
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by complex pathological and biological features. Notably, extracellular amyloid-β deposits as senile plaques and intracellular aggregation of hyperphosphorylated tau as neurofibrillary tangles remain the primary premortem criterion for the diagnosis of AD. Currently, there exist no disease-modifying therapies for AD, and many clinical trials have failed to show its benefits for patients. Heme oxygenase 1 (HO-1) is a 32 kDa enzyme, which catalyzes the degradation of cellular heme to free ferrous iron, biliverdin, and carbon monoxide under stressful conditions. Several studies highlight the crucial pathological roles of HO-1 in the molecular processes of AD. The beneficial roles of HO-1 overexpression in AD brains are widely accepted due to its ability to convert pro-oxidant heme to biliverdin and bilirubin (antioxidants), which promote restoration of a suitable tissue redox microenvironment. However, the intracellular oxidative stress might be amplified by metabolites of HO-1 and exacerbate the progression of AD under certain circumstances. Several lines of evidence have demonstrated that upregulated HO-1 is linked to tauopathies, neuronal damage, and synapse aberrations in AD. Here, we review the aspects of the molecular mechanisms by which HO-1 regulates AD and the latest information on the pathobiology of AD. We further highlight the neuroprotective and neurodystrophic actions of HO-1 and the feasibility of HO-1 as a therapeutic target for AD.
INTRODUCTION
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder of dementia characterized by memory deficit and cognitive decline [1]. Most cases of AD occur past the age of 65 and its incidence rate over the age of 85 is nearly 50%. Previous research indicates that the AD patients inevitably succumb within 5–12 years post-diagnosis [2, 3]. Given the rising incidence of AD, there is an increasing need for corresponding prevention, early diagnosis, and treatment. Heme oxygenase 1 (HO-1), a member of heat shock protein family [4], was originally identified in the endoplasmic reticulum (ER) and later found to be expressed in other subcellular organelles. By binding to NADPH cytochrome P450 reductase, HO-1 functions by catalyzing the degradation of cellular heme to free ferrous iron, biliverdin, and carbon monoxide [5, 6]. Notably, HO-1 is sensitive to be induced by inflammatory stimuli such as heme, amyloid-β (Aβ), H2O2, or UV light [7, 8]. In the AD brains, HO-1 protein can be detected in the neurofibrillary tangles, senile plaques, astrocytes, neurons, some vascular smooth muscle, choroid plexus epithelial cells, endothelial cells, ependymocytes, and corpora amylacea [9–11]. Aging-related neurodegenerative disorders including, AD, Parkinson’s disease, multiple sclerosis, and dementia with Lewy bodies, among others, share several premortem features among them, induced oxidative stress protein, excessive deposition of iron, damaged mitochondrial membrane, and mitophagy in the affected tissues [12, 13]. In these aging-related neurodegenerative disorders, reports have confirmed upregulated HO-1 as a primary transducer of deleterious stimuli of these cytopathological signatures. However, only a handful of research has verified the beneficial roles of HO-1 [14–16].
In this article, we first outline key aspects of molecular mechanisms in HO-1 regulation and review the latest information on pathobiology and treatment approaches of AD. Thereafter, we refer to detailed literature exposing the protective role of HO-1 in AD. Further, we demonstrate the dark side of HO-1 in human degenerative CNS disorders, specifically in AD. Some studies also elucidate the involvement of HO-1 overexpression in primary culture neurons as a sufficient cause of reactive oxygen species (ROS), damaged dendritic morphology, and tauopathies. In the end, this paper reviews the feasibility of HO-1 as a therapeutic target for AD.
THE REGULATION MECHANISMS OF HO-1
HO was identified by Tenhunen et al. in 1968 and was described as a unique microsomal enzyme [17]. The first two different isoforms of HO, namely HO-1 and HO-2, were discovered in mammalian cells [18, 19]. Later, the third isoform was identified in rats, which was shown as a pseudogene [20]. In humans, HO-1 and HO-2 showed over 40% amino acid sequence homology which exhibits identical substrate specificities and cofactor [21]. Notably, HO-1 and HO-2 are different from tissue distribution, molecular weight, antigenicity, electrophoretic mobility, and regulation [22]. Due to the destabilization of proline-glutamic acid-serine-threonine sequence in carboxy terminus, HO-1 is sensitive to degradation [23]. The constitutive HO-2 protein is abundant in hippocampal pyramidal cells, cerebellar Purkinje, olfactory epithelium, olfactory bulb, dentate gyrus, and granule cell layers [24, 25]. A considerable overlap exists between the topography of HO-2 expression and the distribution of soluble guanylate cyclase in the brains of rats [26]. The HO-1 protein is a 32 kDa enzyme located within the ER and other subcellular organelles such as the nucleus [27]. Together with NADPH cytochrome P450 reductase, HO catalyzes the degradation of cellular heme to free ferrous iron, biliverdin, and carbon monoxide. Subsequently, the biliverdin reductase metabolizes biliverdin to bilirubin, one of bile pigments [28] (Fig. 1).
