide-sensitized DCs vaccine to non-aggregated Aβ peptide. DCs vaccine is dendritic cells sensitized by E22W42, WT42, or DC-only control. b) The same sera bound to pre-aggregated Aβ peptide (three days). c-e) Western blot results. Lane 1 is hexafluoroisopropanol (HFIP)-treated wild Aβ1-42 (WT42, monomer), lane 2 is HFIP-treated WT42 aggregated for 3 days (oligomer), lane 3 is E22W42-treated with HFIP (assumed monomer), lane 4 is E22W42 peptide treated with HFIP under aggregation for 3 days (oligomer). Descriptions are as follows: c) Probed with mouse monoclonal anti-Aβ antibody named A8 (an oligomer specific antibody), d) Probed with purified antibody from plasma collected from E22W42 DCs vaccinated mice, and e) Probed with 6E10 antibody. f) Antibody duration post-vaccination. T1 is the blood collected 10 days after the second immunization. T2 is the blood collected 10 days after the 5th immunization. T3 is the blood collected 2 months after T2. There is no significant difference in antibody titers after 2 months, between T2 and T3 (p > 0.05).
The immature DC cell-based vaccine retains the properties of DCs
DCs are considered a bridge of the immune system, connecting innate immunity and adaptive immunity. A DC vaccine can induce a moderate antibody response that helps avoid adverse effects. Table 2 shows the correlation analysis results of the cytokine profile post-vaccination among all vaccinated groups. The horizontal rows are correlation results of IL-6 versus GM-CSF, IL-10 versus GM-CSF, and IL-6 versus IL-10. The vertical columns are the DCs control and WT42 and E22W42-treated groups of mice. The correlation results of IL-6 versus GM-CSF, IL-10 versus GM-CSF, and IL6 versus IL-10 results indicate that the WT peptide group is significantly different from the DCs only and E22W42 groups. This indicates that the WT42-sensitized DCs vaccine can change the DCs’ properties. However, the E22W42-sensitized DC vaccine retains the property of DCs (p < 0.01). This means that the natural dendritic cell immune function will remain the same, which makes the E22W42 DC vaccine a better approach for AD treatment.
The Correlation Results Between Cytokines of Different Groups
A table showing correlation analysis results of the cytokine profile post-DCs vaccination administered to APP/PS1 mice. The horizontal rows are correlation results of IL-6 versus GM-CSF, IL-10 versus GM-CSF, and IL-6 versus IL-10. The vertical columns stand for treatment groups (DCs control, WT42 DCs, and E22W42 DCs treated mice groups). The number values stand for correlation efficiency and p-value level of significance. E22W42 DCs have a similar pattern to the DCs control group (p < 0.01 for all three groups). However, cytokine correlation in WT42 DCs has no significance for all tested cytokines in the table (p > 0.05). Column 1: The anti-Aβ antibody level is inversely correlated to the ratio of Aβ40/42. The 40/42 ratio level is a benefit indicator of immunotherapy targeting Aβ, therefore the negative correlation demonstrated the efficacy of the E22W42 DCs vaccine.
The DC vaccine promotes a Th2 response
Due to the failure of many Aβ-based vaccines in clinical trials, we need to further characterize the immunotype of the vaccine by using the IgG1/IgG2 ratio. Our result indicated that the E22W42-sensitized DCs vaccine initiates the Th2 type response (Fig. 3). It is well known that Th1 cells are involved in cellular immunity and a pro-inflammatory response. Th2 cells can help B cells differentiate into antibody-secreting cells and normally generate a less inflammatory response. A study showed IFN-γ production by Aβ-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of AD [48], and Aβ-specific Th2 cells have been shown to reverse cognitive impairment [49]. Meanwhile, Th1 cytokines and Th2 cytokines induce isotype switching to IgG2a (Fig. 3a) and IgG1 (Fig. 3b), respectively [50, 51]. In order to exclude individual differences, we used the ratio of post/pre IgG1 or IgG2a. The results show that the E22W42 groups only increased the ratio of IgG1 level compared to the WT42 and control group. However, the ratio of post/pre IgG2a level remained the same, indicating that the effect of the E22W42 DC vaccine generated a favorable immune response.
