
Abstract
The neurovascular unit (NVU) is responsible for synchronizing the energetic demand, vasodynamic changes, and neurochemical and electrical function of the brain through a closed and interdependent interaction of cell components conforming to brain tissue. In this review, we will focus on cyclin-dependent kinase 5 (CDK5) as a molecular pivot, which plays a crucial role in the healthy function of neurons, astrocytes, and the endothelium and is implicated in the cross-talk of cellular adhesion signaling, ion transmission, and cytoskeletal remodeling, thus allowing the individual and interconnected homeostasis of cerebral parenchyma. Then, we discuss how CDK5 overactivation affects the integrity of the NVU in Alzheimer’s disease (AD) and cognitive impairment; we emphasize how CDK5 is involved in the excitotoxicity spreading of glutamate and Ca2+ imbalance under acute and chronic injury. Additionally, we present pharmacological and gene therapy strategies for producing partial depletion of CDK5 activity on neurons, astrocytes, or endothelium to recover neuroplasticity and neurotransmission, suggesting that the NVU should be the targeted tissue unit in protective strategies. Finally, we conclude that CDK5 could be effective due to its intervention on astrocytes by its end feet on the endothelium and neurons, acting as an intermediary cell between systemic and central communication in the brain. This review provides integrated guidance regarding the pathogenesis of and potential repair strategies for AD.
THE NEUROVASCULAR UNIT: AN INTEGRATED PERSPECTIVE ON BRAIN FUNCTION
The neurovascular unit (NVU) behaves as a guardian of cerebral homeostasis, and the components that make up the NVU coordinate homeostasis in the cerebral microenvironment, regulate blood flow, regulate interchange across the blood-brain barrier (BBB), contribute to immune vigilance, and provide trophic support to the brain. Studies consider the brain as an integrated organ, in which a network of various cell types, including astrocytes, oligodendrocytes, microglia, pericytes, and microvasculature endothelial cells [1–3], work interdependently to conform to the NVU responsible for brain function [4, 5], transcending the neuronal concept as the main contributor to brain function and its pathologies.
Therefore, most of the progress made over the past decade regarding the preservation of neurons as the nearly “unique” valuable cell target to be rescued has shown unsuccessful translational potential. A possible therapy directed only to neurons is apparently ineffective for regulating the morpho-functional homeostasis of the brain, which is necessary to address the NVU as a therapeutic tissue target for the prevention and recovery of neurological dysfunction because 1) neurotransmission and neurotransmitter uptake in the synapses are mediated by signal transduction between neurons and astrocytes, 2) BBB integrity depends on the signals between astrocytes and endothelial cells, and 3) neuronal activity requires coupling of the vascular response to provide the energy needed [6, 7].
Astrocytes form structures known as astrocyte end-feet, which communicate with neurons, pericytes, and endothelial cells. This cell population has acquired great importance because it has been demonstrated to modulate synaptic transmission and neuronal plasticity processes [7–9]. Also, astrocytes in AD patients have been reported to present a state of hyperactivity called astrogliosis, characterized by mitochondrial dysfunction, impairment of neurotransmitter recycling, and release of pro-inflammatory factors, disrupting neurovascular unity and neuronal function [4, 11]. These cells and blood vessels play a fundamental role in NVU, to such an extent that alterations in the communication of those elements influence the ability to provide the necessary flow for the regions that are active, triggering brain dysfunction, as in Alzheimer’s disease (AD) [12, 13].
Thus, an in-depth understanding of the pathophysiological mechanisms that govern NVU dysfunction and the role of astrocytes and the factors released by these cells, as actors in tauopathy and β-amyloidosis in dementia, to subsequently propose therapeutic strategies is needed. Therefore, in this review, we focus on cyclin-dependent kinase 5 (CDK5) targeting, a protein involved in key central nervous system (CNS) processes, but its deregulation is related to tau hyperphosphorylation and neurodegeneration [14, 15]. The role of CDK5 has been extensively studied in neurons, but this is not exclusive to these cells; some reports have shown their participation in vascular endothelium and astrocytes [16–18], and based in our previous studies of CDK5 targeting in astrocytes is being of greater interest for proposing NVU therapy for AD.
THE NVU IS CRUCIAL FOR AN UNDERSTANDING OF NEURODEGENERATIVE PROCESSES
In neurodegenerative processes, the function and structure of cells associated with the NVU are affected, thus reducing the capacity of brain parenchyma repair due to the loss of regulation of cerebral blood flow, loss of neuronal connections, excitotoxicity, and apoptosis. Most studies on dementia are geared toward understanding the molecular and physiopathological mechanisms of neuronal death, recovery, and pharmacological intervention strategies, as well as clinical and epidemiological behavior, without considering the other NVU components (astrocytes, endothelium, pericytes, extracellular matrix) as central actors [19, 20].
Vascular causes of dementia have gained attention in basic and clinical studies because a large proportion of dementias are caused by vascular pathology [21, 22]. Notably, the coexistence of cerebral ischemia and neurodegenerative pathologies has been shown to have a profound impact on the development of dementia [23–25]. These observations have redirected the interest in neurovascular factors as fundamental elements in the pathogenesis of neurodegenerative diseases [26–28].