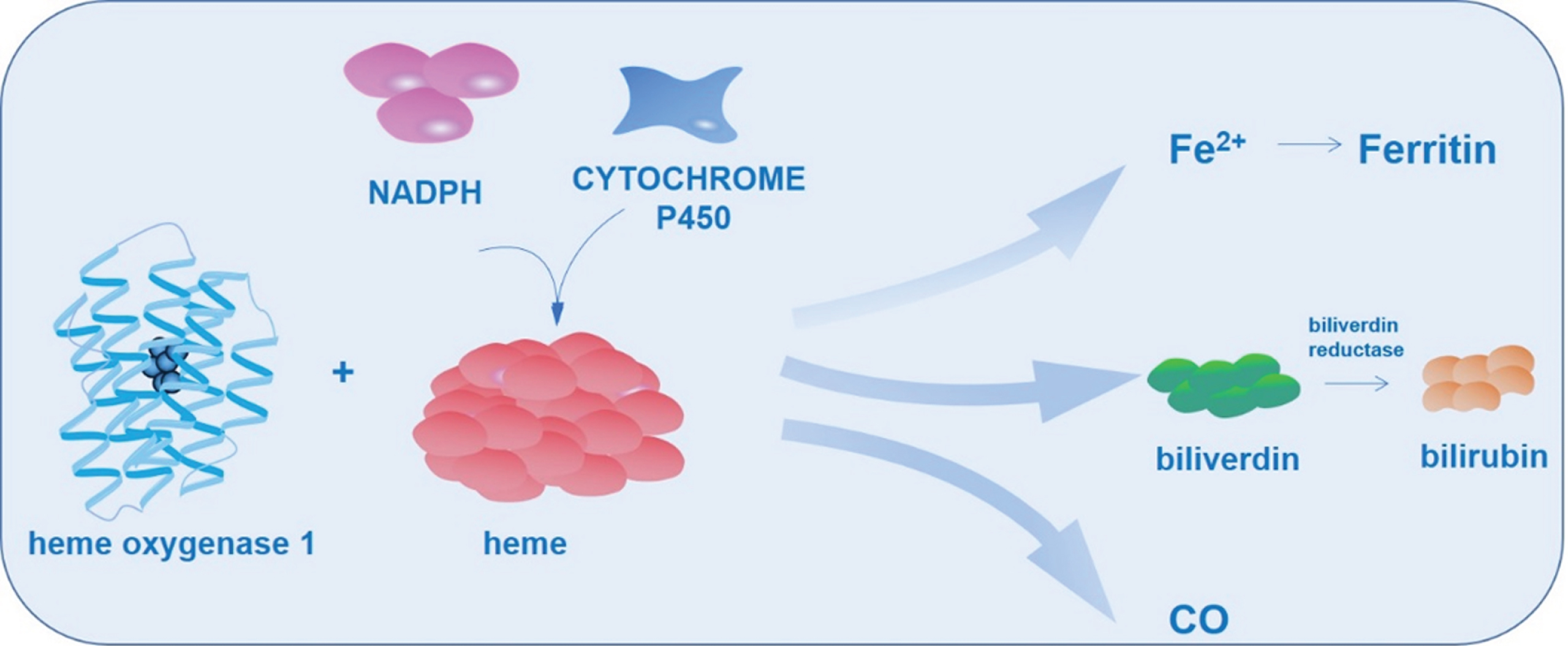
Heme oxygenase 1 catalyzes heme metabolism. HO-1 catalyzes the degradation of cellular heme in association with NADPH cytochrome P450 reductase to free ferrous iron, biliverdin and carbon monoxide. The biliverdin reductase further metabolizes biliverdin to bilirubin.
Human HO-1 is encoded by HMOX1 located in the chromosome 22q12 with 5 exons, 4 introns, a 500 bp promoter region, two or more distal enhancers, and one proximal enhancer (Fig. 2). The regulatory region contains hypoxia-inducible factor 1, nuclear factor kappa B (NF-κB), activator protein 1 (AP-1) binding sites, activator protein 2 (AP-2) binding sites, metal response elements (MtRE, CdRE), stress response elements, and heat shock consensus sequences [29, 30]. All these different binding sequences and diverse elements render HO-1 the target of many transduction pathways. They also make it sensitive to inflammatory stimuli such as heme, ROS, dopamine, H2O2, UV light, nitric oxide, TH1 cytokines, Aβ, heavy metals, radiations, modified lipids, peroxynitrite, growth factors, hyperoxia, inflammatory cytokines, peroxynitrite, and oxidized lipid products [31, 32]. The expression of HO-1 is regulated by many transcription factors. For instance, nuclear factor E2-related factor 2 (Nrf2) is a regulator of cellular redox homeostasis, which promotes the protection of cells against oxidative damage [33, 34]. By binding to the HO-1 promoter of Maf response elements, Nrf2 triggers the expression of HO-1 [35–38]. Of note, the activators of HO-1 expression are numerous while the negative regulators are unknown. Bach1 is a heme-regulated protein that dimerizes with sMafs to prevent ARE-dependent gene transcription. Keap1 is a negative regulator of Nrf2 and triggers the ubiquitination and proteasomal degradation of Nrf2. Bach1 and Keap1 are confirmed to induce the repression of HO-1 [39–41].