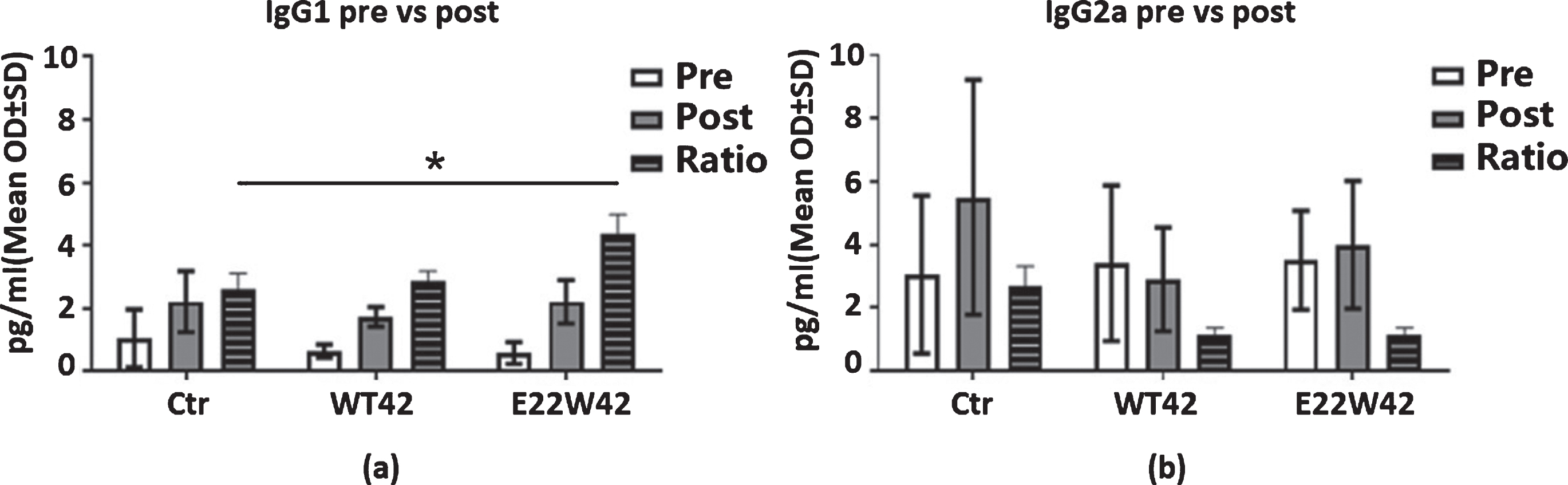
E22W42 DCs vaccine promotes Th2 response. Ig Isotyping changes post-DCs vaccination injection. The IgG1 (a), IgG2a (b) levels in the mice plasma of control, WT42, and E22W42 DCs vaccination pre- and post-injection, respectively, and the post/pre ratio level. There is increased IgG1 in the E22W42 vaccinated group post-vaccination versus pre-vaccination, but there are no differences among all groups in the ratio of IgG1/IgG2a.
This DCs vaccine does not cause inflammation
Active immunotherapies (anti-Aβ vaccines) have failed in the past because of the abnormal immune responses [17, 19]. AN-1792 (an Aβ-based adjuvant vaccine) exhibited an increased risk of Th1-mediated meningoencephalitis [52]. Meanwhile, increased inflammation is a primary symptom of AD, so treatment will need to avoid causing any inflammation. Th1 (pro-inflammatory) cytokines IL-2, TNF-α, and IFN-γ cause stimulation of CD8-positive cytotoxic T lymphocytes [53]. We tested the IL-2, IFN-γ, and TNF-α levels in the plasma of E22W42 vaccine-treated mice and found no significant differences. This indicates that the E22W42-sensitized DCs vaccine has little potential for over priming the immune system (Fig. 4).
