
Abstract
The ɛ4 allele of the Apolipoprotein E (APOE) gene in individuals infected by Herpes simplex virus type 1 (HSV-1) has been demonstrated to be a risk factor in Alzheimer’s disease (AD). APOE-ɛ4 reduces the levels of neuronal cholesterol, interferes with the transportation of cholesterol, impairs repair of synapses, decreases the clearance of neurotoxic peptide amyloid-β (Aβ), and promotes the deposition of amyloid plaque, and eventually may cause development of AD. HSV-1 enters host cells and can infect the olfactory system, trigeminal ganglia, entorhinal cortex, and hippocampus, and may cause AD-like pathological changes. The lifecycle of HSV-1 goes through a long latent phase. HSV-1 induces neurotropic cytokine expression with pro-inflammatory action and inhibits antiviral cytokine production in AD. It should be noted that interferons display antiviral activity in HSV-1-infected AD patients. Reactivated HSV-1 is associated with infectious burden in cognitive decline and AD. Finally, HSV-1 DNA has been confirmed as present in human brains and is associated with APOE ɛ4 in AD. HSV-1 and APOE ɛ4 increase the risk of AD and relate to abnormal autophagy, higher concentrations of HSV-1 DNA in AD, and formation of Aβ plaques and neurofibrillary tangles.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is devastating and the most common neurodegenerative disorder for the aging population worldwide, doubling the risk every five years for individuals over the age of 65 and affecting up to 50% over the age of 85 [1, 2]. AD is consistent with clinical syndromes, such as cognitive profiles and classical symptoms and now with a specific biological neuropathological presence: extracellular deposition of diffuse neuritic plaques and intraneuronal neurofibrillary tangles (NFTs) resulting from aggregated hyperphosphorylated tau protein (P-tau) [3, 4]. However, an endless parade of mid-to-late-stage clinical trial failures has frustrated pharmaceutical development for AD and no disease modifying therapies are currently available, which are direly needed to prevent or slow the rate of disease progression [4].
Late-onset AD, accounting for approximately 95% of AD cases, appears to have multiple susceptibility genes, especially the ɛ4 of the Apolipoprotein E (APOE ɛ4) gene [5, 6]. Apolipoprotein E (APOE) protein could influence neurite outgrowth, synaptic plasticity and amyloid accrual, cholesterol metabolism, and particularly the risk of AD [7–10]. Interestingly, the negative effect of APOE ɛ3/ɛ4 on AD progression may increase AD risk about 4-fold in women in contrast with men, as compared to the ɛ3 allele of APOE (APOE3) [11, 12].
Herpes simplex virus type 1 (HSV-1) DNA resides in the brains of elderly people at a high proportion, whereas some individuals infected by HSV-1 do not develop AD [13]. Herpes viruses evade, manipulate, and exploit host immune responses and have been found to leave a footprint of the host immune system resulting from an arms race between viral evasion and host immune defense [13]. HSV-1 specific neutralizing ability of AD sera was reduced because HSV-1 specific immunoglobulin G (IgG) 3 were more frequently detected in mild cognitive impairment (MCI), compared to AD and healthy controls groups, but the presence of IgG3 was at a relatively high quantity [14]. New data from the World Health Organization indicated that almost 3.7 billion people, who were under the age of 50, or 67% of the population, had HSV-1 infection (oral or genital) in 2016, and prevalence of the infection was highest in Africa (88%) and lowest in the Americas (45%) [15]. The inhibition of HSV-1 autophagy in host cells and viral induced amyloid-β protein precursor (AβPP) processing alterations could result in accumulation of intraneuronal amyloid-β (Aβ) [16]. The virus interacts with oxidative stress to significantly enhance the effects of accumulation of Aβ and autophagy impairment induced by HSV-1 [17].
HSV-1 can directly access the central nervous system (CNS) bypassing the brain stem by olfactory bulb (OB), and the levels of latency-associated transcripts isolated from the OB were marginally above uninfected controls during HSV-1 latency [18]. There is a strong need to demonstrate the route of HSV-1 infection in animal models and humans, and to explore the contribution of herpes simplex viruses to AD etiology and pathogenesis. Here we present recent discoveries relevant to methods of HSV-1 entry and regions of HSV-1 infection (Table 1). It is likely that HSV-1 infection is more prevalent in older adults, because HSV-1 DNA presents in brain autopsies of non-AD elderly at a significant frequency [19]. The levels of HSV-1-specific IgG and IgM in blood are not elevated in all studies of AD, which may due to altered immunoglobin levels caused by old and new viral infections and other factors [20, 21]. A viral etiology of AD may be confounded by the result that HSV-1 cannot be the sole cause of AD. Conversely, the bulk of the evidence shows that HSV-1 infection may contribute to AD etiology and pathogenesis.
Studies relating entry of HSV-1
This review focuses on elucidating the relationship between APOE polymorphism and AD and further demonstrates APOE ɛ4 as an AD-associated risk factor to accelerate pathological characteristics in AD. In addition, the role of herpes simplex virus type 1 in AD is described. In this paper, a cooperative role between the APOE ɛ4 and HSV-1 infection in the etiology of AD is presented.