AD and cerebrovascular disease are the most common causes of cognitive deficit and dementia. Although the pathogenesis of these two conditions is traditionally separate [21, 30], following brain damage, a wide spectrum of physiopathological mechanisms arise at the NVU. Common mechanisms are divided into excitotoxic, free radical, and pro-apoptotic pathways, in which a Ca2+ imbalance could bind or cover the majority of these multifactorial signals. It has been assumed that the accumulation of intracellular Ca2+ and generalized ionic imbalance are the main mediators of cell death in a neurodegenerative process induced by ischemia or AD [31–35]. At the cellular level, the generation of reactive oxygen species (ROS), nitric oxide, peroxynitrites, mitochondrial dysfunction, and endoplasmic reticulum (ER) stress lead to ionic dysregulation and metabolic alteration. A subsequent activation of the MAPK pathway, such as ERK, p38, and JNK, is carried out as a dual mechanism (pro-survival or pro-death) depending on the context, the intensity of the toxic stimulus, or the state of cell differentiation—in other words, in the development, maturation states, and adult function. The intracellular activation of proteases such as calpains, cathepsins, and caspases can specifically degrade cellular components in an advanced state of tissue alteration [31, 35–37]. In the case of vascular dementia, the interruption of blood flow to the brain during ischemia results in oxygen and glucose deprivation, which reduces the energy available for the function of brain cells [38–40]. Neurons particularly become incapable of maintaining the necessary ionic gradients for cell function and homeostasis [41, 42]. This implies excessive neuronal depolarization, an increased release of excitatory neurotransmitters, and a reduced recapture of these neurotransmitters from the extracellular space. These pathological events induce an excessive accumulation of sodium ions and Ca2+ simultaneously with the dysregulation of multiple signaling pathways, thereby activating the catabolic processes mediated by proteases, lipases, and nucleases, which affect each of the cellular components and the integrity of the NVU [38, 43]. The increased concentration of glutamate in the CNS is present in various pathologies, including ischemia/reperfusion and AD, after acute and chronic imbalance, respectively, resulting in the hyperactivation of glutamate receptors, excess flow of the aforementioned ions, and subsequent cell death [44–46]. Such processes are grouped in the event known as “glutamate-mediated excitotoxicity” [47, 48]. The various NVU components are susceptible to this toxicity process; in the case of neurons, it is known as excitotoxicity; in the case of glial cells, it is known as gliotoxicity [48–53]. For endothelial cells, there is evidence demonstrating that high levels of glutamate are specifically toxic in the brain microvasculature [54–57]. Particularly in neurons, excitotoxicity by glutamate results in reduced ATP synthesis, increased ROS, calpains, ER stress, MAPK activation, increased nitric oxide synthase, and subsequent deterioration of the actin cytoskeleton and microtubules, dendrite loss, and cell death [34, 59]. In vivo and in vitro studies have demonstrated that glutamate-induced gliotoxicity facilitates intracellular Ca2+ entry and the exit of ER reservoirs and mitochondria, thus increasing LDH release, generating ROS, and reducing glutathione and ATP, which are necessary for coupling with neuronal activity [11, 61]. In vivo studies have also demonstrated that endothelial cells of the brain microvasculature (adult or neonatal) exposed to glutamate can permeabilize the BBB, which induces the activity of the tissue plasminogen activator [54]. In addition, in an in vitro proteomics study conducted in cultures of human brain endothelial cells treated with glutamate, there was a different actin cytoskeleton protein expression pattern with respect to control cells and those treated with MK801, an NMDA (N-methyl-D-aspartate) inhibitor [57]. In another study, however, a Rho GTPase inhibitor (Rho GDP-dissociation inhibitor) decreased following toxicity by glutamate and was not reverted by MK801, which suggests that the remodeling of the glutamate-induced actin cytoskeleton in the endothelium was not dependent on NMDA receptors [55]. However, in studies conducted in bEnd.3 cells, it has been demonstrated that this cell type expresses NMDA receptors, such as GluN1 and GluN2B-D, which apparently play a fundamental role in BBB integrity [62]. Nonetheless, exposure to high concentrations of glutamate increases ROS (peroxynitrites) and reduces cell viability, which is reversed by MK801 [57] (Fig. 1).

A) Glutamate toxicity in the NVU: Glutamate toxicity affects each of the components of the NVU, such as excitotoxity in neurons, gliotoxicity in astrocytes and toxicity in the endothelium (purple lines). Intracellular Ca+2 imbalance is a common process in neurodegenerative disorders, such as ischemia and AD. Extracellular Ca+2 sources from mitochondria or ER lead to the final increase in ROS, reactive nitrogen species, proteases, and pro-apoptotic pathways [31, 115]. B) Participation of CDK5 and Ca+2 in glutamate excitotoxicity in the postsynapses. CDK5 under pathological conditions is apparently involved in the generation of ROS, mobilization of Ca+2 from the ER to mitochondria and dysregulation of cytosolic Ca+2 during neurodegeneration. Astrocytes uptake glutamate excess from neurons, inducing astrocyte hyperreactivity and stress on endothelium, which loss adherent proteins triggering a vicious circle of NVU impairment [52–55, 236].
From a different perspective, AD, as a chronic neurodegenerative disorder associated with a progressive loss of learning and memory, has principally been considered a neuronal pathology. In AD, overproduction and deposits of amyloid-β (Aβ) are initiation factors of various neurotoxic pathways, including excitotoxicity, oxidative stress, energy imbalance, inflammation, and apoptosis [19, 64]. The decreased levels of primary substrates of energy, such as ATP and NAD+ in neurons, are involved in cognitive dysfunction related to age and AD. These energy substrates are particularly critical for neuron function and survival, as they need to consume large quantities of energy to maintain the homeostasis of the ionic gradients after synaptic activation and the generation of action potentials. When energy levels are reduced during aging and neurodegeneration, the levels of intracellular Ca2+ remain elevated as a result of the influx sustained throughout the glutamate pathways or voltage-dependent channels [65–67]. On the other hand, the degeneration caused by Aβ in vivo has been linked to excessive activation and altered distribution of NMDA receptor subtypes in AD [68]. During the process of excitotoxicity, neuronal morphology is characterized by spine loss or the presence of aberrant spines. Therefore, the dendritic spine, as a basic functional unit of the neuron integrated circuit and a site of functional and structural synaptic plasticity, suffers a drastic deterioration following excitotoxicity by glutamate [69–71]. Dendrite degeneration directly involves cytoskeletal disassembly, which is observed as swelling or beading in arborization and lengthening or loss of spines [72, 73]. At a fundamental level, complex AD pathology is based on aberrant Ca2+ signaling, which leads to cell death [74–78]. Different strategies have been proposed to block Ca2+ dysregulation at the neuronal level; however, the poor efficacy of the treatments [79] suggests that it is necessary to explore the development of new therapies oriented toward intervening with other NVU components and analyzing their effectiveness. Over the past decade, a relationship has been demonstrated between the disruption of NVU and AD because the Aβ peptide directly breaks the tight junctions of the endothelium and alters BBB integrity [80, 81]. Additionally, a new concept has been established regarding the gliovascular unit, made up of astrocytes and endothelium, which is susceptible to alteration by the addition of Aβ and increased Ca2+ influx [82]. Likewise, it has been demonstrated in murine models of AD that Ca2+ increases abnormally in astrocytes [83], which suggests the abnormal gliovascular coupling of the vasoactive substances supplied by astrocytes to the endothelium [84]. These observations support the hypothesis of decoupling of the NVU in AD, triggered by a Ca2+ imbalance, which may be associated with toxicity processes that are mediated by glutamate or NMDA receptors, thereby generating cognitive impairment.
CDK5 NORMAL AND PATHOLOGICAL FUNCTION
Dementias such as AD, general tauopathies, and neurodegeneration after stroke, are characterized by hyperphosphorylation of tau protein [85–87]. Tau protein is dissociated from microtubules when it is the target of an abnormal action by kinase through hyperphosphorylation. Hyperphosphorylated tau accumulates in the cytosol in packages known as paired helical filaments. These aggregates are characteristic of AD and form neurofibrillary tangles [87, 88]. A main kinase involved in the rate of phosphorylation of tau and neurofibrillary tangle formation is CDK5.
