
Abstract
Alzheimer’s disease (AD) is a chronic neurodegenerative disease that has been recognized as one of the most intractable medical problems with heavy social and economic costs. Amyloid-β (Aβ) has been identified as a major factor that participates in AD progression through its neurotoxic effects. The major mechanism of Aβ-induced neurotoxicity is by interacting with membrane receptors and subsequent triggering of aberrant cellular signaling. Besides, Aβ transporters also plays an important role by affecting Aβ homeostasis. Thus, these Aβ receptors and transporters are potential targets for the development of AD therapies. Here, we summarize the reported therapeutic strategies targeting Aβ receptors and transporters to provide a molecular basis for future rational design of anti-AD agents.
INTRODUCTION
Alzheimer’s disease (AD) is a chronic neurodege-nerative disease that destroys memory and other important mental functions, with the most common early symptom of short-term memory loss, and eventually leads to bodily dysfunction and death [1, 2]. AD is the cause of 60% to 70% of cases of dementia, and the incidence increases exponentially with age. In 2015, approximately 29.8 million people worldwide had AD, and this number is predicted to be quadruple by the year 2050 [3]. AD has been recognized as one of the most intractable medical problems with heavy social and economic costs.
AD is characterized by the loss of neurons and synapses in the cerebral cortex and certain subcortical regions [3]. The main neuropathological features of AD are extracellular deposits of amyloid-β peptide (Aβ) in senile plaques [4]. Over the decades, many hypotheses have been proposed to explain the pathophysiology of AD. According to the amyloid cascade hypothesis [5], the imbalance between Aβ production and clearance leads to gradual accumulation and aggregation of the peptide in the brain, thereby initiating a neurodegenerative cascade that involves tau pathology [6, 7], inflammation [8], oxidative stress [9], and neuronal injury and loss [10, 11].
The Aβ peptides are cleaved from the much la-rger precursor glycoprotein named amyloid-β protein precursor (AβPP), which is a natural membrane glycoprotein present in neurons. The sequential enzymatic processing of AβPP by the aspartyl proteases, β- and γ-secretase, result in the rise in the production of Aβ peptides [12]. The procession of AβPP is found to include amyloidogenic and nonamyloidogenic pathways which lead to different outcomes [13]. In general, AβPP is cleaved by α-secretase and then γ-secretase, a process called nonamyloidogenic process; however, in amyloidogenic pathway, AβPP is initially cleaved by β-secretase (BACE1, β-site APP cleaving enzyme 1) to release a 99 amino acid fra-gment (C99), which then undergoes a series of cleavages by γ-secretase to produce different lengths of Aβ peptides, of which Aβ38, Aβ40, and Aβ42 are the most abundant in the cerebrospinal fluid (CSF) [14, 15].
Following cleavage from the membrane, Aβ mon-omers aggregate into various types of assemblies, including oligomers, protofibrils, and amyloid fibrils. Compared with monomers and fibrils, Aβ oligomers (AβOs) are widely regarded as the most toxic and pathogenic form of Aβ. Recently, growing evidence has pointed to AβOs rather than amyloid plaques as playing a central role in pathogenesis [16, 17]. Synaptic loss was present in APP transgenic models of AD in the absence of plaque formation [18]. AβOs were increased in the AD brain with respect to controls and were correlated with severity of cognitive impairment and loss of synaptic markers [11].
The molecular mechanism of extracellular Aβ damaging neurons, which existing in CSF and interstitial fluid, has been the focus of intensive clinical and basic research in recent years. Interact with membrane receptors and subsequent triggering of aberrant cellular signaling has been identified as a major mechanism. Several Aβ receptors and transporters have been reported and can been classified according to their hypotheses and mechanisms (Fig. 1).