DIFFERENT STRUCTURAL DOMAINS AND FUNCTIONAL ISOFORMS OF APOLIPOPROTEIN E PROTEIN IN HUMAN
The human APOE gene, located on chromosome 19, is a polymorphic gene with three analogous allelic forms designated ɛ2, ɛ3, and ɛ4, which respectively encode E2, E3, and E4 protein isoforms [22]. APOE protein coded by the APOE gene is a glycoprotein with 299 amino acids, and is a major component of very-low density lipoproteins [23]. Related to human APOE isoforms (E2, E3, and E4), only amino acid positions 158 and/or 112 of these three isoforms of APOE differ (Fig. 1). APOE3 protein contains an arginine at position 158 and a cysteine at 112, whereas APOE4 contains arginine at both positions and APOE2 contains a cysteine at both sites [24, 25]. APOE protein has two main regions: the lipid-binding region at the C-terminal domain and the receptor-binding region at the N-terminal domain, where a flexible hinge joins them [26].
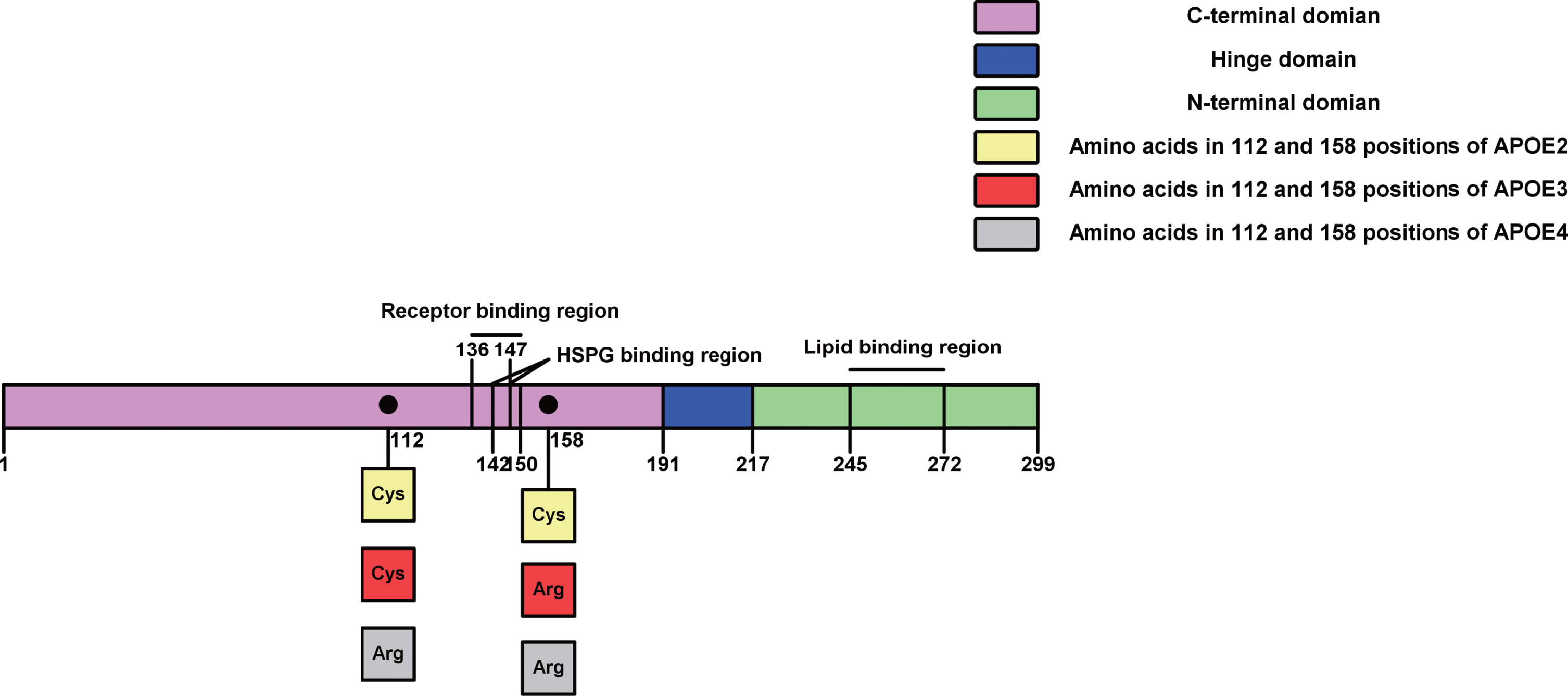
APOE with different protein isoforms, structural domains, and functional regions. APOE protein has 299 amino acids divided to three main domains, including N-terminal domain (purple), hinge domain (blue) and C-terminal domain (green). The hinge domain (blue) is from position 191 to 217. The C-terminal domain (green, 217–299) has a lipid binding region from 247 to 272. The N-terminal domain (purple) involves the receptor binding region (136–150) containing the heparan sulfate proteoglycan (HSPG) binding region (142–147). There different isoforms of APOE protein in the N-terminal domain only have one/two difference of amino acids in 112 and 158 positions, such as APOE2 (two Cys), APOE3 (one Cys and one Arg) and APOE4 (two Arg).