CDK5 is a serine-threonine kinase that participates in neuronal function and development. This kinase is involved in the regulation of different processes, such as neuritogenesis, synapse formation, and synaptic transmission. CDK5 also plays an important role in neural connection control and is involved in cognitive functions such as memory and learning. CDK5 also has broad functions in neuronal and nonneuronal cells. Activators CDK5, p35, and p39 are highly expressed in the CNS, particularly in postmitotic neurons, astrocytes, and oligodendrocytes, in which another CDK is minimally active or expressed. During embryogenesis, CDK5 plays a central role in brain development, neuronal migration, neurite growth, axon guidance, and synapse formation. Additionally, CDK5 functions in cells other than neurons, including functioning in the lymphatic system and in the vascular system [15, 89–92].
Dysregulation of CDK5 activity is also associated with neuronal death in neurodegenerative diseases [89, 93]. CDK5 phosphorylation at Ser159 and Tyr15 increases its activity [90, 94]. Cleavage of p35 by calpains results in the formation of the CDK5/p25 complex, which gives way to considerable and sustained CDK5 activity in an abnormal manner [95]. In particular, it has been demonstrated that CDK5 and its respective activators phosphorylate tau epitopes that are related to AD. An excessive increase in the activity of CDK5 induced by p25 binding contributes to neurodegeneration processes [18, 96–98].
Little is known about the role of CDK5 in astrocytes; however, two activity contexts have been suggested. First, CDK5 and its specific activator in astrocytes, p35, have been associated with the regulation of the microtubule cytoskeleton and the formation of the glial scar following a “scrape wound” [18]. Second, the activity of CDK5 is increased in astrocytes in a mouse model of accelerated senescence (SAMP8); this increase leads to tau phosphorylation in S396, which is reversed by CDK5 inhibition with roscovitine. Furthermore, a close relationship between CDK5 activity and the reduced potential of the mitochondrial membrane in SAMP8 astrocytes suppresses the recapture of glutamate, which is critical for neuroprotective mechanisms [98]. This suggests that CDK5 is an adequate target for astrocyte protection and neuronal survival. Additionally, CDK5 seems to regulate the coupling of neurons and astrocytes during neurotransmission. NMDA activation in astrocytes triggers the release of Ca2+ from the ER and favors the phosphorylation of Nrf2 by the CDK5/p35 complex. The phosphorylation of CDK5-Nrf2 induces antioxidant gene expression and neuroprotection against H2O2 [99]. Nfr2 and trophic factors (brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), vascular endothelial growth factor (VEGF), nerve growth factor (NGF)) seem to combine to induce neuroprotection [100, 101]; however, the specific mechanism of the protective effects of CDK5, Nrf2, and BDNF remain unclear.
Not much is known about the role of CDK5 in endothelial cells, but it is suggested that, under normal conditions, CDK5 participates in 1) the regulation of angiogenesis and the migration of these cells and 2) the formation of lamellipodia in actin cytoskeleton dynamics [92, 102]. In 2015, Liebl et al. generated conditional lymphatic endothelial cell-specific knockout (KO) mice to determine the role of CDK5 in the endothelium. The authors reported that CDK5 plays an essential role in the regulation of the development of lymphatic vessels, although CDK5 suppression seemingly did not have a great impact on blood vascular endothelial cells, supported by cell adhesion markers, such as EphB4, endomucin, and CD31 (PECAM-1) [92, 103]. In contrast, in a pathological context associated with cell proliferation and angiogenesis in cancer, CDK5 inhibition has been suggested as a target for anti-angiogenic therapy [16, 17]. These findings warrant further research that explores the potential role of CDK5 silencing in the brain microvascular endothelium. In in vivo studies and in humans (postmortem), it has been suggested that CDK5 activity and its activating proteins in the peri-infarct zone increase following brain injury; the phosphorylation of NMDA receptors is also induced, causing death in the NVU [104–107] (Table 1).
CDK5 functions in each NVU components
CDK5 TARGETING IN THE PROMOTION OF NEUROPROTECTION AND NEUROPLASTICITY
CDK5 suppression has been proposed as a protective strategy against toxicity by glutamate or kainate [104, 107–112]. In primary neurons with high concentrations of glutamate, increased cytotoxicity and nuclear condensation are associated with increased Ca2+ influx in the soma, calpain activation, an increase in cleavage from p35 to p25, CDK5 hyperactivation, and tau hyperphosphorylation [113, 114]. Calpain activation mediated by Ca2+ is crucial to generate CDK5/p25 as a neurodegeneration complex. The intracellular Ca2+ imbalance occurs due to excessive NMDA- and AMPA-mediated influx as well as the release of Ca2+ from the mitochondria and ER, which are favored by CDK5 activation [106, 115–117]. Additionally, CDK5 is a calpain target; specifically, CDK5-silenced neurons avoid cleavage from p35 to p25 mediated by calpain, suggesting that CDK5 shRNA-miR facilitates the stability of p35 in neuroprotection [113].
CDK5 silencing on in vitro neurons induces neuroprotection associated with BDNF increase, phosphorylation of CaMKII, ERK1/2, and pCREB, and p35 increase [113, 114]. Different peptides derived from p35, which reduces CDK5 hyperactivity, have been shown to prevent neurodegeneration in AD models [118–120]. Specifically, p10, a peptide originating from the N-terminal domain of p35, is essential for survival, and loss of p10 itself results in CDK5/p25 toxicity [121]. Furthermore, CDK5 acts upstream from ERK to inhibit p35 production [122]. CDK5 silencing increased ERK1/2 phosphorylation, possibly triggering a potential p35 increase. Additionally, p35 phosphorylation, which is favored by CDK5, is a negative signal for degradation by the ubiquitin system, suggesting that CDK5 silencing prevents the degradation of p35 [123, 124].
p35 plays a critical role in survival induced by CDK5 silencing involving Rac GTPase activity. p35 can associate with various cytoskeletal proteins, such as Rac1 and PAK1, without CDK5 [125], favoring PAK1 function in the membrane to regulate neuronal morphology [126]. Additionally, CDK5/p35 and Rac1-PAK1 regulate the hippocampus-dependent extinction process [127]. Additionally, there is a convergent role of Rac activity in neuronal plasticity and neuroprotection induced by CDK5 silencing. Rac-GTP is capable of inducing survival dependent on the activation of PAK and PI3K. PAK signaling stimulates neuritic growth, synapse formation, and Ca2+-dependent signal activity, leading to cell survival, and PI3K induces Akt activation, which inhibits various pro-apoptotic proteins and stimulates the transcriptional activity of CREB to induce anti-apoptotic Bcl-2 expression [128, 129]. In contrast to Rac activity, RhoA-GTP leads to the downstream activation of p38α MAPK, which induces apoptosis [130] and promotes the increase of Aβ42 by the regulation of AβPP processing [131, 132]. In addition, a RhoA/ROCK inhibitor prevents apoptotic neuronal death by Akt activation during brain ischemia [133]. In our investigations, pharmacological inhibition of RhoA-ROCK (Y27632) decreased the formation of the CDK5/p25 complex and improved cognitive deterioration produced by brain ischemia [134]. Therefore, the therapeutic effect of CDK5 silencing involves Rac activation and RhoA/ROCK inhibition. Last, p35 silencing blocks Rac activity on one side and facilitates RhoA activity, suggesting that p35 plays a determining role in survival by modulating the activity of Rho GTPases [113] (Fig. 2).