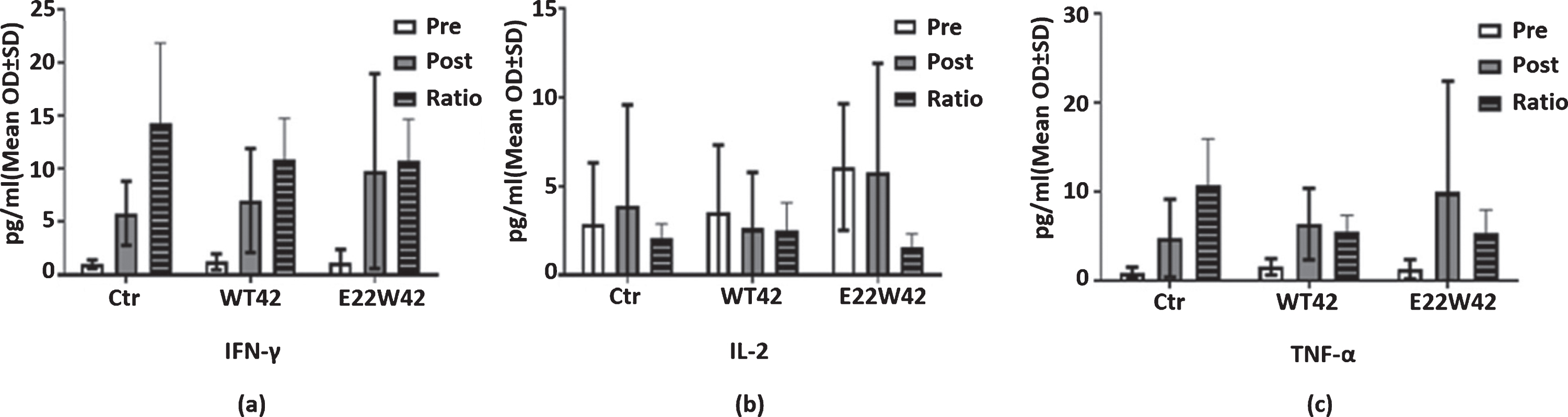
E22W42 DCs vaccine has no inflammatory response. Cytokine expression profile detection post-vaccination. Levels of IFN-γ (a), IL-2 (b), and TNF-α (c) in mouse plasma of the control, WT42 DCs, and E22W42 DCs vaccine for pre and post-vaccination, respectively, and also the post/pre ratio level of each group. There is no significant difference across all inflammatory cytokines (a-c).
E22W-sensitized DCs vaccine can benefit memory in APP/PS1 mouse model
Memory benefit is the major readout for any AD treatment method. We have tested the change in spatial working memory and showed that this vaccine generates significant memory benefits compared to non-sensitized DC-injected transgenic mice (Fig. 5). The E22W42 DC vaccine-treated old APP/PS1 mice showed memory improvement when tested by a radial arm water maze. Figures 5a and 5b show that the working memory performance of old APP/PS1 mice was significantly worse compared to non-transgenic mice (NTG) controls for both Errors and Latency, respectively. This demonstrates that the APP/PS1 transgenic mice showed memory loss as characteristic of AD. Working memory performance lost in old APP/PS1 mice is similar to AD patients. Meanwhile, errors of working memory are reduced in the E22W42 group mice. The latency of working memory is shortened in WT42 group mice compared to the DC control group. This implies that this type of DCs vaccine may be an ideal treatment for AD patients.
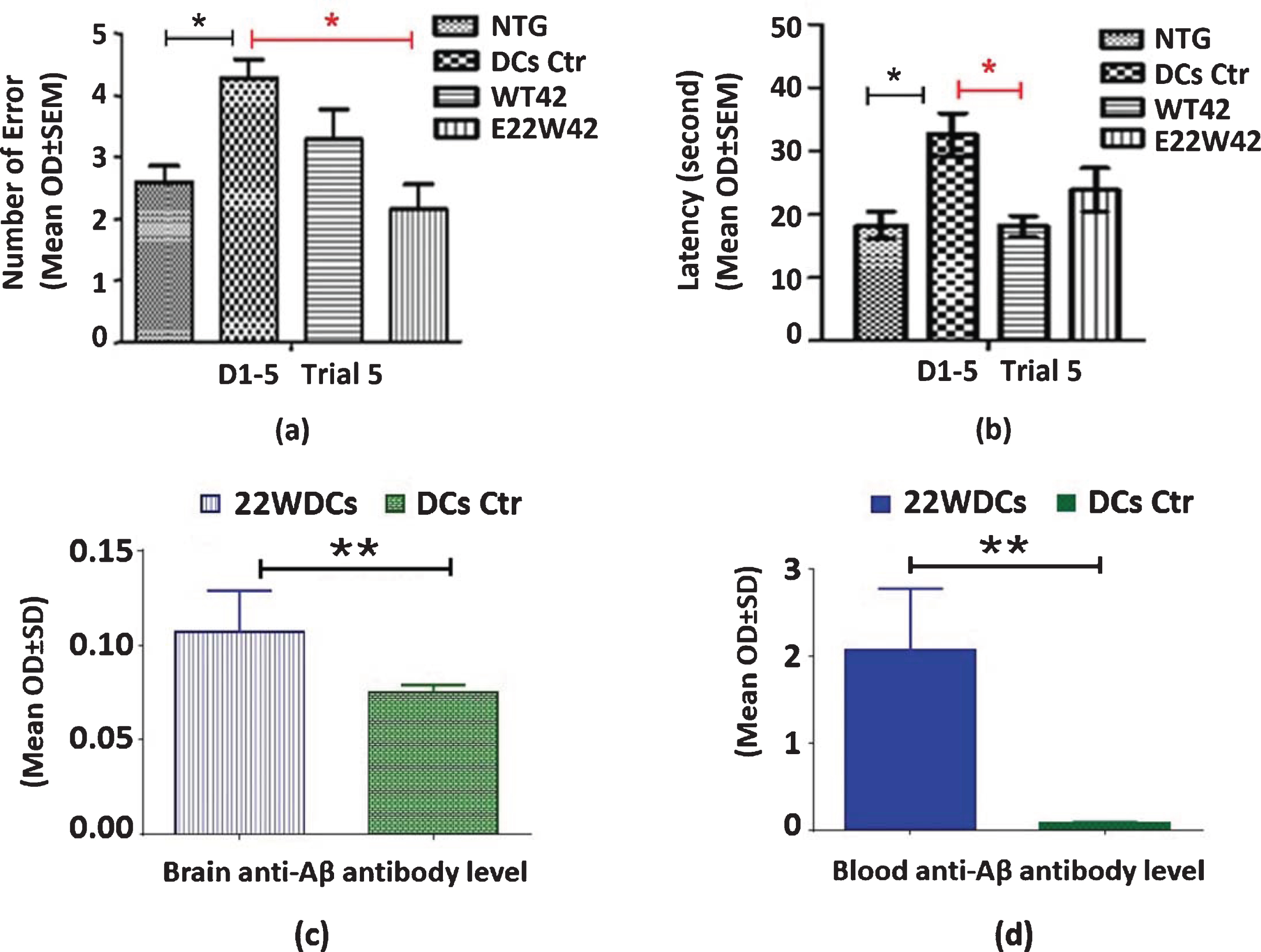
E22W42 DCs vaccine improved memory in APP/PS1 mice. Spatial Working Memory improved after post-vaccination (bar graphs). In both a and b, the first bar (from left to right) is the non-transgenic mice, the second (from the left) bar is the DC control group, the third bar (from the left) is the WT42 DCs vaccine treated group, and the last bar (from the left) is the E22W42 DCs treated group. a) Number of errors that mice made in testing working memory