Highly conserved regions representing approximately 47 amino acids (of 299) appear to be related to the primary APOE protein functions by reflecting ligand binding sites and regions involved in the propagation of the arginine-cysteine change at position 112 to distant regions in the N- and C-terminal domains of the protein [27]. Taken together, APOE dimer tandem repeat peptide was found to have activity against HSV-1 and 2 [28]. In addition, an N-terminal fragment of APOE has antiviral activity due to this receptor-binding domain and may cause innate immunity to infection of virus by inhibiting attachment of virus and/or direct disruption of viral particles, subsequently reducing viral entry, suggesting that the peptide participates in preventing viral attachment, interfering with the earliest phase of viral infection [28]. This may structurally explain that APOE protein influences infectivity of HSV-1.
INDEPENDENT FUNCTIONS OF APOE ɛ3, ɛ2, AND ɛ4 GENE POLYMORPHISMS, ESPECIALLY THE AD SUSCEPTIBILITY GENE APOE ɛ4 GENOTYPE
APOE ɛ4 as genetic risk variant in AD
APOE gene in brain is also involved in amyloid accrual, cholesterol metabolism, and AD risk [10]. APOE ɛ4 is linked to a variety of diseases including Lewy body disorders, Parkinson’s disease, stroke, sleep apnea, multiple sclerosis, and AD [29]. APOE3 protein maintains lipid homeostasis and thus shows protection against cardiovascular diseases, while APOE2 protein is connected to dysbetalipoproteinemia [30]. In many studies with APOE ɛ3 as the comparator, APOE ɛ4 has emerged as the greatest genetic risk variant for AD with risk for homozygous carriers increasing over 15-fold, whereas APOE ɛ2 decreases risk 4-fold, regardless of geographical location or race [31]. Intracranial aneurysms with hypertension were associated with the ɛ2 allele and the ɛ2/ɛ3 genotype of APOE in a case-control study [32]. It has been shown previously that the carriage of ɛ4 genotype affects prognosis, pre-symptomatic risk and treatment response for various diseases, such as AD [33]. Indeed, the clearance of soluble Aβ in brain interstitial fluid depended on the expression of APOE protein isoforms [34]. Possession of human APOE ɛ4 caused the greatest accumulation of amyloid in models of AβPP transgenic mouse, compared to APOE ɛ3 and APOE ɛ2 transgenic mice [35]. Nevertheless, the ɛ4 allele of APOE alone is neither sufficient nor necessary for the development of AD [36, 37].
Role of APOE ɛ4 in neuron-associated functions in AD
APOE ɛ4 may be involved in deficiency of neuroprotective functions and toxicity [31, 38]. It is hypothesized that APOE ɛ4 allele is consistent with an increased AD risk-associated hippocampal neurotoxic lifetime exposure and the highest transportation of macromolecules to neurons, such as lipids and proteins [39]. APOE ɛ4 elevated proteolytic degradation reducing the levels of neuronal cholesterol, interferes with the transportation of cholesterol, and impairs function and repair of synapses [40]. Furthermore, future therapies to slow down pruning of synapses might meditate differential effects based on APOE allele subtype [41].
Idiosyncrasies of APOE polymorphisms in women
Interacting differently in women, the genetic risk factor APOE ɛ4 for AD may display an elevated risk of adverse clinical changes from healthy aging to disease [42]. Current findings indicate that the carriage of APOE ɛ4 shows connection with late-life Aβ deposition and midlife dyslipidemia in women primarily [43]. These factors interact resulting in a compounded risk for Aβ deposition twenty years later [43]. Low-density lipoprotein (LDL) cholesterol associated with Aβ notably increases in midlife after adjusting for age, cholesterol medication, cognition, and education, but adjustment for APOE4 protein attenuates LDL cholesterol [43].
The interactive effects of estrogen therapy (ET) and APOE protein on AD pathology are suggested by preclinical studies [44]. ET treatment may be particularly effective at cognition improvement in APOE ɛ4 carriers, even though the published data are inconsistent [45, 46]. In a study with placebo-control, ET was associated with more cognitive decline in APOE ɛ4 positive women than in APOE ɛ4 negative carriers [47, 48]. The Nurses’ Health Study indicated that ET provided no benefits in older women, regardless of APOE genotype. In contrast, hormone replacement therapy (HT) associated with less cognitive decline was significantly pronounced with APOE ɛ4 carriers in follow-up analysis [46, 49]. It has also been demonstrated that HT attenuates the risk of dementia linked with APOE ɛ4, and elevated markers of aging show acceleration in APOE ɛ4 carriers [50].