Pathways modulated by CDK5 silencing in neuroprotection, neuroplasticity, and NVU integrity. A) The neuroprotection and neuroplasticity induced by CDK5 shRNA-miR. CDK5 silencing decreased the CDK5/p25 complex and prevented tau hyperphosphorylation in neurons and consequently protected against glutamate excitotoxicity. The neuroprotection mechanism is related to the increase in p35 and p120ctn, activation of Rac1 and consequent decrease in RhoA. In addition, CDK5 silencing increased dendritic protrusion morphogenesis and postsynaptic PSD95 and activated the CaMK/pERK1/2/pCREB/BDNF pathway, which favors synaptic plasticity, LTP, Ca+2 facilitation, and consequently cognitive improvement [113, 114]. B) NVU integrity induced by astrocyte-CDK5 knockdown (astrocyte-CDK5-KD). Astrocytes silenced for CDK5 generated glioprotection after glutamate excitotoxicity. The activation of Rac1, astrocyte stellation, and BDNF release are indicative of survival and neuroprotection (green lines) [52]. Finally, astrocytes-CDK5-KD induce cell adhesion and increase BDNF release, which are associated with endothelial protection against glutamate toxicity, producing cell adhesion and recovering of brain parenchyma [186, 195].
Complementarily, in vitro and in vivo CDK5 silencing is a good strategy to promote neuroplasticity, such as dendritic spine morphogenesis, facilitation of Ca2+ in neurites, long-term potentiation (LTP), and spatial memory. Several studies based on CDK5 suppression have shown to favor synaptic plasticity in adult models [135–137]. CDK5 silencing increases LTP in young mice and recovers LTP in adult mice in an AD model. In primary cultures, CDK5 silencing increases the density of dendritic spines by modulating Rho GTPases, promoting the activity of Rac and Cdc42, which is associated with the F-actin polymerization necessary for dendritic spine morphogenesis. These findings are controversial because Lai and collaborators have shown that CDK5 silencing in neurons has a deficiency in spine formation because of decreased phosphorylation of TrkB [138, 139]; however, other models suggest that the suppression of CDK5-KO-mediated phosphorylation of δ-catenin increases the density of dendritic protrusions [140].
Additionally, CDK5 favors dendritic development and gene expression as a BDNF transcription inducer in the nucleus [141]. These data contrast with in vitro and in vivo findings, in which CDK5 silencing increases BDNF levels and CREB activation, indicating that this effect depends on the context of intrinsic plasticity during development [142–144] or neuroprotection [145]. In addition, it is important to clarify that CDK5 silencing shows a knockdown effect near 50% of the total protein, suggesting that the total remaining CDK5 protein could be functional and active [113, 146]. Membrane localization of CDK5 specifically occurs when it is associated with p35, and its action in the nucleus was observed by Liang et al., in which CDK5 was dissociated from p35 [147]. Analyzing our findings, CDK5 silencing favors an increase in p35 and a reduction of the CDK5/p35 complex, supporting the findings that p35 exerts an additional role as it is dissociated from CDK5, specifically in survival and neuroplasticity [113].
CDK5 silencing generates a range of spine lengths with intermediate or filopodia-like morphologies [114, 148], which present in a transition state, where it can seek new postsynaptic targets to return to a mushroom shape to promote mechanisms of synaptic plasticity [149, 150]. According to our results, the pharmacological inhibition of Rac blocks the plasticity effects in the dendritic spine shown by CDK5 silencing [114]. In addition, phosphorylation of Trio, a CDK5-mediated Rac activator, impedes the function of Rac [151]. Also, WAVE1, a Rac effector, has been identified as a CDK5 substrate [152]. These last two observations show that CDK5 negatively regulates the formation of the dendritic spine through Rho GTPase signaling in immature neurons. Furthermore, in vitro CDK5 silencing induced fine PSD95 clustering in dendritic spines, suggesting the enhancement of the synaptic contacts necessary for improvement in spatial learning and memory. The function and localization of PSD95 is also regulated by CDK5 phosphorylation in its N-terminus [153], which inhibits the multimerization of PSD95 and the anchoring of PSD95-mediated potassium channels. The PSD95 clustering induced by CDK5 silencing favors the recruitment of postsynaptic proteins involved in the dynamic remodeling of the spines in mature neurons. Since excitatory synaptic contacts predominantly occur in dendritic spines, these have been proposed as sites where glutamate-mediated excitotoxicity begins [154]. In the context of glutamate-mediated excitotoxicity in mature neurons, there is an increase in large-size aberrant PSD95 aggregates, which are diminished by CDK5 inhibition or p35 overexpression [113].
AD patients show a reduced density of synapse hallmarks and dendritic spines in the cortex and hippocampus [155, 156], which is supported by our observations in familial AD patients, who showed a marked decrease in p120ctn and associated synapse proteins [157]. Interestingly, CDK5 silencing increased p120ctn and increased the p120ctn location in dendritic arborization in 3×Tg-AD mice in CDK5 shRNA-miR neuronal cultures. This effect has been associated with increased dendritic spine density and coincides with what was reported by Poore et al. (2010) using a δ-catenin mutant to impede the phosphorylation of CDK5 [140]. In addition, it has been demonstrated that the overexpression of δ-catenin makes it possible to induce Rac and Cdc42 activity to increase spine density [158]. Additionally, Elia et al. showed that the overexpression of p120ctn in neurons induces variable spine morphology, taking on filopodia-like morphologies. This type of spine is induced by the overexpression of p120ctn [159]. Furthermore, our findings demonstrate that the activity of Rac induced by CDK5 shRNA-miR required p120ctn. Together, these observations suggest that the Rho GTPases are downstream from p120ctn in the morphogenesis of the dendritic spine. These findings are concordant with previous studies conducted by our group, where we reported that pharmacological therapy by means of treatment with statins recovers the levels of αN-catenin and p120ctn [160]. This shows that p120ctn is necessary for neuronal recovery following excitotoxicity.