performance. b) Latency of mice to find the platform in testing working memory performance. c) ELISA are the results of brain anti-Aβ antibody level for both E22W42 DC and DC control. The plate is coated with re-aggregated Aβ, and then brain tissue lysates are loaded as samples at a 1:50 dilution (protein concentration is 1mg/ml). E22W42 DCs vaccine group is significantly higher than the DC control group (n = 6 per group, p < 0.01). d) The blood anti-Aβ antibody level for both E22W42 DCs vaccine and DC control is demonstrated. The plate is coated with pre-aggregated Aβ1-42, and plasma is diluted at 1:800 then detected with an anti-mouse antibody. The Anti-Aβ antibody level in E22W42 DC vaccinated group is significantly higher than the DCs control group (n = 6 per group, p < 0.01).
Significantly, the E22W42 DC vaccine treatment elevated the level of brain Aβ antibody, compared to DC control (Fig. 5c). In addition, the E22W42 DC vaccine treatment elevated the blood Aβ antibody level, compared to the DC control (Fig. 5d).
The vaccine can increase brain LC3-II level
Mapping the mechanism is very critical for a safe and effective therapeutic, so we further analyzed autophagy in brain tissue and discovered that LC3-II levels increased in the vaccinated group compared to the control group (Fig. 6). LC3 is a protein involved in autophagy. In normal conditions, the LC3 protein exists in the cytosol as type I (LC3-I). LC3-I is converted to LC3-II by lipidation and is recruited to autophagosomes when autophagy is activated. A study showed that LC3-II is also associated with endocytosis, facilitates Aβ clearance, and mitigates neurodegeneration in murine AD [54]. E22W42 group mice show a higher level of LC3-II (Fig. 6a) and LC3-II /LC3-I (Fig. 6c) in APP/PS1 mice compared with the WT and DC only group (control group). E22W42 can enhance autophagy activity and neuroprotection; no differences were observed in the LC3-I levels among the groups (Fig. 6b).