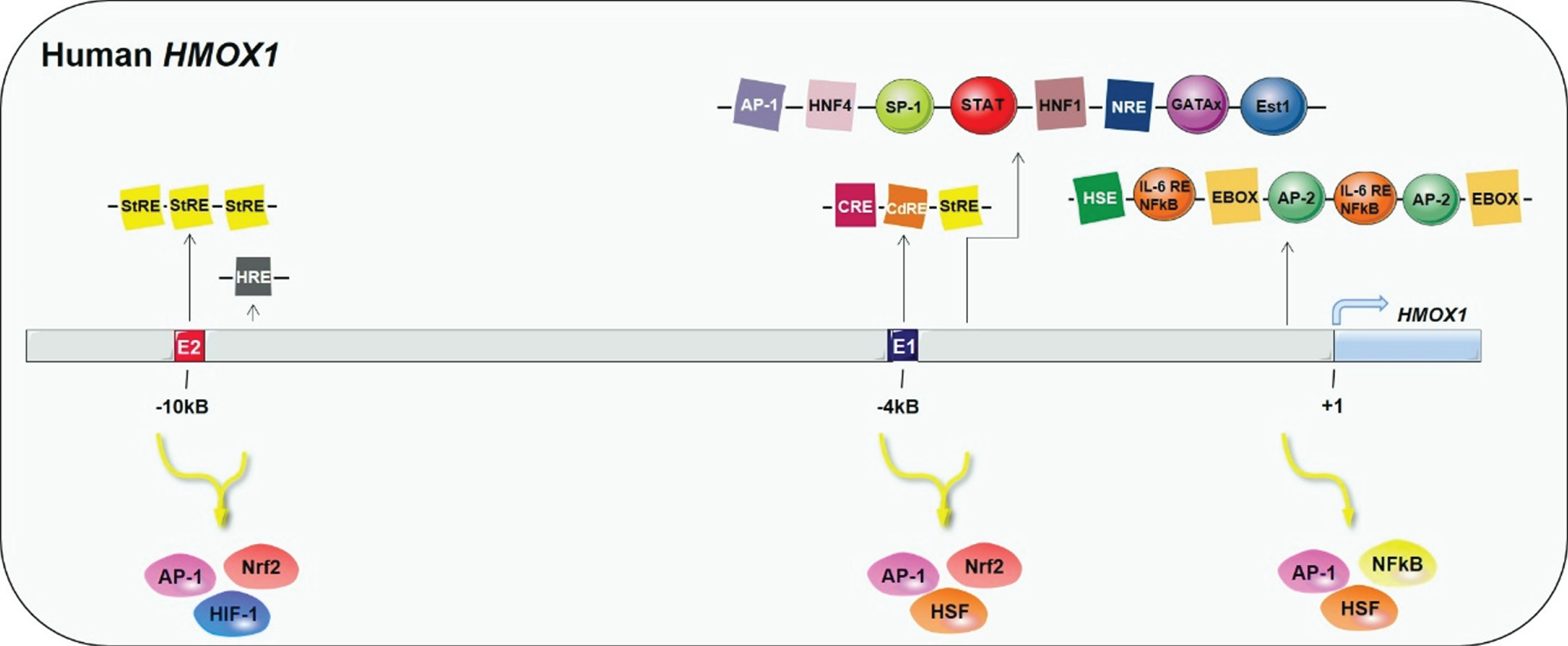
5’ flanking regulatory domains and three main enhancer clusters of human HMOX1 gene. AP-1, activator protein 1; AP-2, activator protein-2; CdRE, cadmium responsive element; CRE, cAMP-responsive element; E1 and E2 are binding regions for Nrf2; GATAx, GATA binding proteins; HNF1/4, hepatocyte nuclear factor 1/4; HRE, hypoxia-responsive element; HSE, heat shock element; NF-κB, nuclear factor κ-B; Nrf2, nuclear factor E2-related factor 2; NRE, negative regulatory element; SP-1, specificity protein-1; STAT, signal transducer and activator of transcription; StRE, stress responsive element. The blue arrow shows the transcription initiation site of HO-1.
HO-1 was originally identified in the ER through a single transmembrane segment of its carboxyl terminus [13]. However, accumulating evidence suggests that HO-1 is located on other subcellular organelles. In vitro studies on mitochondria show that the translocation of HO-1 to mitochondria regulate the metabolism of mitochondrial heme and protects the gastric mucosa from oxidative stress injury [42]. Besides its roles as an enzyme, recent studies suggest that HO-1 has a non-enzymatic function. Research on HO-1 localization shows that HO-1 translocates from the ER to the nucleus after proteolysis under stress conditions [43, 44]. Additionally, exposing cultured cells to heme or hypoxia promotes nuclear translocation of HO-1. The signal peptide peptidase, an ER-associated intramembrane protease, has been confirmed to catalyze the intramembrane cleavage of secretory signal peptides and membrane protein of ER, can also catalyze the proteolytic cleavage of HO-1 thereby promoting nuclear localization of HO-1 [43]. Several studies support the noncanonical functions of HO-1 in the nucleus. The nuclear HO-1 is susceptible to acetylation and deacetylation which occurs at its K243 and K256 residues. Therefore, the post-translational modification of nuclear HO-1 contributes to cell proliferation [45]. This non-enzymatic form of HO-1 elevates catalase and glutathione enzymes, implying that the noncanonical HO-1 might regulate the transduction of cellular signaling. For example, HO-1 has been proved to downregulate NF-κB activity by interacting with the p65 subunit of NF-κB, triggering cell apoptosis and cell cycle arrest [46].