Possible survival advantages in youth with APOE ɛ4, as an ancestral genotype
The APOE ɛ4 was the first gene in evolution, whereas the ɛ2 and the ɛ3 mutations were respectively produced 80,000 and 300,000 years ago [51]. APOE ɛ3 is the most common allele [52]. However, the driving force for the transformation from ɛ4 to ɛ3 predominance is currently still obscure [41]. APOE ɛ4 may be less prevalent due to the ancestral advantages of the APOE ɛ4 and the evolutionary trade-off of superior performance in youth [41]. Historical lifespans were brief such that advantages in youth associated with the APOE ɛ4 allele might have increased fitness compared to the ɛ3 allele [41].
The ancestral predominance of the ɛ4 allele might have been influenced by protective effects on fertility and reproduction [53, 54]. Furthermore, APOE ɛ4 genotype showed a protective role via a decrease in risk of intracranial aneurysms in a Chinese population [55]. In prehistoric populations with a high carriage of ɛ4 allele, endemic infectious diseases show resistance associated with the predominance of APOE ɛ4 [56, 57]. Studies suggest that carriers of the APOE ɛ4 might display higher mental performance in young healthy subjects [58–60] with fewer neurons in certain brain regions allocated to memory processes than in non-carriers [61, 62].
EVOLUTION OF HERPES SIMPLEX VIRUS 1 HYPOTHESIS OF ALZHEIMER’S DISEASE CAUSATION
The concept of effects of HSV-1 in AD was first proposed more than thirty years ago [63]. HSV-1 emerged as a likely candidate virus due to a very rare but extremely serious and acute infection of herpes simplex encephalitis (HSE) in the CNS, where the earliest and most severe regions showed pathological changes similar to AD [63]. The neuropathological changes corresponding to the hippocampus and mesial temporal cortex are associated with long-term inflammation both in animal models and in human HSE survivors [64]. HSV-1 DNA resided in latent form in a high proportion of elderly brains from both normals and AD patients. In contrast, HSV-1 infections were present in only a small proportion of brains of AD patients and elderly normals [65–68]. Viral DNA was present in only a very small proportion of pediatric and brains of the young, in contrast with the high frequency of HSV-1 DNA found in elderly brains [69]. HSV-1 DNA co-localized strongly with amyloid plaques in AD brains but not in normal brains [70]. Further, HSV-1 might recurrently reactivate in brain and subsequently may cause a productive infection, leading to severe damage, cell death, and eventually to the development of AD [71–73]. Infections with microbes like bacteria and spirochetes, which generate nitric oxide, free radicals, and further induction of apoptosis as proinflammatory cytokine activators, may be considered as a risk factor to cognitive changes or pathophysiological changes of AD [74]. The infections of other viruses like the measles virus and HIV implicated in neurological disease can result in subacute sclerosing panencephalitis [75]. HIV-associated dementia [76–79] were associated with the formation of characteristic pathological features in AD, including NFTs, phosphorylated tau protein, and neurodegeneration in brain [80].
CHARACTERISTICS OF HSV-1 INFECTION IN BRAIN
Structural advantages facilitating HSV-1 entry into the host cell and into relevant regions of AD
HSV-1 is an enveloped virus surrounded by an icosahedral shaped nucleocapsid, composed of a 152 kB core double-stranded DNA genome [81]. The viral envelope is composed of a lipid bilayer decorated with various glycoproteins. Viral glycoprotein B (gB) and glycoprotein C (gC) participate in attachment of virus to the receptor for heparin sulfate proteoglycan (HSPG) in the host cell. It has been identified as necessary for viral entry into the host cell via host cellular receptor protein interaction with HSV-1 glycoproteins, including gB, gD, gH, and gL [82]. HSV-1 gB had a significant homologous region in common with Aβ and interacted with lipoproteins, particularly APOE protein which may stimulate molecular interaction and further neurodegeneration [83]. The data supports the conclusion that the peptide of gB could emerge as a potent initiator for the cascade of fibril formation and deposition of Aβ if cleaved appropriately, and suggests a link between gB and APOE [84]. HSV-1 viral envelope gD interacts with herpesvirus entry as a mediator when the viral envelope fuses into the host cell membrane [85]. The tegument, located between the viral envelope and the capsid, contains 26 viral proteins. These proteins are relevant to the HSV viral lifecycle, including transporting viral DNA to the nucleus of the host cell, transcripting viral genes, and overturning a variety of host cellular processes [82]. After the virus fuses to the host cell, nucleocapsid and tegument proteins enter the host cytoplasm. The nucleocapsid leaves from cytoplasm to the nucleus, wherein viral DNA is separated and circularizes. A specific tegument protein blocks off host cell protein synthesis [86].