Increased level of LC3-II and LC3-II /LC3-I level in E22W42 DCs vaccine group may have a role in neuroprotection. The detection of protein expression levels in the brain by western blot. There is a total of 28 APP/PS1 mice that are individually label-located above in three different groups in (a), (b), and (c). DCs control, WT42 DCs, and E22W42 DCs vaccine groups are labeled in the blot (DC ctr=9, WT42 DCs=9, E22W42 DC=10). The first horizontal row of bands is detected with the anti-mouse LC3-I antibody and the second horizontal row of bands is probed with LC3-II antibody. The third horizontal row of bands is probed with β-actin (loading control) antibody. (d), (e), and (f) are the quantification data of (a)-(c). (d) is the relative quantitative level of LC3-II (the second row versus the third row of (a)-(c). The first bar from the left is the E22W42 DCs treated group, the second bar from the left is the DC control group, and the third bar from the left is the WT42 DCs treated group. The LC3-II expression level in the E22W42 group is significantly higher than the other two groups (E22W42 DC versus WT42 DC p < 0.05, and E22W42 DC versus DC control p < 0.01). (e) is the quantitative level of LC3-I (the first row versus the third row of (a)-(c), the same order as (d), and there are no significant differences among all three groups. (f) is the level of LC3-II /LC3-I ratio (second row versus the first row of (a)-(c), the same order as (d). The E22W42 DC group is significantly higher than the other two groups (p < 0.05).
Brain GSK-3β level
We have analyzed pGSK-3β and total GSK-3β, as well as the ratio of pGSK-3β/GSK-3β (Fig. 7). Increased GSK-3β activity has been proposed to play a central role in the development of AD, and its overexpression has been shown to induce neuronal death [50, 51]. The p-GSK3β level is associated with memory decline in AD, so it is pivotal to determine any changes during anti-Aβ treatment. The E22W42 DC vaccine-injected mice did not show an effect on the level of p-GSK-3β, but longer treatment times need to be conducted. The average GSK-3β level in brain tissue of E22W42-vaccinated mice stayed the same as the DC injected group. The average GSK-3β level of the WT42 group is 98% of that of the DC control; the average p-GSK-3β levels of E22W42 and WT42 are 91% and 95% of that of the DC control, respectively. Meanwhile, the average pGSK-3β/GSK-3β level of E22W42 and WT42 is 93% and 99% of that of the DC control.

E22W42 DC vaccine mice effects on the level of p-Gsk-3β. Brain GSK-3β expression post-vaccination. There were a total of 28 APP/PS1 mice, with group labels located in three different blots in (a), (b), and (c). GSK-3β, β-actin of GSK-3β, and p-GSK-3β bands are clearly labeled. In both (d) and (e), bar 1 from left is the E22W42 DCs group, bar two from left is the DC control group, and bar three is the WT42 DCs group. (d) is the quantification of the level of p-GSK-3β/actin ratio. (e) is the quantification of the level of GSK-3β/actin ratio. There are no significant differences among all three groups (p > 0.05).
DISCUSSION
Immunotherapy against neurodegenerative diseases has opened a new era for treatment and has brought new hope for AD patients. However, the initial vaccine trials were suspended by the FDA due to T cell infiltration into the brain that caused encephalitis [55]. The major obstacle is finding a method to produce a safe and effective vaccine for AD patients. There is a desperate need to create and implement effective strategies for AD. The history of immunotherapies against AD reveals that AD patients are aged subjects with decreased immune systems. Therefore, a normal vaccine strategy will not work on an aged population unless strong adjuvants are used to stimulate and reactivate their immune systems. Unfortunately, such adjuvants will lead to over-activation of the immune system that induces an unwanted response. Aged subjects have a difficult time overcoming such an overreaction, and this will likely lead to disastrous results for the patients. There has been evidence suggesting that a therapeutic vaccination strategy targeting the Aβ protein (i.e., Aβ1–42) may have some efficacy. To overcome the major immunotherapy issues seen in clinical trials, our laboratory has generated several different derivatives of the T cell epitope in Aβ1–42. We have introduced several mutations in the T cell epitope of Aβ and showed that a peptide with a mutation can sensitize dendritic cells. These cell-based therapies can have memory benefits when tested on aged APP/PS1 mice [33]. We have identified a novel peptide, E22W42, that may be the best candidate for the development of a safe and effective therapeutic vaccine for AD.