In mammals, including humans, promoter polymorphisms in the lengths of the single nucleotide polymorphism and GT sequences in HMOX1 have been hypothesized to influence the magnitude in expression profiles of HO-1 [47, 48]. Furthermore, long GT repeats have been confirmed to reduce the transcriptional activity of HO-1 and susceptibility to oxidative injury. In contrast, short GT repeats are linked to the robust activity of HO-1 in different types of cardiovascular diseases [49]. Generally, most multiexonic genes are alternatively spliced in mammalian cells. HO-1 has been proved to express a 14 kDa novel alternative splice isoform protein other than the widely reported 32 kDa protein. The 14 kDa HO-1 is highly expressed in immortalized cells and generated by the exclusion of exon 3. The two isoforms of HO-1 are different from cellular sub-localization and expression induction. Both UV and H2O2 induce the two isoforms of HO-1 but more 14 kDa isoform is produced under UV irradiation. Moreover, upon UV irradiation the 32 kDa isoform of HO-1 migrate from the cytoplasm to the nucleus leaving the 14 kDa isoform of HO-1 in the cytoplasm. Previous studies on cell proliferation argue that 14 kDa isoform of HO-1 overexpression increased the number of cells in S and G2 phases compared to 32 kDa isoform of HO-1. The 14 kDa isoform of HO-1 increases the relative telomere length of the immortalized cells thereby promoting cell cycle [50].
The molecular mechanisms in dysregulation of HO-1 induction are still puzzling. Recent reports on post-transcriptional modifications of Bach1, such as ubiquitination shows that they regulate their binding capacity to the promoter of HO-1 [51]. In addition, microRNAs (miRs) have been proposed in regulating the expression of Nrf2, Bach1, and HO-1 in neuronal cells [52, 53]. For instance, miR-153 and miR-424 prefer neuron survival by up-regulating the Nrf2/HO-1 axis [54, 55]. Moreover, miR-494 induces the upregulation of HO-1 under oxidative stress in neuroblastoma cells [56]. Notably, heme metabolism occurs in all mammalian cells [22, 57]. In cell stress, the upregulated HO-1 catalyzes the conversion of pro-oxidant heme to biliverdin and bilirubin with significant free radical scavenging ability which provides protection [58–62]. In these cells, the joint refinement of iron storage proteins and ferritin limits free radical damage, which might occur due to the release of heme-derived ferrous iron. However, in some instances, iron and CO released by heme cleavage can exacerbate oxidative tissue damage by enhancing the formation of ROS in mitochondria or other subcellular [42, 64]. The intensity and duration of HO-1 induction and the chemical properties of the redox microenvironment might determine whether HO-1 behaves as a pro- or antioxidant exposed to any given conditions [65, 66].
THE PATHOGENESIS AND PATHOLOGY OF AD
AD is the most common cause of dementia. Presently, the presence of extracellular Aβ deposits as neuritic plaques and intracellular accumulation of hyperphosphorylated tau as neurofibrillary tangles remain the major premortem criterion for the diagnosis of AD [67, 68]. Patients inevitably succumb within 5–12 years since the onset of AD symptoms [69]. Most cases of late-onset AD occur beyond the age of 65. The early-onset AD begins before 65 years of age, which accounts for less than 5% of all the cases [70]. In recent years, significant progress has been made in clarifying key aspects of potential pathology which inform the biology of AD. So far, basic findings have exposed the important pathological roles of other key cellular and molecular processes [71–74]. However, the clinical manifestations of AD are progressing over time. Typical features include early learning and memory impairments, followed by complex attention, visuospatial function, executive function, praxis, language, gnosis, behavior, and/or social impairment [75–78].
Diagnosis of AD premortem utilizes cerebrospinal fluid (CSF) or positron emission tomography (PET) imaging biomarkers as surrogate markers for Aβ and tau deposition in brains [79, 80]. Recent studies have demonstrated the ability to detect CNS Aβ deposition using plasma assessments of Aβ species [81, 82]. Further studies on cognitive function and changes in CSF, as well as neuroimaging biomarkers in early-onset AD and late-onset AD, found that the disease is at an important preclinical stage at least 10 to 20 years prior to the onset of clinical symptoms [83, 84]. It is characterized by early deposition of Aβ in early-onset nerves and other cortical areas, including the default pattern network, followed by regional cortical hypometabolism, decreased hippocampal volume, accumulation of tau pathology, and the onset of symptomatic cognitive impairment. Plasma neurofilament light chain and CSF are the upcoming biomarkers that seemly track the general level of neurodegeneration in all forms of neurodegenerative dementia [85–88].
Aβ oligomers have been shown to “seed” in a prion-like manner. The intraperitoneal or intracerebral injection of misfolded Aβ extracted from AD brain or APP transgenic mice triggers cerebral amyloidosis, and evidence has revealed that amyloid pathology can spread from human-to-human [89, 90]. Moreover, a recent study showed that amyloid reduces the metabolic activity of distant neurons projecting to the area where amyloid is deposited. Although amyloid accumulation might be critical in the onset of AD pathogenesis, other downstream events, such as tau accumulation or neuroinflammation, might be the primary driving force for neurodegeneration [91]. In the past decade, several studies on tauopathy have shown that the aggregated human tau fibers exhibit a prion-like ability to self-propagate [92–97]. These aggregated tau fibers spread through the synapse to the remotely, anatomically connected brain regions, thereby triggering further seeding and gathering. Therefore, prion-like seeding and transmission might represent a potential mechanism of AD pathogenesis [98]. ApoE might affect amyloid pathology by directly binding to Aβ in the plaque, thereby largely regulating AD risk [99]. ApoE has been shown to have a regulatory effect on tau pathology and tau-related neurodegeneration. Also, ApoE can independently influence neurons and neuronal networks [100, 101]. Researchers agree that reactive astrocyte hyperplasia and microglia hyperplasia are prominent pathological features of the AD brain and the activation of the immune system regulates AD pathology [102, 103]. Ferroptosis is characterized by iron-dependent lipid peroxidation that ultimately causes oxidative stress and cell death. Recent studies also found that iron, a metabolite of HO-1, plays a key role in ferroptosis. In AD brains, the long-noted accumulation of iron dyshomeostasis and lipid peroxidation are two essential conditions of ferroptosis, which might trigger neuronal loss [104, 105].