Complementarily, young healthy mice treated with CDK5 shRNA-miR exhibited increased LTP as a form of synaptic plasticity [114]. Studies involving CDK5 inhibition using roscovitine showed the role of CDK5 in synaptic plasticity, triggering LTP [161]. Furthermore, CDK5 conditional KO adult mice models have shown that a 50% decrease in total CDK5 increases LTP and induces hippocampal-dependent spatial memory [135]. These LTPs have been attributed specifically to the increased GluN2B availability in the membrane [136, 162]. However, although CDK5 conditional KO improves learning and synaptic plasticity initially, this mouse model has been susceptible to seizures, suggesting a progressive increase in excitability [162]. This observation is explained by the absence of calpain regulation by CDK5, which avoids the proteolysis of NMDA receptor subunits, such as GluN2B [135].
In vivo CDK5 silencing reduces neurofibrillary tangles and Aβ plaques and recovers cognitive deterioration in the 3×Tg model for AD [113, 163], and this effect was sustained until a year [163]. However, the reduction of these two neuropathological hallmarks does not completely explain cognitive recovery in AD pathology. In an AD model with APPswe/PS1Δ9 mice, CDK5 shRNA-miR rescues synaptic deterioration, which present synaptic dysfunction and a reduction of dendritic spines [114, 164]. This presynaptic dysfunction in APPswe/PS1Δ9 mice is due to altered Ca2+ caused by mutations in APP and PS1, which induce an increased release of Ca2+ from the ER [165–167]. AD models show an exaggerated Ca2+ signal and increase neuronal death, which supports the hypothesis that the release of intracellular Ca2+ is one of the targets to be blocked in neurodegeneration [165]. The transfer of neurotoxic Ca2+ from the ER to the mitochondria is triggered by CDK5 overactivity [115, 168]. In an in vitro model of membrane depolarization with K+, CDK5 was demonstrated to increase external Ca2+ influx, increase the release of Ca2+ from the ER to the cytoplasm, and decrease the rate of Ca2+ removal [169]. This allows us to suggest that CDK5 silencing would regulate intracellular Ca2+ – specifically intramitochondrial Ca2+. This relationship requires further research.
On the other hand, paired pulse facilitation is known as a short-term form of synaptic plasticity, which is facilitated by residual presynaptic Ca2+ and release of vesicles loaded with neurotransmitters. APPswe/PS1Δ9 mice have demonstrated presynaptic dysfunction regarding the diminished facilitation of the postsynaptic response [164]. Interestingly, CDK5 silencing recovered the presynaptic dysfunction that was altered in APPswe/PS1Δ9 mice. Although the impact of CDK5 silencing on Ca2+ levels was not determined in the in vivo tissue slice model, neurons cultured with CDK5 shRNA-miR showed a Ca2+-dependent increase in neurites [113, 114], which could be linked to the facilitation of residual Ca2+ necessary for presynaptic function.
CDK5 silencing promotes plasticity through the increase in BDNF in in vivo and in vitro models [114, 170]. Glutamate excitotoxicity restricted the increase in Ca2 +, especially in the soma, whereas CDK5 silencing redirected it toward neurites; concomitantly, there was an increase in BDNF/pCREB in the first minutes and at 24 h following glutamate. During LTP, the phosphorylation of CAMKII and ERK is required for the early activation of CREB and memory formation [143, 171–173], and the decrease in CDK5 following NMDA treatment facilitates the activation of CAMKII, inducing LTP [174]. In our findings, CDK5 silencing increased LTP in young mice and recovered diminished LTP in adult APPswe/PS1Δ9 mice, which was associated with an increase in the pro-plasticity BDNF/TrkB/pCaMKII/pERK/pCREB pathway (Fig. 3).
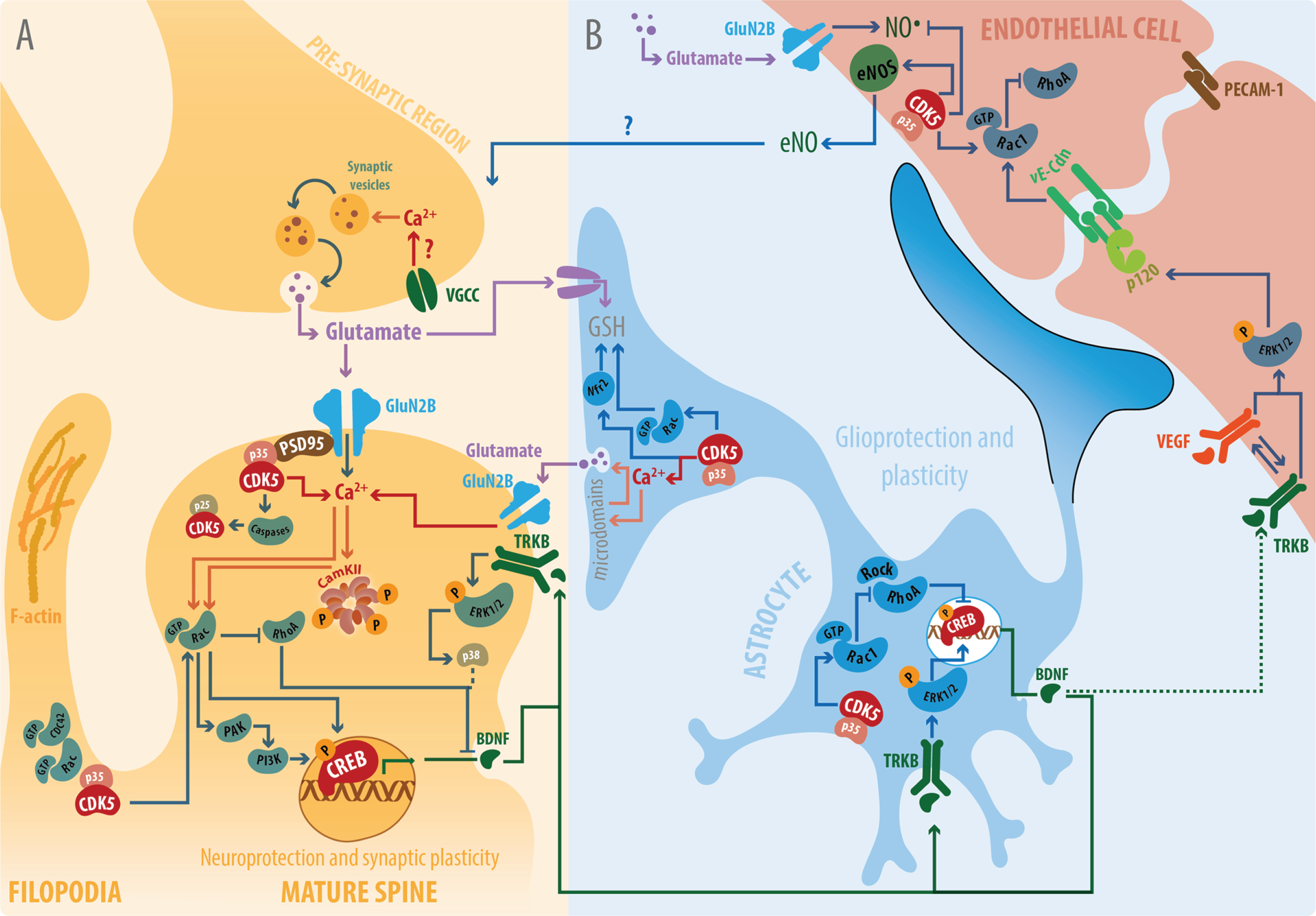
Possible cellular targets involved in the neuroprotection, neuroplasticity, and integrity of the NVU induced by CDK5 RNAi. A) Neuroprotection and neuroplasticity. Activation of Rac1, increase of p35, and activation of the TrkB/BDNF pathway have been implicated in the activation of the PI3K survival pathway. Consequently, there is a decrease in RhoA and the MAPK/p38 inactivation pathway, which have been observed to be protective. In addition, dendritic spine morphogenesis has been related to an increase in p35 and p120ctn and the activation of Rac1 and Cdc42. The presynaptic mechanisms, such as increased likelihood of neurotransmitter release favored by Ca+2 influx through voltage gated Ca+2 channels and paired pulse facilitation. The increase in PSD95, the facilitation of Ca+2 in dendrites, and the activation of CaMKII/pERK1/2/pCREB/BDNF support the increase in LTP [114, 170]. B) NVU integrity. Astrocyte-CDK5-KD could facilitate gliotransmission through the activation of GluN2B, activation of Nrf2, and consequent glutathione release to balance neurotransmission and facilitate synapse potentiation. It is possible that astrocyte Rac1 activation facilitates pCREB pathway activation that consequently allows BDNF release, which becomes a key factor in protection. The possible BDNF/TrkB activation and VEGFR cross-linking signaling mediates ERK1/2 activation and the production of adhesion proteins such as E-cadherin and p120ctn [179].