During latency, when HSV-1 occurs reactivation in the trigeminal ganglion (TG), anterograde transport of HSV-1 particles can cause orofacial lesions [87]. Retrograde transport of viral particles between axons can facilitate entry into the locus coeruleus and thus infiltrate into the temporal lobe, especially the entorhinal cortex and hippocampus [87]. HSV-1 infection progressed from the nose to the TG and then reemerged in the facial skin [88]. Blood monocytes infiltrated into the CNS after intranasally infecting mice with a sub-lethal dose of HSV-1 during experimental herpes simplex virus encephalitis [89]. HSV could transmit in the anterograde direction from olfactory receptor neurons to mitral cell neurons in the OB and could extend to amygdala, hippocampus, and the entorhinal cortex [90]. The OB connection to the entorhinal cortex and the entorhinal cortex itself show neurodegeneration characteristic of AD pathology at an early age [91–93]. HSV-1 DNA was detected from almost all olfactory and trigeminal ganglia of postmortem tissue samples in HSV-1-infected patients [94]. In 90% of all patients diagnosed antemortem with clinical AD, HSV-1 DNA was identified in the TG [95]. Following intranasal administration of HSV-1 to the 17Syn+strain of BALB/c mice, inflammatory cells including macrophages, lymphocytes, neutrophils, and plasma cells, were confirmed via immunochemical antibodies reactive to Mac2 from macrophages, CD3 from T lymphocytes, and myeloperoxidase from neutrophils [96]. During the chronic phase, the presence of microglial activation seems to persist in the damaged area. These cells intensely immunostain with Mac2, known to be a marker upregulated in macrophages participating in phagocytosis [64]. Further, the Morris water maze test, used to detect spatial memory ability, showed deficits in BALB/c mice [96]. This spatial memory deficit seen in HSV-1 infected mice might be similar to the spatial disorientation observed in early-stage AD patients [96]. The molecular logic of vomeronasal organ ligand detection was better understood due to functional expression of vomeronasal receptors which deciphered the neural mechanisms controlling behavior in herpes virus-based amplicon delivery system [97]. In subepithelial myeloid cells, HSV-1 spread from the olfactory epithelium to TG neurons [98]. Further, HSV-1 entered the tree shrew brain following ocular inoculation and HSV-1 transcripts were detected during acute infection [99]. The aforementioned animal studies and others, support the entry of HSV-1 into the brain through the olfactory bulb [100], structures of the limbic system along nerve pathways including the entorhinal cortex and hippocampus, i.e., structures adversely impacted in AD [101]. Clinical olfactory function impairment is connected with increased incidence of MCI and AD [102–104].
Lifecycle of HSV-1 in viral infections
Viral latency has been established in the TG of the peripheral nervous system (PNS) after initial infection. Life-long HSV-1 infection escapes from the immune system in host cells [105]. HSV-1 infections appear to undergo subclinical and clinical reactivation, including keratitis, recurring oral ulcers and severe encephalitis [106]. The virus has both a latent and productive lifecycle. During the productive lifecycle, the presence of newly produced virions can cause host cell death. Viral genes, such as α-genes (immediate-early), β-genes (early), or γ-genes (late), respectively express α proteins to modulate the viral genome, β-proteins to participate in DNA synthesis of virus, and γ-proteins to emerge as viral structural proteins [86, 107]. During the latent lifecycle, the viral genome exists with no formed virus and the expression of viral gene ceases in the host cell, except for latency-associated transcripts termed LATs facilitating the latency and inhibiting host cellular apoptosis [105].
The weight-of-evidence suggests that HSV-1 reactivation occurs in merely a small number of neurons infected in latency during reactivation phase [108]. Periodic viral reactivation during the latent lifecycle could be induced by diseases, immunodeficiency, and other stressors [109]. HSV-1-infected reactivation was detected in explant cultures of brain tissue in mice, suggesting that mice might be an appropriate animal model for future viral studies [110, 111]. Massive doses of immunosuppressive drugs and radiation in HSV-1-infected mice influenced viral reactivation in vivo. However, the latent reactivation of virus could not be found from brains in explant cultured mice with HSV-1 infection [112]. T cell immune system impairment associated with age is identified as immunosenescence, and is related to increased peripheral reactivation from infected herpes virus including HSV-1 in elderly people [113–115]. Early neurodegeneration and neuroinflammation are found in the cerebral cortices and trigeminal ganglia of mice after reactivation of HSV-1 from latency [116]. An immune response, mediated by CD8+ T lymphocytes infiltrates mouse and human HSV-1-infected TG ganglia. This immune response is in part mediated by secretion of IFN-γ, and suppresses reactivation of virus in infected trigeminal ganglion during latency [117, 118].
ROLE OF HERPESVIRIDAE FAMILY (EXCEPT HSV-1) ALONE OR IN CONCERT WITH APOEɛ4 IN DEVELOPMENT OF AD
Infectious agents, such as HSV-2 and cytomegalovirus (CMV), have been examined for associations with cognitive decline and AD by increasing Aβ production [119–122] and evoking tau phosphorylation [123, 124]. Furthermore, spirochetes and Chlamydophila pneumonia (C. pneumoniae) might evade the host immune response thereby achieving high burdens in the brains of AD patients. In vitro studies and animal models suggest that this brain burden could induce formation of P-tau and amyloid plaques [125–129]. In aged APP/PS1 mice, respiratory infection exacerbated Aβ pathology and increased glial activation, suggesting an adverse effect on peripheral regions of AD [130]. Human astrocytoma cells were bound by C. pneumoniae elementary bodies, with the APOE ɛ4 having 3-fold higher levels than cells bearing the non-APOE4 allele [131]. This result suggests that attachment of C. pneumoniae elementary bodies to target host cells is enhanced by APOE4 expression.