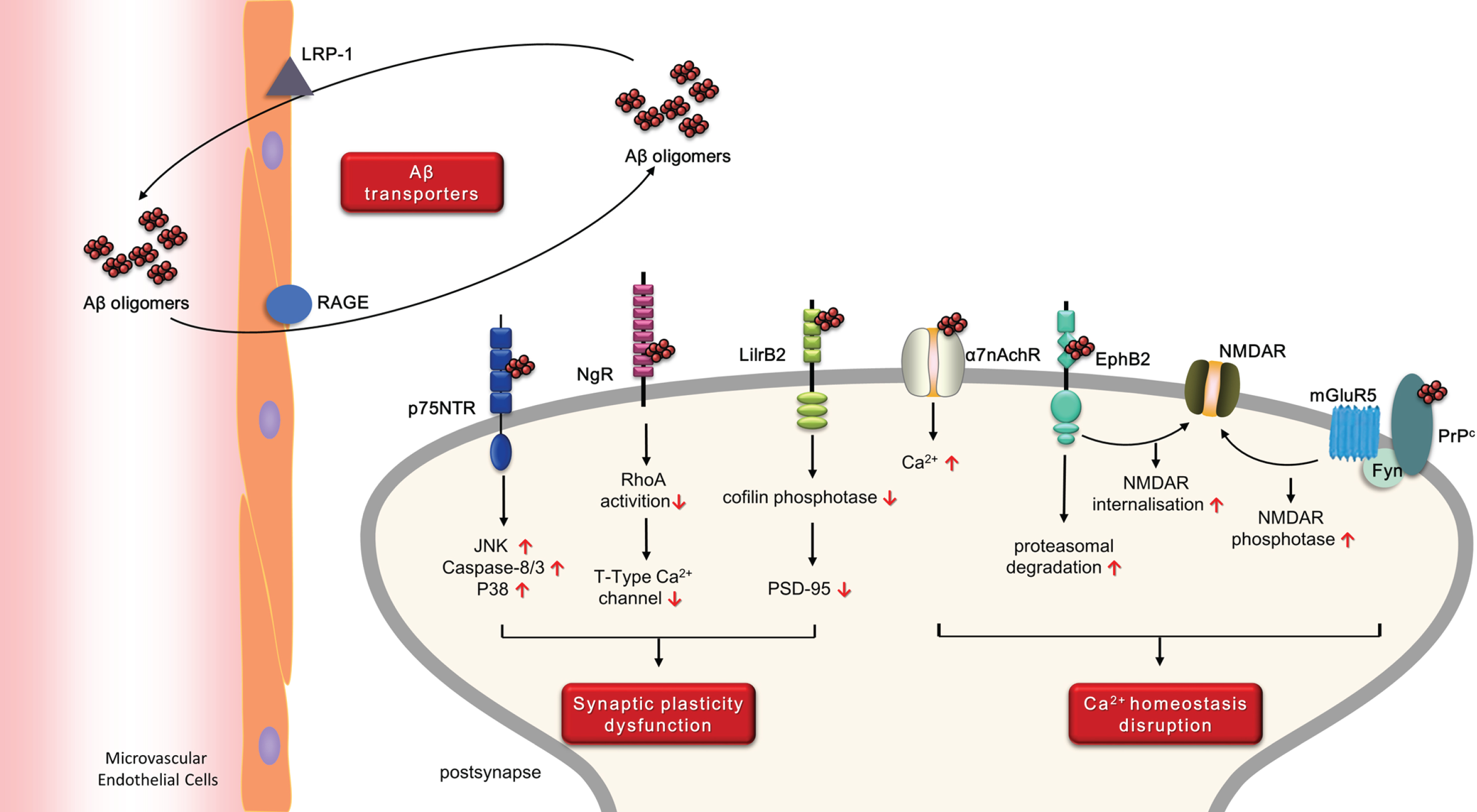
Overview of the receptors and transporters of Aβ and their potential pathogenesis in AD.