While acknowledging significant progress in understanding the pathology of AD, the discovery of disease-relief therapies effective in humans is lacking. Despite biochemical analysis, genetic analysis in vivo and in vitro, as well as longitudinal imaging studies provide strong supports for the roles of many potential proteins in the pathogenesis of the disease; nonetheless, the clinical trials conducted have not been successful [2, 106–108]. Notably, the pathological biology of AD is complex. It has been reported that the older the age, the greater the possibility that other age-related diseases and AD pathology will thus cause cognitive decline. The ongoing in-depth investigation in this area is critical to birthing key discoveries that will eventually reveal novel treatment strategies for managing the disease.
THE EXPRESSION OF HO-1 IN AD
In the normal, unstressed mammalian brain, the expression of HO-1 is limited, and only a small population of neurons or scattered neuroglia in the dentate gyrus, cerebellum, hypothalamus, cerebral cortex, and thalamus are immunoreactive with HO-1 [62, 109–112]. The number of HO-1-immunoreactive cells increases with the increase in age; however, under basal conditions, the number of these cells remain small. In the AD brain, the expression of HO-1 detected by western blots show approximately 4-fold more intense compared to the age-matched normal specimens and post-mortem intervals [113]. The HO-1 protein is co-localized with neurofibrillary tangles, senile plaques, some vascular smooth muscle, choroid plexus epithelial cells, endothelial cells, ependymocytes, corpora amylacea, neurons, and astrocytes [9, 113]. In the AD hippocampus, 86% of GFAP+ astrocytes were immunolabeled with HO-1, whereas in the normal, age-matched control subjects, only 6–7% astroglia in hippocampi expressing HO-1 were immunolabeled with HO-1. Studies on mild cognitive impairment (MCI), a frequent harbinger of incipient AD, showed that the overexpression of HO-1 in astrocyte was observed in MCI and associated with the neurofibrillary pathological burden in patients [11, 113]. The expression of astrocyte HO-1 in the temporal cortex of MCI was confirmed to trigger a reduction in working memory, semantic memory, and global cognition, while the immunoreactivity of hippocampal astrocytes is associated with semantic memory, global cognition, and perceptual speed [114]. These studies indicate that induction of glial HO-1 is a relatively early event in the pathogenesis of sporadic AD. Oxidative stress engendered by the amyloid burden, pro-inflammatory cytokines, and infidelity of electron transport in senescent mitochondria might promote the upregulation of HO-1 in AD and MCI brains [115, 116]. However, studies conducted in Schipper laboratory have shown that the protein or mRNA levels of HO-1 were suppressed compared to control values in plasma, CSF, blood mononuclear cells, and choroid plexus epithelium of AD patients [117]. The decrease in the levels of CSF HO-1 in AD might reflect a decrease in the synthesis and secretion of the choroid plexus, or impaired HO-1 exudation from the peripheral circulation [48, 118]. In comparison with the age-matched normal subjects, the HO-1 promoter polymorphisms in AD patients showed no aberrant, indicating that the low levels of HO-1 protein or mRNA in the blood, CSF, and choroid plexus were not caused by genetic variants of the HMOX1 [48, 118]. Cerebral amyloid angiopathy (CAA) is a vascular lesion present in up to 95% of AD patients and produces MRI-detectable microbleeds in many of these patients which in turn triggers expression of HO-1 [119]. Studies have shown that HO-1 is only increased in AD patients with CAA, and not without CAA, indicating that the heme degradation pathways are potentially induced by vascular injury and/or microhemorrhagic changes [120].
THE PROTECTIVE EFFECTS OF HO-1 IN AD
Through inflammation modulation, mitigation of oxidative stress, and regulation of apoptosis, the reaction products of HO-1 in CNS neurodegeneration might confer protective effects [121, 122]. In brains of AD patients, prolonged exposure of neurons and non-neuronal brain cells to stressed stimuli and HO-1 shows a strong response in astrocytes, microglia, neurons, and oligodendroglia, which might offer cytoprotection to cells [123]. Notably, the formation and aggregation of Aβ is one of the pathological manifestations of AD. Ferulic acid potentially increases the production of bilirubin and CO which counteract ROS induced by Aβ protein [124]. Also, through disrupting the activation of AMP-activated protein kinase, Aβ-induced toxicity could be inhibited by CO [125]. The proteasome activity reduction is linked to protein aggregates, which has been confirmed to promote the pathogenesis of various neurodegenerative diseases. Reports from α-synuclein and tau studies show that HO-1 promotes the degradation of α-synuclein and tau by the proteasome pathway and reduce the aggregation of toxic protein in Parkinson’s disease or AD, which exert cytoprotection to CNS [9]. Furthermore, the neuroprotective function of HO-1 might be attributed to the enhanced generation of neurotrophic factors other than its anti-inflammatory and antioxidant activity. For example, overexpression of HO-1 has been shown to up-regulate the production of glial cell-derived neurotrophic and brain-derived neurotrophic factors in neurons and astrocytes [126–129]. Also, the neuroprotective function of HO-1 has been witnessed against oxidative damage in AD animal models. Microinjection of cocaine- and amphetamine-regulated transcript peptide into the hippocampus of rat activates the Nrf2/HO-1 signaling pathway thereby attenuating the oxidative stress damage [130]. The overexpression of HO-1 in the brains of AD patients represents a cell adaptation to stress. Therefore, the cytoprotective roles of HO-1 are primarily due to its metabolic products, which contribute to anti-inflammatory, antioxidant and anti-apoptotic properties [10]. The major pathways involved in HO-1 neuroprotection effects on AD are shown in Fig. 3.