Human herpesvirus 6 (HHV6) and HHV7, in a manner similar to HSV-1, were present in elderly normals and AD brains, with AD levels significantly higher than the control groups [132, 133]. Studies by others had shown that HHV6 worsened the damage caused by APOE ɛ4 in AD and HSV-1 in cell cultures and animal tissue [134, 135]. The proportion of HHV6 present in AD patients was high, while the proportion in age-matched normal controls was significantly lower than in patients with HSV-1 [135]. HHV-6A infection was found to enhance APOE ɛ4 mRNA synthesis with a time dependent correlation, indicating that the increased production in HHV-6A RNA correlated to the enhancement in APOE ɛ4 mRNA [136]. APOE4 expression by APOE ɛ4 allele was observed to be upmodulated by HHV-6A infection [137] and protein kinase A (PKA) activity was increased by APOE and in a PKA-dependent pathway the secretion of APOE is operational [138]. Stronger specific humoral responses with HSV-1 infections correlate with protection of cortical volumes and AD conversion, reinforcing the hypothesis referring to a role for HSV-1 in AD pathogenesis [139].
HSV-1 AS A PREDISPOSING FACTOR FOR THE DEVELOPMENT OF AD
Location of HSV-1 in AD-related amyloid plaques and antiviral functions of Aβ peptide
In AD brains, 72% of HSV-1 DNA was associated with plaques and 90% of the plaques contained the viral DNA [140]. In aged normal brains, only 24% of the viral DNA was found, with a lower frequency to amyloid plaques although 80% of plaques contained HSV-1 DNA [140]. These results suggest that viral infection was a major cause resulting in amyloid plaques and thus is probably significant in AD. HSV-1-infected neurons in vitro display tau protein hyperphosphorylation [141]. Specific sites of P-tau inhibiting Aβ toxicity might relate to Aβ antiviral action. Abnormal P-tau presumably caused by HSV-1 infection might produce aggregated Aβ based on anatomic considerations [142–144]. HSV-1 infection in SH-SY5Y cells and cortical neurons of rat activates amyloidogenic pathways of the host cell, causing multiple AβPP cleavages with accumulation of secreted intracellular and extracellular Aβ1–40, Aβ1–42, and additional neurotoxic fragments of AβPP [145]. AβPP level could be reduced in HSV-1-infected neuronal cells [146].
The Aβ peptide has been identified as playing a role in the formation of amyloid plaque and defense against multiple microbes [147–149]. Amyloid acts protectively by encapsulating pathogens—a once heretical notion that was either ignored or derided [150]. Moreover, studies have found that HHV6 and HSV-1 can induce the production of amyloid plaque after 24–48 hours of infection in mice, and that Aβ oligomers attenuate HSV-1 infection in models of 3-D neural cell culture from human, and protect from acute viral encephalitis in 5×FAD transgenic mice [151, 152]. They also indicated that Aβ can inhibit infection in brain cells and accelerate amyloid fibrilization [151, 152]. While evidence suggests an association of herpesvirus with AD, causation has been difficult to establish [153].
Antiviral activity of IFNs relevant to HSV-1-infected patients in AD
The cytokines including interferons (IFNs) could inhibit infection caused by HSV-1, stimulate the immune system, and activate pathways of neuroinflammation modulated by T-cells particularly [154]. However, IFN treatment adversely affected mood transiently over the first months, while the placebo with antidepressant effect might counterbalance the reduction in cognitive decline rate [155]. IFN and IFN-β1a inducing cognitive decline rate reduction appeared to be well tolerated and safe in AD patients at early phase [155]. Mounting evidence suggests that the antiviral action of IFN displays a beneficial effect in AD patients by its anti-proliferative and immunomodulatory properties. IFNs showed potential efficacy via blocking latent HSV-1 reactivation in a later study [156]. The function of IFN has been identified in TG cell cultures infected by HSV-1 by treating with supernatant enriched with IFNs [157]. Transgene expression in HSV-1-infected TG cells with IFN-β or IFN-γ in explant culture suppressed and delayed reactivation of HSV-1 [154]. Several other studies have identified that IFN-β and IFN-γ have similar effects on blocking HSV-1 replication in immunocompetent mice and cell culture [158–160]. IFN-λ, a key cytokine participating in innate antiviral defenses, has a particularly perceptible antiviral activity in HSV-1-infected individuals [161]. HSV-1-seropositivity measurements showed that INF-λ serum concentration and anti-HSV-1 antibody titers were higher in MCI individuals and AD than in healthy controls [161].