The significance of our study is that the mutated peptide can generate new T cell epitopes, which may explain our previous discovery that E22W42 can activate DCs from 30-month-old mice [36]. Another major benefit of this mutation is that it leads to the silencing of some T cell epitopes in the wild type peptide, which can dampen the autoimmune response. In addition, the new epitopes can stimulate a specific T cell response that activates the immune system. In this reported study, we first demonstrated that our novel E22W42 DC-based therapeutic vaccine applied to middle-aged APP/PS1 mice can generate a proper and durable antibody response. This vaccine approach was shown to be efficient and safe in memory protection [33 , 56]. Further, our data showed that the E22W42 vaccine targets only the soluble oligomer Aβ, instead of monomers. In addition, our vaccine can generate an isoform-specific antibody response (Fig. 2a–c) without causing T cell infiltration into the brain. This is significant because oligomer-specific antibodies are important for the treatment of AD pathogenesis [57 –60].
We discovered that this vaccine did not change the natural properties of the dendritic cells. The E22W42 DCs vaccine, acting as a natural adjuvant, can help prevent potential vaccine related adverse effects in AD. Furthermore, when we used the E22W42 DCs vaccine in APP/PS1 transgenic mice, the memory impairment of the mice was delayed compared to the DCs control group (Fig. 5). Another significant result was that the E22W42 DCs vaccine increased LC3-II/I ratio compared to the other two groups. However, the E22W42 DCs vaccine did not change the levels of ERK, p-ERK, or p-Gsk3β. Moreover, we were unable to see a difference in the level of Aβ with the E22W42 DCs vaccine. This may partially be due to the antibody mainly targeting oligomeric Aβ and not targeting monomeric Aβ. Pathologically, the Aβ-induced cascade response takes a long time to cause memory impairment. Therefore, a longer treatment may be required to observe a more significant change in the AD mouse model. Overall, our results strongly indicate that there are many changes following the E22W42 DCs vaccination. Since aging is the most important risk factor for AD, an effective vaccine must address age-related impairments in the immune system. The damaged immunity can be a risk factor for many diseases, such as AD and PD. Our previous data also demonstrated that E22W42 stimulated DCs from 30-month old DCs co-cultured with splenocytes and led to increased CD8 + T cell counts and IFN gamma levels in vitro. If this also occurs in vivo, it could potentially provide benefits against AD in elderly patients.
Conclusion
Our results lead to the following important conclusions: 1) Only Aβ peptides with mutations in the T cell epitope, such as E22W42, can sensitize DCs as a vaccine. The major advantage of using E22W42 is that it can generate several novel T cell epitopes and silence auto T cell epitopes. These novel T cell epitopes may lay a foundation for identifying T cell activators for cell-based immunotherapy. 2) E22W42 can sensitize DCs to target biologically relevant oligomeric forms of Aβ. The sensitized DCs vaccine mediated the amelioration of memory deficits that occurred in the mouse AD model. 3) This is the first time that the E22W42 DCs vaccine has been tested on APP/PS1 transgenic mice. The E22W42 DCs-based vaccine has immunomodulatory effects that target oligomeric Aβ. 4) The E22W42 DCs helped to maintain dendritic cells’ immunomodulatory properties. Though the E22W42-sensitized DC vaccine is being developed for AD patients, it can potentially help strengthen the immune system of elderly patients as well. E22W42 may possibly lay the foundation for future immunotherapies for aging-related disorders.
Footnotes
ACKNOWLEDGMENTS
Thank you to Paul Sinu for his generous help with the T cell epitope analysis. Also, thank you to Alzamend Neuro Inc. for providing some of the peptides for this research.
This research has been supported by NIH R01 AG056569, Florida High Tech Corridor matching funds, and MegaNano Biotech Inc.
The patent on E22W42 was awarded on May 29th, 2012, and the patent No. is US8,188,046 B2.