Aβ receptors selectivity bound to Aβ with high af-finity and can transduce extracellular events into in-tracellular changes. According to their pathologic effects, Aβ receptors can briefly be divided into two categories that are not mutually exclusive: 1) recept-ors related to synaptic plasticity dysfunction, including leukocyte immunoglobulin-like receptor B 2 (LilrB2), P75 neurotrophin receptor (p75NTR), and Nogo-66 receptor (NgR); and 2) receptors related to Ca2 + homeostasis disruption, including ephrin type-B receptor 2 (EphB2), α7-nicotinic acetylcholine Re-ceptor (α7nAchR), and cellular prion protein (PrPc).
Aβ transporters also plays an important role in Aβ homeostasis. Soluble Aβ can be transported across the blood-brain barrier (BBB) from blood to brain via receptor for advanced glycation end-products (RAGE), and from brain to blood via low-density lipoprotein receptor-related protein-1 (LRP-1) (Fig. 1).
Currently, growing evidence indicated that targeting Aβ receptors and transporters may be a promising strategy for anti-AD drug development by alleviating Aβ toxicity or promoting Aβ clearance. Here, we summarize the latest advances of therapeutic strategies blocking or rescuing these Aβ binding proteins.
THERAPEUTIC STRATEGIES TARGETING Aβ RECEPTORS
Therapeutic strategies that reverse synaptic plasticity dysfunction
Therapeutic strategies targeting LilrB2
LilrB2, which was known as paired immunoglo-bulin-like receptor B (PirB) in mouse, has been tra-ditional recognized as a functional receptor that played a critical role in inhibiting axonal regenera-tion and functional recovery after brain injury [19]. Very recently, LilrB2 was reported as a receptor for Aβ oligomers with nano-molar affinity [20]. A dual polarization interferometry study revealed a 37.3 nM Aβ-LilrB2 affinity [21]. Mice lacking PirB were im-mune to the damaging effects of Aβ in hippocampal LTP and recognition memory, as well as to alterations in cofilin signaling and PSD-95 synaptic loss. Preliminary evidence indicated that calcineurin, a PP2B serine/threonine phosphatase typically involved in synaptic depression and Aβ-mediated synaptotoxicity, is the mediator of the toxic effects [22].
Designing peptide that contains the binding do-main for Aβ is an efficient strategy of blocking the Aβ-LilrB2 interaction. The extracellular segment of PirB is predicted to consist of six tandem Ig-like domains, referred to as D1-D6, while for LilrB2 are D1-D4. In vitro binding to Aβ oligomers revealed that the two most N-terminal Ig domains (D1D2) of PirB and of LilrB2 are critical [20]. TAT-PEP is a recombinant soluble PirB ectodomain fused with TAT domain for blood-brain barrier penetration. TAT-PEP can reverse the axon outgrowth inhibition induced by PirB over-expression. In an in vivo study, intraperitoneal administration of TAT-PEP was capable of enhancing motor capacity and spatial learning and memory in mice, which appeared to be mediated th-rough the regulation of brain-derived neurotrophic factor secretion [19] (Table 1).
The potential therapeutic strategies that reverse synaptic plasticity dysfunction
Several small molecule Aβ-LilrB2 interaction inhibitors have been identified based on the analysis of the Aβ-LilrB2 complex. The Aβ segment 16KLVFFA21, which is also widely considered to be a key element of Aβ aggregation, represented as antiparallel dimer rather than a single copy as a minimal Aβ oligomer and the core epitope for LilrB2 binding. In the crystal structure of LilrB2 D1D2 complexed with Aβ, the two pockets can accommodate the phenylalanine side chains of KLVFFA, which was confirmed to be 16KLVFFA21 binding sites by mutagenesis [23]. Based on these facts, 12 candidate inhibitors have been identified by structure-guided selection. Among them, fluspirilene was the most potent with the affinity of 1.09μM [23]. In primary neurons, fluspirilene blocked LilrB2-induced cell attachment and inhibited Aβ toxicity. The further study of fluspirilene on the downstream pathway of LilrB2 showed that it protected neurons from Aβ-induced changes in the cofilin signaling pathway (Table 1).