CDK5 TARGETING: LIMITATION AND CLINICAL TRIALS PERSPECTIVE FOR AD
CDK5 KO mice and specific CDK5 silencing and inhibition have shown deleterious and beneficial effects on the CNS, depending on the type of suppression and the short term (acute) or long term (chronic) effect. Specifically, CDK5 is essential in development, and embryonic CDK5 KO mice show neuronal migration defects and perinatal lethality [175]. On the other hand, CDK5 KO-conditional models have shown CDK5 suppression in neurons and endothelial cells are related to spontaneous epilepsy, neuroinflammation, and neurodegeneration [162, 177].
RNAi, which targets CDK5 messenger RNA (mRNA), reduces partially CDK5 expression levels by suppressing RNA transcription. Early CDK5 silencing in immature neuronal culture impair axon guide and dendrite branching, therefore CDK5 silencing should be considered in adult age against CDK5 overactivation state for a neuroprotective and synaptic plasticity effect [113, 141]. CDK5 silencing strategy is based on the use of an endogenous microRNA (miR30) that carries the CDK5 shRNA (exogenous), which is transduced into cells using an adeno-associated viral vector AAV 2.5. This strategy has several points of advantage because the miR-30 environment confers low toxicity, high processing and expression in the cell compared to the use of only the CDK5 shRNA [178]. Furthermore, the use of AAV, specifically serotype 2.5, confers: 1) tropism to neural cells, astrocytes and neurons; 2) decreased inflammatory response or response to interferon, 3) low insertion of exogenous DNA into host cell DNA, which reduces the probability of tumor formation, 4) stable expression of CDK5 RNAi for up to 1 year in mice, and 5) ease of monitoring expression with the reporter eGFP (enhanced green fluorescent protein) [146, 163]. A partial CDK5 deletion using CDK5shRNA-miR on 3×Tg-AD mice under short-term (6 months) and long-term (12 months) silencing conditions has shown that does not cause tumor, even decreases neurodegeneration markers [163]. Accordingly, on cerebral ischemia model using rats, also CDK5 shRNA-miR neuroprotects in acute (one month post ischemia) and chronic (long term in a 4 month post ischemia) silencing [145, 179]. However, it may be a long time before RNAi are in widespread clinical use; possible disadvantages of silencing are the high cost of producing viral vectors, delivery to the CNS, and cranial stereotaxic requirement in the specific neuroanatomical location [180, 181]. In addition, the neurologists questions about the specific region of the gene therapy injection in a diseases with several brain affected areas; we consider that the neuropathology studies suggest an area with major glutamatergic hyperexcitability from where arise the spread overload of calcium waves [182]; for example, cortical layer III and V, dorso-ventral striatum, subiculum, CA1 area in a mild cognitive impairment stage, and also considering the clinical manifestations.
Finally, CDK5 inhibition has been achieved using competitive inhibitors that affect the ATP binding site to the kinase; different companies have developed drugs in this regard to affect kinase activity completely or partially [183,184, 183,184]. Roscovitine (Seliciclib) is inserted into the lipophilic pocket mainly delimited by Val64 and Phe80 residues from CDK5. There are currently no reported clinical trials using CDK5 as a target for AD treatment; however, CDK inhibitors are an intriguing clinical therapy for cancer treatment. R-roscovitine, that affects CDK2 and CDK5, is in Phase I-II for treatment of advanced solid tumors, non-small cell lung cancer (NCT00999401, NCT00372073), and Phase II for Cushing disease (NCT02160730 NCT03774446) opening a new panorama for the treatment of CNS diseases. So far no side effects have been shown in these clinical trials; however, due to functions of CDK5 in different tissue types and the pan-CDK inhibitory effect in other family members, the undesirable effects of a CDK5 inhibitor drug can be produced [185].
CDK5 SILENCING IN ASTROCYTES: A LINK BETWEEN NEUROPROTECTION AND NEUROPLASTICITY
A new novel approach to CDK5 targeting in CNS is cell therapy using astrocytes CDK5-silencing. In vitro and in vivo CDK5 silencing or inhibition is a good therapeutic strategy because it reverts the cell alteration generated in a process of glutamate-mediated gliotoxicity in astrocytes. Additionally, Rac1 activation and the subsequent formation of lamellipodia and BDNF release are involved in CDK5 shRNA-miR astrocyte-induced neuroprotection [52, 186] (Fig. 3). Gliotoxicity is due to large quantities of glutamate generating an ionic imbalance in the astrocyte because of an excessive exit of K+, an uncontrolled expense of energy due to the activation of K+ Na+ ATPase and the activation of mGluR receptors and subsequent cytosolic Ca2+ propagation, which entails extracellular glutamate release [61, 187–189].