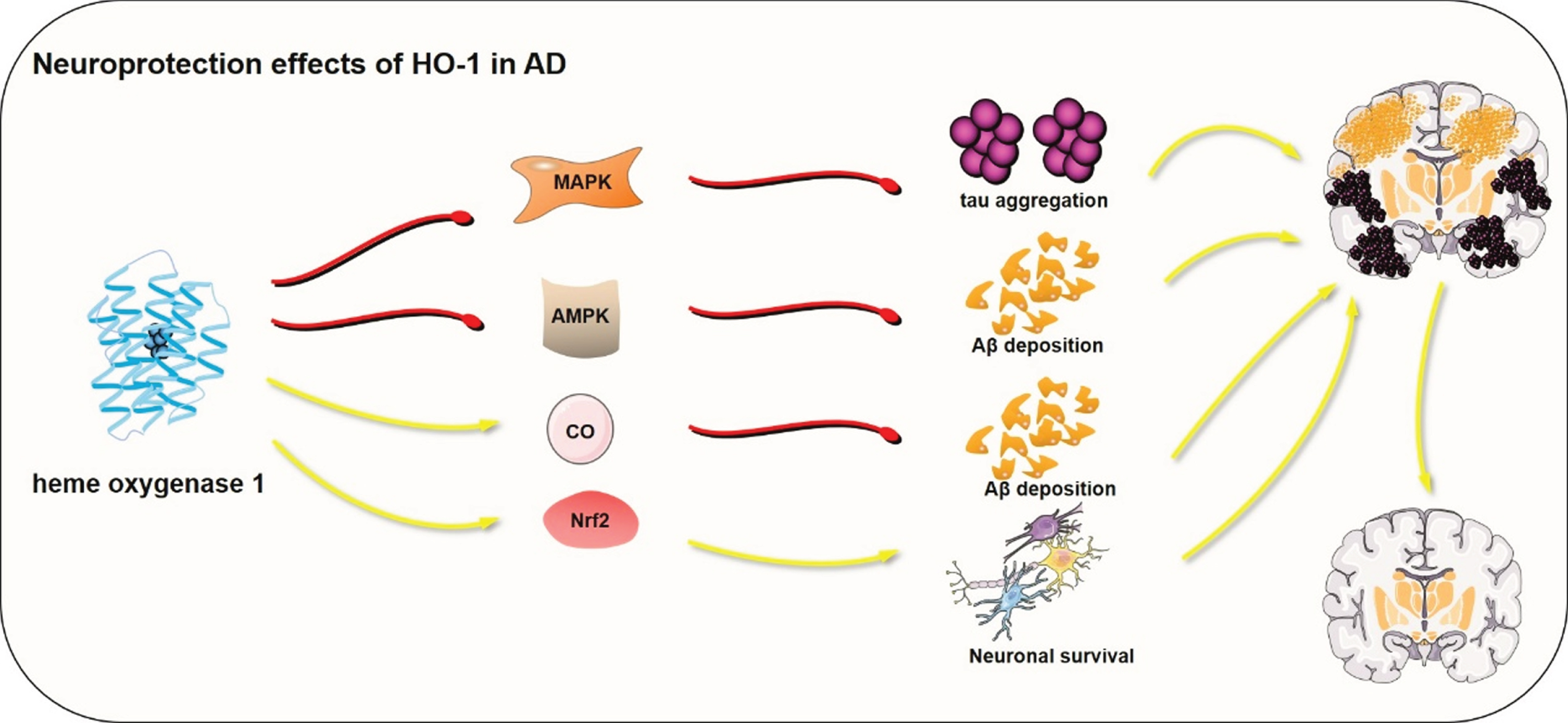
The main pathways involved in HO-1 neuroprotection effects on AD. MAPK, mitogen-activated protein kinase; AMPK, AMP-activated kinase; CO, Carbon monoxide; Nrf2, nuclear factor E2-related factor 2. The red lines represent suppression and the yellow arrows represent promotion. The red or yellow lines represent signal pathways involved in HO-1 regulation in neuroprotection.
THE SIDE EFFECTS OF HO-1 IN AD
In the previous section, this paper focused on the neuroprotective action of HO-1 in AD. Notably, studies on the neuronal damage and degeneration effect of HO-1 have matured. Known as “oxidative stress responder”, HO-1 is highly susceptible to oxidative stress, inflammation, and other factors indicating that high levels of oxidative stress such as AD, engenders prolonged upregulation in the expression of HO-1 [131]. In this state, the function and mechanism of HO-1 might differ from the conventional concept of protection which is dangerous for the brain tissues in AD [10].