Therapeutic strategies targeting p75NTR
p75NTR is a TNF-family low-affinity receptor for neurotrophins and brain-derived neurotrophic factor. Cholinergic neurons in the basal forebrain which are severely affected in AD express p75NTR throughout adult life [24]. Aβ accumulation has been shown to be accompanied by an increased level of p75NTR in hippocampal [25, 26]. Aβ has been demonstrated directly responsible for activating p75NTR-mediated cell death in AD, given that it is a ligand for the receptor and can stimulate p75NTR-mediated death signaling cascades [27]. Aβ binding to p75NTR activates p38 and JNK in a DD-dependent manner, followed by NF-κB translocation and p53 activation [28]. Unfortunately, the deletion of p75NTR exacerbates AD pathologies such as increased amyloid plaques and microgliosis, which suggests that simply reducing expression of p75NTR may not be a good therapeutic strategy for AD [29].
Interestingly, the extracellular domain of p75NTR (p75ECD) after shedding by TACE from the membrane may bind and suppress Aβ aggregation and reduce Aβ deposition in the brain, making it a possible therapeutic strategy [30]. p75ECD consists of four cysteine-rich repeat domains that account for ligands binding [31]. It was found that CRD 2 and CRD4 are Aβ binding domains of p75NTR and capable of antagonizing Aβ neurotoxicity [32]. The feasibility and efficacy of peripheral administrated of AAV-p75ECD on the brain amyloid burden and associated pathogenesis have been studied. Intramuscular delivery of AAV-p75ECD increased the level of p75ECD in the blood, significantly improved the behavioral phenotype of APP/PS1 transgenic mice, and reduced brain amyloid burden, tau hyperphosphorylation, and neuroinflammation. Thus, peripheral delivery of p75ECD represents a safe and effective therapeutic strategy for AD [33] (Table 1).
LM11A-31, a water-soluble amino acid derivative similar to the NGF loop 1 domain, was reported to interact with p75NTR. LM11A-31 was shown to re-duce the hyperphosphorylation and misfolding of tau, decrease neurite degeneration, and attenuate microglial activation in mouse model of AD. Thus, LM11A-31 is a novel therapeutic under Phase II clinical evaluation for AD treatment (Table 1).
Therapeutic strategies targeting NgR
The NgR signaling pathway plays an important role in limiting neuronal plasticity after trauma to the adult central nervous system [34]. Recently, NgR was identified as a surface neuronal molecule that modulates AβPP and Aβ metabolism and that may participate in Aβ pathology [35]. NgR1 was shown to be involved in Aβ-induced synapse assembly, plasticity, and learning inhibition. RhoA- and ROCK- mediated Aβ-NgR1 synaptic block in new synapse assembly on the dendritic shaft may contribute to LTP loss. T-type calcium channels were also been defined as a target of Aβ-NgR signaling that mediated Aβ inhibitory effects on calcium, synapse assembly, plasticity, and learning. In addition, the NgR family has been shown to be related to Aβ production. Park et al. [36] revealed that NgR1 overexpression decreases Aβ production by sequestering AβPP from secretase activity; however, the interaction between AβPP and NgR2 favor AβPP processing by BACE1 though a small region adjacent to the BACE1 cleavage site, which resulted in promoting Aβ plaque load. What the functional significance of opposing effects of NgR1 and NgR2 in APP cleavage remains unclear.
In vitro binding studies indicated that the sAPP fragment (APP597) and Aβ1–28 fragment both exh-ibit affinities for NgR with a Kd of 60 nM and 62 nM, respectively [37]. The high affinity of NgR for the central domain of Aβ suggests that it might promote peripheral clearance if delivered outside of the CNS. The NgR(310)ecto-Fc fusion protein, which contains the entire leucine-rich repeat ligand-binding domain of the NgR fused to the Fc portion of IgG, was de-signed and administered subcutaneously to enter the brain of mouse (Table 1). In transgenic mice, subcutaneous NgR(310)ecto-Fc [37] treatment reduces brain Aβ plaque load while increasing the relative levels of serum Aβ. These changes in Aβ were correlated with improved spatial memory in the radial arm water maze. The benefits of peripheral NgR administration are evident when therapy is initiated after disease onset.