HSV-1 induces neurotropic cytokine expression with pro-inflammatory action and inhibits antiviral cytokine production, associated with IL-6, cGAS, UL37, and RIG-I
Subacute and acute inflammatory responses induced by HSE in humans were mediated by interleukin 6 (IL-6), IFN-γ, and tumor necrosis factor-α (TNF-α) [162]. Elevated IL-6 in the CNS of mice has been found in association with cerebral neurodegeneration thought to be caused by over expression of IL-6 near amyloid plaques [163]. Mouse trigeminal ganglia infected by HSV-1 led to expression of major histocompatibility complex II antigens and increased pro-inflammatory cytokines TNF-α, IFN-γ, and IL-6 [164, 165]. Cyclic GMP-AMP (cGAMP) synthase (cGAS) can function as a DNA sensor. It plays an essential role in catalyzing cGAMP synthesis, detecting cytosol double-stranded (ds) DNA, and inducing IFNs and inflammatory cytokines [166, 167]. Mouse or human cGAS deamidated by UL37 promotes lytic replication of HSV-1, indicating that viral pathogen uses variations from natural sequence of innate immune components to destroy host defenses [168].
Murine gamma herpesvirus 68, one of the gamma herpesviruses similar to human Kaposi’s sarcoma-associated herpesvirus, evades host cytokine production [169, 170]. Site-specific deamidation of retinoic acid-induced gene I (RIG-I) in the helicase 2i domain was characterized by impaired ATP hydrolysis and RNA detection of RIG-I [171]. RIG-I induced by HSV-1 prevents RIG-I activation through sensing viral ds RNA and inhibits production of antiviral cytokine, thereby increasing HSV-1 replication [171, 172]. The UL37 as a protein deamidase, targets RIG-I to block activation induced by RNA and its tegument protein was sufficient for deamidation of RIG-I in vitro, suggesting the role of deamidation of viral protein in immune regulation [173]. The cytosolic RIG-I receptor, a genuine RNA sensor response to infection of virus, activates interferon regulatory factor 3 and nuclear factor κB by the mitochondrial antiviral signaling protein [174–176].
IB along with CMV or HSV-1 associated with cognitive decline and AD
The first study on total infectious burden (IB), as systemic microbial burden, investigated 383 home-dwelling individuals and found a history of stroke or coronary heart disease, and 18% had type 2 diabetes [177]. Viral IB, a composite serological measure of serum antibody levels correlated to common pathogens such as CMV and HSV-1, leads to stroke [178] and cardiovascular diseases and is associated with cognitive decline in epidemiological studies [179, 180]. IB and cognitive decline by using a composite serologic measure detect exposure to both viral pathogens (HSV-1, HSV2, and CMV) and bacterial (Helicobacter pylori and C. pneumoniae) [180]. The elevation of IB may increase Aβ levels directly and subsequently cause AD development. Cell culture production of Aβ increased by HSV-1 infection by upregulating β- and γ-secretase levels, suggests that Aβ is a pore-forming antimicrobial peptide [181, 182]. In a previous study, bacterial IB showed no connection with cognitive impairment [183]. Adjusted for age, gender, education, APOE genotype, and other comorbidities, AD-associated higher total IB, viral IB (CMV and HSV-1), and bacterial IB (C. pneumoniae), supports a role for infection and inflammation in the etiology of AD [184]. Rates of positivity for HSV-1 in older Blacks and Whites were much lower than for CMV [185]. It indicates that CMV but not HSV-1 increases more risk of AD, highlighting that another vital factor is assay sensitivity. The protein expression from infections of common pathogens had significant AβPP and Aβ homology [186, 187]. This expression of proteins may trigger autoantibodies with cross reaction to AβPP bound with membrane, cause neuronal and synaptic dysfunction, and final cognitive decline [186, 187]. IB index of weighting risk of quantitative stroke was inversely related to cognitive decline of memory domain and executive function from baseline, suggesting that common infections with subsequent inflammatory responses interact with vascular damage, neurodegeneration, and aging [188].
ASSOCIATION OF HSV-1 INFECTION WITH CARRIAGE OF THE APOEɛ4 GENE
Studies measuring HSV-1 DNA from brain or HSV-1 antibody serology showed that the risk of AD was significantly increased by infection with HSV-1 alone and in concert with the APOE ɛ4 [66]. AD risk-associated genes emerged as transcriptional regulators such as Gamma-secretase Subunit Presenilin-1, Beta-Site APP Cleaving Enzyme 1, Clusterin, and Phosphatidylinositol Binding Clathrin Assembly Protein could be altered by viral infection [189]. It should be noted that virus in brain of carriers of the APOE ɛ4 allele, accounting for 60% of cases, confers a strong risk factor for AD [190]. The presence of the HSV-1 genome in the brain plus the possession of APOE ɛ4 shows more risk toward developing AD relative to the carriage of APOE ɛ4 alone [191, 192]. Furthermore, APOE has been found to modulate susceptibility to microbial infections caused by HSV-1 and HSV-2, e.g., herpes labiali and genital herpes [190, 193].