The overexpression of HO-1 in AD patient brains causes mitochondrial insufficiency and pathological iron sequestration. Therefore, treatment with heme oxygenase activity, competitive inhibitor tin mesoporphyrin (SnMP), or HO-1 transcriptional suppressor dexamethasone in stressed astroglia, significantly reduces the deposition of mitochondrial iron. In addition, treatment through transient transfection of SnMP or DEX on overexpressed HO-1 in astroglia decreased the pathological deposition of mitochondrial 55Fe [132, 133]. After HO-1 transfection in astroglia, the concentrations of 8-OHdG (nucleic acid oxidation), protein carbonyls (protein oxidation), a synthetic redox reporter molecule, and 8-epiPGF2a (lipid peroxidation) were significantly elevated, implying that overexpression of HO-1 exacerbates intracellular oxidative stress in astroglia [134, 135]. α1-antitrypsin can be detected in AD choroid plexus epithelial cells, neurons, and astrocytes and exhibits close spatial proximity with HO-1 [10]. As a result, α1-antitrypsin might promote negative regulation of HO-1 in the AD brain to attenuate iron-dependent damage [136, 137]. Emerging evidence stipulates that the production of oxysterol and dysregulation of sterol might promote the degenerative processes in AD. Furthermore, overexpression of HO-1 in astrocytes significantly decreases the concentration of intracellular cholesterol and increases the concentration of several oxysterol species [13, 138]. Gupta et al. reported that selectively inhibiting the activity of HO-1 by OB-28 intraperitoneal injections in adult APPswe/PS1ΔE9 TG mice (a model of familial AD), reduced neuroinflammation compared to saline-injected controls [139]. Moreover, the OB-28 treatment on AD mice performed significantly better on complex maze learning tasks compared to saline-injected controls [140]. The cytotoxic outcome of HO-1 overexpression might be associated with signaling pathways. AP-1 or NF-κB related HO-1 activation exerts neurotoxic effects, while activation of HO-1 by Nrf2 is mainly linked to the protective effects on neurons and glial cells [141].
In vivo studies demonstrated that overexpression of HO-1 decreased the expression of memory-related synaptic proteins and promoted the iron loading, triggering enhanced phosphorylation of tau (Ser199/202/396) in HO-1 transgenic mice [142]. Since upregulated HO-1 co-localizes to AD pathological features including tauopathies, we confirm that HO-1 influenced two pathways for tauopathy. Of note, HO-1 induced the expression of CDK5 by accumulating ROS produced by iron, the downstream products of HO-1. On the other hand, HO-1 promoted the cleavage of tau protein at D421 in HO-1 transgenic mice [143]. Interestingly, overexpression of HO-1 induced the aggregation of Aβ oligomers and tau oligomers [144, 145]. Additionally, the elevated HO-1 damaged the morphology of the synapse and impaired the neural circuit at the hippocampal region of neonatal HO-1 transgenic mice. Expression of HO-1 impaired the dendritic morphology of dendritic intersections, dendritic diameter, dendritic length, spine density, and spine length. Nonetheless, treatment by isoprenaline, an inhibitor of tau oligomers formation, partially restored the damaged dendritic morphology indicating a key role of tau oligomers in dendritic morphology [144, 145]. The main pathways involved in HO-1 neurodegeneration effects on AD are represented in Fig. 4.