However, CDK5 silencing induces protection in astrocytes following gliotoxicity, reverting the morphological atrophy generated by glutamate. In the literature, the presence of tau filaments forming aggregates in astrocytes in human diseases and animal models has been described [98, 191]. This phenomenon is related to a dysfunction of glutamate uptake leading to focal neurodegeneration [191]. The kinase activity of GSK3β and CDK5, which regulate tau phosphorylation, is also increased in astrocytes in a senescence-accelerated mouse model (SAMP8). In addition, the close relationship between CDK5 activity and the reduced potential of the mitochondrial membrane in SAMP8 astrocytes suppresses glutamate uptake, which is critical for neuroprotection mechanisms [98, 193]. This suggests that CDK5 is an adequate target for the protection of astrocytes and neuronal survival [52, 195].
Additionally, CDK5 silencing in astrocytes induces lamellipodia and protrusions, increasing the cell area, related to Rac1 activation, which favors stellation or the generation of new protrusions in astrocytes [196]. Rac1 activation by PI3K has been associated with the activation of the AKT pathway, which promotes cell survival [197, 198]. In a similar manner, the Rac1 activator Tiam carries out a function in astrocyte polarization and migration during cell development or repair through the reorganization of actin microfilaments and microtubules [199]. A study in a senescence model demonstrated that the increase in CDK5 activity reduces Rac1 and PAK activity, which is related to the structural loss of F-actin and is required for the senescent phenotype, indicating that this phenomenon is involved in cell death [200].
Astrocytes promote neuronal survival due to the release of trophic factors such as BDNF [144, 201–206], which induce neuroprotection [142, 208]. Astrocytic BDNF requires Rac1 activity to recover the dendritic structure after glutamate gliotoxicity and to promote neurogenesis [138, 209]. Additionally, BDNF treatment triggers Rac1 activation to induce axonal recovery [210]. Interestingly, RhoA-ROCK inhibition, which promotes Rac1 activation, increases glutamate uptake and produces stellation through BDNF [196, 211]. Accordingly, CDK5 silencing on astrocytes promotes stellation throughout Rac1 activation and BDNF release, which are necessary for neuroprotection [52, 186] (Fig. 3).
CDK5 shRNA-miR astrocytes under excitotoxic conditions release BDNF as a neuron-astrocyte signal to generate protection. CDK5/p35 activation phosphorylates Nfr2 as a part of the antioxidant activity in the presence of excitotoxic stress in astrocytes [99]. Nrf2 is considered the master regulator of the antioxidant response through glutathione biosynthesis. Rac1 seemingly has antioxidant effects through Nrf2 activation [212–214]. KO mice for Nfr2 decrease BDNF levels [213], and BDNF/Nfr2 mediate neuroprotection induced by C6 cells [215].
In addition, neuronal plasticity and the tripartite synapse mediated by astrocytes occur through NMDA receptors, which are mainly made up of GluN2B subunits [216, 217]. Studies using CDK5 conditional knockout in adult mouse brains showed better performance in spatial learning tasks mediated by GluN2B [135, 136]. This observation, in association with the finding that CDK5 silencing induces neuroplasticity and cognitive recovery, could suggest that the use of CDK5 RNAi in astrocytes promotes the tripartite synapse and recovery from cognitive deterioration following neurodegenerative pathology; however, this is a topic to be addressed in the future (Fig. 3).
Finally, Sundaram et al. (2012) suggested that CDK5/p25 induces neuroinflammation and triggers neurodegeneration through neuron-astrocyte signaling. Interestingly, the formation of the CDK5/p25 complex in neurons induces phospholipase A2 activation, generating lysophosphatidylcholine, which behaves as a soluble factor that initiates astrocytic hyperreactivity (increasing GFAP) and neuroinflammation [218]. Together, these data suggest a bidirectional relationship in the role of CDK5 in neurons on astrocytes or vice versa, which supports our findings that CDK5 silencing promotes astroprotection and neuroprotection. This is also supported because transplantation of CDK5-knockdown astrocytes in the somatosensory region of ischemic rats induced neuroprotection, recovering astrocytes and the endothelial population [186, 195]. These in vivo observations suggest the possibility of a cell therapy based on CDK5-silenced astrocytes in neurodegenerative pathology.
CDK5 DOWNREGULATED ASTROCYTES IN THE ENDOTHELIUM: ASTROCYTE-ENDOTHELIUM COUPLING
On the other hand, we also suggest CDK5 shRNA-miR astrocytes as mediators of a greater protective effect of adhesion on endothelial cells in the presence of glutamate-mediated toxicity associated with BDNF release, which underlies this recovery of cell adhesion and protection (Fig. 3). Some studies conducted on humans and endothelial cell cultures show an increase in CDK5 following brain ischemia or hypoxia in the NVU [102, 219]. Additionally, recent studies have suggested CDK5 suppression as a therapeutic strategy to recover the endothelium, favoring angiogenesis following hypoxia and avoiding leukocyte infiltration [102, 220]. Therefore, CDK5 inhibition or silencing in endothelial cells is a good therapeutic strategy to recover endothelial adhesion [186].
Our in vivo and in vitro results have shown that treatment with the CDK5 inhibitor recovers the endothelium following glutamate-mediated toxicity, reverting the decrease in CDK5 levels, the formation of intracellular gaps, and recovering adhesion proteins [186]. Inhibition of CDK5 reduced endothelial motility and angiogenesis, suggesting an effect that depends on actin cytoskeleton remodeling [92]. Increased expression of CDK5 and its activator p35 are detected in neurons and endothelium in the peri-infarct zone following middle cerebral artery occlusion [221]. In models of ischemic mice, inhibition or silencing of CDK5 has significantly reduced infarct volume [145, 222]. Interestingly, CDK5 inhibition prevents endothelium activation and the endothelium-leukocyte cell interaction that is generated during leukocyte infiltration in the brain parenchyma [220].
The recovery of cell adhesion by CDK5 inhibition impacts the integrity of the NVU and is also fundamental in angiogenesis since during this process [186], there is spatial and temporal control of focal adhesion and the activation of cytoskeletal organizers that regulate migration, cell differentiation, and final adhesion [223]. In particular, p120ctn regulates the development and morphology of vascularization [224, 225]. Our observations suggest p120ctn as a molecular target that needs to be studied regarding the recovery of adhesion in the NVU following glutamate-mediated toxicity [157, 226].