Therapeutic strategies that maintain Ca2 + homeostasis
Therapeutic strategies targeting EphB2
EphB2 belongs to the Eph family, which based on their domain structures can be divided into two cla-sses, EphA and EphB receptors [38]. EphB2 receptors have been shown to form a complex with NMDA receptors (NMDARs) and to regulate their trafficking and function [39]. In AD, Aβ promoted a rapid decrease in the membrane expression of both NMDA and EphB2 [40]. EphB2 is diminished in the hippocampi of AD patients and hAPP mice [41, 42]. The Aβ-EphB2 interaction triggered EphB2 degradation in the proteasome [43], thereby impairing the normal functioning of NMDA receptors and resulting in cognitive deficits. In nontransgenic mice, EphB2 knockdown reduced NMDA receptor currents and impaired LTP in the dentate gyrus, which are important for memory formation. The surface depletion of EphB2 and NMDARs by Aβ oligomers might imp-air downstream Kalirin7/Rac1/PAK signaling, there-by inducing actin network disorganization, spines shrinkage and loss [44]. This cascade ultimately leads to neuronal network dysfunction and cognit-ive deficits. On the other hand, a very recent study showed that the protective effect of EphB2 depends on its PDZ-binding motif and association with GluA2, but not on its kinase activity [45].
The Eph receptor extracellular domain contains the ligand binding domain (LB), a cysteine rich domain (CRD) and two fibronectin type III repeats (FNIII) [46]. The FN domain has been suggested as the binding domain of Aβ [43]. A formaldehyde cross-linking assay has further confirmed that ADDLs interacted directly with FN to form complexes [47]. Based on these facts, a peptide array method to iden-tify four 10-mer peptides were designed to repre-sent the FN domain and could mediate ADDLs binding [47]. Among them, Pep63 effectively interfered with the EphB2-ADDL interaction and affected the EphB2 levels, GluN2B phosphorylation, and GluN1/GluN2B surface expression [48]. As a therapeutic strategy, Pep63 can improve both the hippocampus-dependent and amygdala-dependent memory loss in a concentration-dependent manner (Table 2).
The potential therapeutic strategies that maintain Ca2 + homeostasis
What’s more, some small molecules that blocked the EphB2–ADDL interaction have been identif-ied utilizing a cell free screening assay [49]. The four compounds (dihydroergotamine, bromocriptine, cepharanthine, and levonorgestrel) did not affect the binding between EphB2 and ephrinB2, an endogenous ligand for EphB2, suggesting that the compou-nds selectively inhibited the binding of Aβ to EphB2 (Table 2).
Therapeutic strategies targeting α7nAchR
α7nAChR is a type of nicotinic acetylcholine receptor (nAChR), which is the major receptor subtype with clinical and pharmacological implications for AD. Physiologically, α7nAChR modulates calcium homeostasis and release of the neurotransmitter acetylcholine, two important parameters involved in cognition and memory [50, 51]. In the brains of AD patients, the impairment of cholinergic transmission and decreased numbers of nicotinic binding sites are well known features [52]. There is a dual effect of Aβ and α7nAChR interaction [53]. Aβ at low concentrations (pM) modulates synaptic plasticity and enhances cognitive functions by acting as an agonist of nAChRs to upregulate of the receptor selectively in amyloid plaque burdened area [50]; however, at high concentration, Aβ may suppress synaptic plasticity and results in α7nAChR dysregulation, synaptic plasticity suppression and memory impairment via interaction with α7nAChR [54]. Aβ is thought to interacts with α7nAChR with apparently high affinity (KD of 9 nM) [55, 56]. The binding mechanism involves residue AβK28 which forms cation/π interactions in the acetylcholine binding site, and residues AβG29-AβI32 that form an intermolecular b-sheet with residues α7F189–α7E191 of AChR [57].