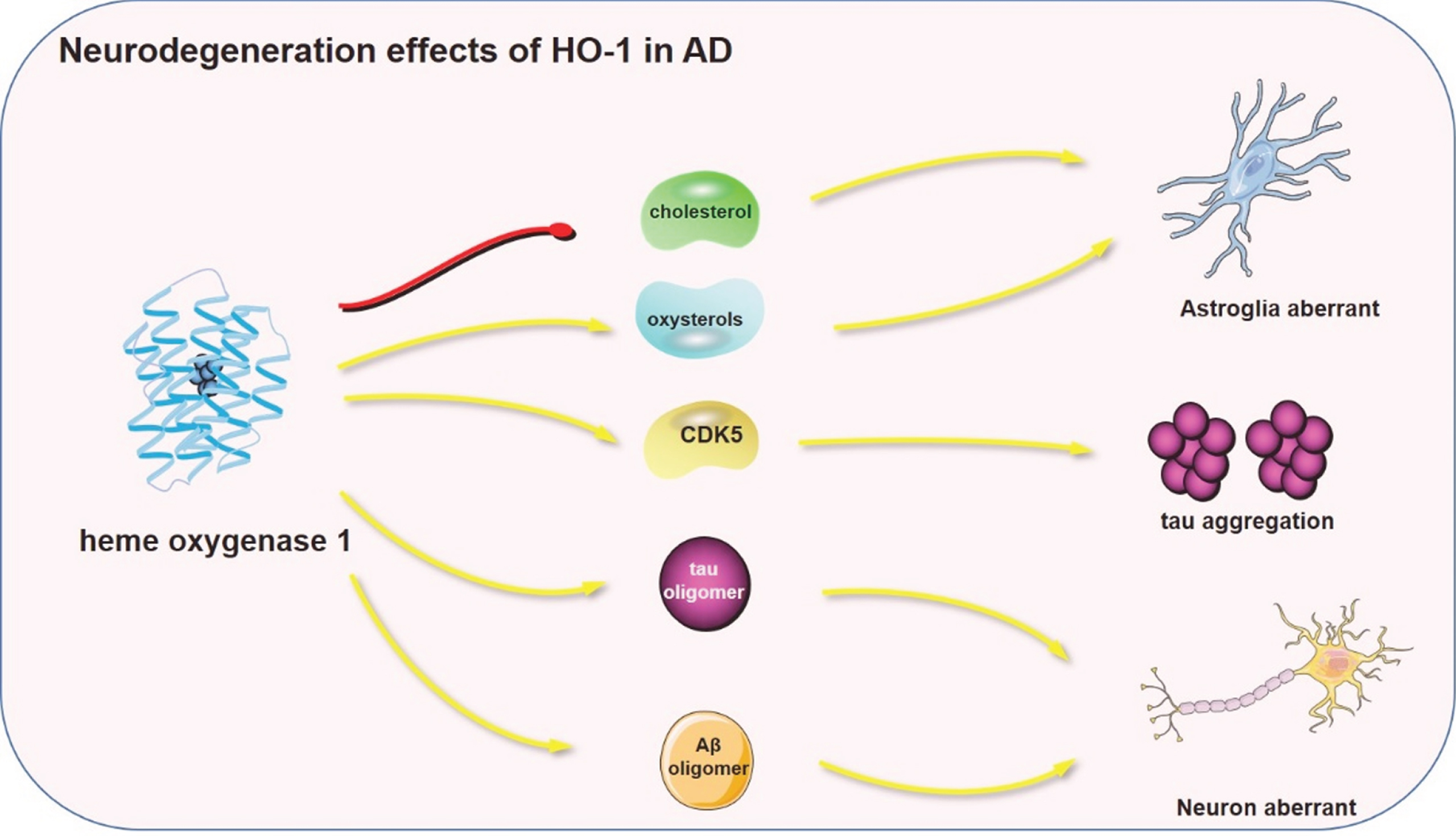
The main pathways involved in HO-1 neurodegeneration effects on AD. CDK5, Cyclin—dependent Kinase 5. The red lines represent suppression and the yellow arrows represent promotion. The red or yellow lines represent signal pathways involved in HO-1 regulation in neurodegeneration.
HO-1 THERAPEUTIC CONSIDERATIONS FOR AD
Studies have confirmed that a significant proportion of redox-active iron pathologically deposited in the aging and degenerating CNS is HO-1-dependant. This means that the efforts on targeting transferrin receptor, IRP1/2, ferritin, etc., the classical iron regulatory mechanisms, might be futile in diminishing iron-associated neurotoxicity [146–148]. It has been suggested that HO-1 is a potential adjunct to iron chelation therapy. The metalloporphyrins can competitively block the activity of HO-1 and HO-2 and the permeability of these agents to the blood-brain barrier is poor. This means they are unsuitable as neurotherapeutic agents. Nevertheless, this issue could be circumvented by OB-24 and OB-28, two novel small-molecule HO inhibitors. Both OB-24 and OB-28 are non-peptide imidazolyl molecules with efficient blood-brain barrier permeability [10]. The chemical structure of OB-28 is (2R, 4R)-2-[2-(4- chlorophenyl) ethyl]-2-[(1H-imidazol-1-yl) methyl]-4-methyl-1, 3-dioxolane hydrochloride). It has a higher selectivity for HO-1 compared to HO-2 [32]. Furthermore, in the spleen of rats, the IC50 of OB-28 for HO-1 inhibition is 0.25 μM, whereas the IC50 value for HO-2 inhibition in the brain of rats is 6.70 μM, confirming that OB-28 is a selectively inhibits the activity of [32, 140]. When the familial AD mice model (the APPswe/PS1ΔE9 TG mice) were intraperitoneally injected with OB-28 every day for 6 months, the outcomes showed an inhibition of HO-1 activity in brain regions. After OB-28 treatment, the cognitive decline in the AD mice model was ameliorated without any adverse effects [139]. Furthermore, OB-28 treatment in HO-1-transfected astrocytes in vitro reduced oxidative damage of mitochondrial components [149]. OB-24 was identified by Moulay et al., with its chemical structure being 2-[2-(4-bromophenyl) ethyl]-2-[(1H- imidazol-1-yl) methyl]-1, 3-dioxolane hydrochloride. OB-24 is a selective small-molecule inhibitor of HO-1 but not HO-2 [140]. In a cancer mice model, the OB-24 administration showed no toxicity [140]. The pharmacotherapies for AD discussed are only symptomatic or supportive, therefore the discovery of definitive disease-modifying therapies will resolve the unmet medical, economic imperative, and psychosocial hurdles [150–152]. Although the aberrant expression of HO-1 might not be the ‘cause’ of AD, its overexpression and cytophysiological consequences might serve as a key mechanism in CNS senescence and neurodegeneration in AD patients. For AD treatment, effective neuroprotection is popularly applied than exclusive symptomatic treatment. Moreover, HO-1 inhibitors as potential neuroprotective agents in AD might be more effective than symptomatic treatment. Based on convincing preclinical data, human trials are essential to assess whether the strategic inhibition in the response of HO-1 to significant stimuli can prevent or ameliorate the pathogenesis of AD.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0720r1).