In summary, CDK5 silencing or inhibition in astrocytes is a good therapeutic strategy because it reverts the cell alteration generated in a process of glutamate-mediated gliotoxicity. The Rac1 activation, formation of lamellipodia and BDNF release underlying CDK5 shRNA-miR astrocytes [52, 186]. CDK5 silencing in astrocytes favors BBB integrity and generates a trophic source of BDNF for endothelial cells. In vitro experiments have demonstrated that endothelial cells promote neuronal survival via BDNF release [227–230]; this is also supported in in vivo models, where BDNF is crucial for mediating postischemic angiogenesis [231, 232], promoting microvascular survival and endothelial adhesion [233]. Our findings suggest that BDNF released from astrocytes is an important factor to protect and recover cell adhesion in the endothelium in the presence of glutamate-mediated toxicity, which is increased by CDK5 silencing. Increased BDNF is associated with the CREB activation pathway, which is crucial for the recovery of cognitive deficits in a brain ischemia model and AD [145, 234]. Last, the transplant of CDK5 shRNA-miR astrocytes recovers the neurovascular unit in ischemic mice, generating recovery of the vascular endothelium [186]. Other authors have demonstrated that BDNF production is increased in endothelial cells when they have been cocultured with astrocytes; this phenomenon is possibly related to the increase in the adhesion integrity of the BBB [235].
PERSPECTIVES
Silencing of CDK5 after one month of cerebral ischemia protects neurons, decreases microgliosis and astrogliosis, and generates an increase in BDNF in blood vessels. Although these observations suggest a recovery of several components of the NVU, we suggest that the integrity of the BBB should be evaluated. A functional analysis by the Evans blue tracer postischemia and post-therapy is necessary. In addition, these observations suggest that a recovery of the adherens and tight junctions must be demonstrated, which may be supported by ultrastructural analyses. Additionally, astrocyte-CDK5 knockdown is neuroprotective and facilitates trophic support to recover the endothelium. These findings suggest a cellular therapy combined with gene therapy as a way to favor the interrelation of neurons-astrocytes-endothelium. However, we consider that it is necessary to address some essential components in the NVU, such as microglia, oligodendrocytes, pericytes, and the extracellular matrix. This approach requires cellular, functional, and morphological analyses that demonstrate the recovery of NVU integrity.
Several of the findings of the possible effect of CDK5 silencing on synaptic plasticity have been obtained in primary cultures of neurons. Several of the suggestions by the evaluators include strengthening the phenomena obtained in vitro with the in vivo. Specifically, it is important to delve deeper into the analysis of dendritic spine morphology and Ca2+ regulation since they can vary with age and under pathological conditions. The diversity of spines and Ca2+ regulation after CDK5 shRNA-miR in the brain are necessary points to strengthen.
The observations of mature astrocyte CDK5 silencing and their in vivo validation require determining the impact of these factors on cell proliferation and the generation of neural and endothelial progenitors, which would account for the neuroprotective effect and functional recovery. For this type of analysis, it is necessary to validate specific markers of cell types and glial and neuronal progenitors. Finally, from the mechanistic point of view, it is essential to determine which molecular enclave facilitates the production of BDNF. Although we observed the phosphorylation of CREB, it did not necessarily correspond to the silencing of CDK5 inducing this phosphorylation. The balance between kinases and phosphatases could be a mechanism, but a transcriptional profile by RNAseq could reveal which genes are associated with the regulation of pCREB/BDNF.
Consistent with the previously proposed regulation of pCREB/BDNF, it is necessary to determine the direct dependence of BDNF to mediate the neuroprotection and integrity of the BBB. We propose blocking the effect of BDNF from astrocytes-CDK5RNAi with antibodies that sequester their action on the TrkB receptor or by silencing TrkB in neuron-endothelium or inverse. It is important to note that in our laboratory, in in vivo studies, other trophic factors were evaluated (GDNF, VEGF, NGF), but BDNF seems to have the most robust and consistent effect among the models.
CONCLUSION
In general, CDK5 silencing promotes neuroprotection, neuroplasticity, and integrity of the NVU. This review allows the integration of different molecular and cellular events with physiological and behavioral relevance. CDK5 silencing reverts tauopathy and β-amyloidosis and facilitates protection in neurons through a new neuroprotection mechanism in which the increase in p35 and activation of Rac1 are necessary for survival. These results are consistent with the decrease in neuronal loss in the CA1 region of the hippocampus, which supports the reversion of neurodegeneration and improvement of cognitive function in stroke and AD animal models (Fig. 1). Once neuronal survival is recovered, CDK5 silencing promoted neuroplasticity as the neurons increased the density of dendritic protrusions, thus becoming the functional and structural substrate for new synapses. These dendritic spines are enriched by the postsynaptic marker PSD95, which is dependent on Rac1 activation, promoting the anchorage of adhesion molecules, such as p120ctn and NR2B; these facilitate the stability of neurotransmission and synaptic plasticity, respectively. Postsynaptic recovery is associated with an increase in dendritic Ca2+, which, in turn, is associated with an increase in the activation of CAMKII and ERK1/2 and subsequent CREB phosphorylation. CREB activation facilitated the increase in BDNF and positive feedback from the BDNF/TrkB/CREB pathway. These molecular events induced by CDK5 silencing support the synaptic plasticity evidenced by the facilitation of paired pulse facilitation and the increase of LTP as processes that are mainly associated with presynaptic and postsynaptic function, which support the improvement in cognitive function, such as spatial memory and learning in AD mice (Fig. 2). Concomitant with these effects, CDK5 silencing in astrocytes promoted astrocytic stellation and astroprotection, requiring Rac1 activation, which favored BDNF release as a process of neuron-astrocyte communication. This process of Rac1 activation and BDNF release in astrocytes is a cell mechanism that is common to neuroplasticity, neuroprotection, and cognitive improvement in a neurodegenerative process. Related to the neuroprotective effect of CDK5 knockdown astrocytes, these cells also favored BBB integrity through BDNF release and the increase in p120ctn adhesion, which supports an increase in the TEER involved in the endothelial cell-to-cell junction (Fig. 3). These results indicate that the potential therapeutic effect of CDK5 silencing is explained by coupling between NVU components, in which each component plays a recovery role but where all components, in communication and molecular coordination, are important for the recovery of cognitive and behavioral function. We consider that astrocytes, through the production of trophic factors, such as BDNF and, due to their intermediary communication between neurons and the endothelium, play a central role in the regulation of these survival processes needed to re-establish NVU homeostasis. The potential therapeutic effects of CDK5 silencing in neurodegenerative pathologies may involve multiple mechanisms; in our study, we provide the implications of doing this in an astrocyte-directed manner to favor NVU protection. However, currently is not clear the use of CDK5 inhibitors, related to specificity concerns, and a controlled silencing of CDK5 with the reduction of 50% of the overactivation in adult and aged brain, have difficulties inherent to gene therapy practice and very local action.