Different APOE isoforms influence the susceptibility and outcome of several diseases caused by viral infections [194]. APOE protein interacts directly with purified gB from HSV-1 [195] and accumulations of both APOE and HSV-1 inside the cells result from cellular entry via an HSPG receptor [196]. During acute infection, APOE4 was more efficient than APOE3 in promoting HSV-1 colonization of the brain by attaching to HSPG, and may increase susceptibility to processes of neurodegeneration connected to the APOE ɛ4 gene [197, 198]. Variation at the APOE locus may be connected with clinical manifestations of HSV-1 infection, whereas APOE subtype does not appear to associate with herpes simplex viral reactivation in humans [199]. The evidence proved that patients with frequent recurrent herpes labialis carried APOE ɛ4 more often than HSV-1 seropositive individuals who did not experience recurrences of the disease [44, 200].
In a large population-based cohort study, carriage of HSV showed significant association with declining episodic memory function, especially among APOE ɛ4 carriers both in cross-sectional and longitudinal results [201]. HSV-1-seropositive APOE ɛ4 also shows connection with cognitive impairment in age-adjusted cardiovascular patients [202]. When the immune system declines, HSV-1 reaches the brain in aging and resides there in latency, and reactivates upon stress or immunosuppression in the PNS. Reactivation can lead to an acute but localized infection, which can be more serious in an APOE4 carrier [203].
APOE ɛ4 is associated with the HSV lifecycle by involvement in viral infectivity [204]. Furthermore, APOE in the lifecycle of HSV-1 is related to abnormal autophagy in AD [205]. Acute hematogenous HSV-1 infection in a murine model showed that viral neuroinvasion was reduced in APOE gene knockout mice compared to wild-type mice and that the APOE dose was directly associated with the concentration of HSV-1 DNA [206]. APOE ɛ4 can modulate expression of HSV-1 gene or its spread in the brain by utilizing transgenic mice expressing ɛ2, ɛ3, and ɛ4 genotype of APOE gene in astrocytes from the glial fibrillary acidic protein promoter [207]. APOE ɛ4 acting in concert with HSV-1 in AD brains might lead to Aβ accumulation and AD-like tau, causing amyloid plaques and NFTs, and eventually to AD [208]. In genome-wide association study datasets, AD genes are increased to reflect pathogen diversity and pathogen resistance of implied earlier infection prior to the older ages of AD patients [209]. Wild-type APOE+/+mice infected with HSV-1 peripherally had 13.7 times the concentrations of HSV-1 DNA in brain than mice lacking APOE infected by HSV-1 [210].
CONCLUSION
HSV-1 DNA infection in brain alone or in concert with the APOE ɛ4 allele increased the risk of AD and aggravated the pathogenesis of AD. APOE protein, encoded by the APOE gene, has different structural domains and functional isoforms, which may contribute to a differential role of APOE polymorphisms in AD. APOE ɛ4 is a genetic variant for increased risk in AD compared to APOE ɛ2 and APOE ɛ3. APOE ɛ4 connected with AD plays a role in neuron-associated functions, inflammatory responses and pathological changes. The involvement of APOE ɛ4 elevated risk to hippocampal volume loss, cognitive impairment and AD-associated Aβ deposition is greater in women than men, and HT attenuated the risk of dementia linked with APOE ɛ4 genotype. Conversely, APOE ɛ4 was an ancestral genotype, which shows protective effects on fertility and reproduction, lowers the risk of intracranial aneurysms and shows improved mental performance in youth. It is obvious that HSV-1 exerts a strong influence in AD as the hypothesis has been proposed. HSV-1 enters into the host cell and goes into trigeminal ganglia, hippocampus, and so on, although animal and cell models are limited to systematically illuminate effects of HSV-1 propagation on AD. HSV-1 lifecycle goes through a relatively long incubation period. HSV-1 induces neurotropic cytokine expression with pro-inflammatory action and inhibits antiviral cytokine production in AD. IFNs have antiviral activity of HSV-1-infected AD patients. HSV-1 shows connection with IB in cognitive decline and AD. However, HSV-1 with ɛ4 gene variant of APOE was examined to associate with abnormal autophagy, higher concentrations of HSV-1 DNA in AD and formation of pathological features of AD, involving amyloid plaques and NFTs. The construction of new types of animal models combined with HSV-1 and APOE ɛ4 may prove beneficial in understanding the etiology of AD, although not yet developed.
Footnotes
ACKNOWLEDGMENTS
We sincerely thank the contributions and hard work of all the researchers and experts who provided data support. Initiation of the work by Professor Feng-xue Lao and Ying-hui Shang of Beijing Union University are acknowledged.
Financial support from the National Natural Science Foundation of China (31471587), Ten Frontier Research Directions of Discipline Orientation (ZK40201902) and Graduate Funding Project of Beijing Union University are gratefully acknowledged.