This interaction leads to intraneuronal accumulation of Aβ-α7nAChR complexes, rapid tau phosphorylation [58], severe impairment of α7nAChR channels [59], cholinergic neurotransmission defects [60], and neuronal cell death [61]. From that point, whether the α7nAChR antagonist or agonist rescues Aβ-induced cytotoxicity still remains controversial.
Some a7nAChR partial agonists, such as EVP-6124 and ABT-126, are under development in clinical trials [62]. ABT-126 (7) developed by AbbVie is currently in phase II trials. The results from a phase II study indicated that treatment with the 25 mg ABT-126 once daily was associated with a trend for improvement in cognition in subjects with mild-to-moderate AD. For EVP-6124, however, severe gastrointestinal symptoms side-lined its two phase III trials in 2015 (Table 2).
S-24795, a novel 7nAChR partial agonist, was re-ported to interfere with the Aβ-α7nAChR interaction and alleviate AD like pathologies. S24795 facilitates Aβ release from Aβ-α7nAChR and -Aβ complexes by interacting with the Aβ15–20 region [63]. Preincubation with S24795 in vitro reduces Aβ-α7nAChR interaction and Aβ-induced tau phosphorylation. In Aβ-infused mouse brains and Aβ-exposed organotypic cortical slices, S24795 not only reduced Aβ-α7nAChR association and Aβ immunostaining but also normalized Ca2 + fluxes through both α7nAChR and NMDAR channels [64] (Table 2).
IQ, a homologous to most nAChR subtypes, binded with nanomolar affinity to soluble Aβ and blocked Aβ-induced inhibition of carbamylcholine-induced currents in PC12 cells expressing α7nAChRs. Thus, IQ may be a lead novel drugs to block the inhibition of cholinergic function in AD [65] (Table 2).
THERAPEUTIC STRATEGIES TARGETING PrPC
PrPC, a glycoprotein that expressed in the brain, was first reported to be associated with prion disea-ses [66]. Many recent observations have indicated the possibility of a connection between prion diseases and AD [67]. The altered expression of PrPC in aging and the development of AD are associated with disease progression. Some pathological evide-nce indicates that PrPC deposits often accompany Aβ plaques in AD [68]. The PrPC-Aβ interaction has been demonstrated to be involved in regulating LTP in the hippocampus [69] and triggering the disruption of synaptic plasticity in vitro and in vivo. PrPC mediates neurotoxicity by Aβ oligomers via a signaling pathway involving with mGluR5 and Fyn [71]. The extracellular domain of mGluR5 interacts with both PrPC and Aβ which results in the activation of Ca2 + release from intracellular stores, PKC translocation and ERK1/2 phosphorylation. Aβ and PrPC also activate mGluR5, which then stimulates the Fyn kinase-mediated kinase signaling cascade [72, 73].
AZ59 [74] is a fully human monoclonal antibody against PrPC, which selectively targets the Aβo binding site in the amino-terminal unstructured domain of PrPC to avoid any potential risk of direct toxicity. A preclinical study demonstrated that systemic AZ59 therapy rescued central synapses and memory function from transgenic AD pathology, supporting a disease-modifying therapeutic potential (Table 2).
Pretreatment with an antibody against residues 93–109 of PrPC, 6D11, can prevented neuronal cell death by oligomeric Aβ and rescued cognitive deficits in APP/PS1 transgenic mice [75]. Behavioural testing showed a marked decrease in errors in 6D11 treated APP/PS1 Tg mice compared with the non-treated Tg groups. 6D11 treated APP/PS1 Tg mice had significantly greater synaptophysin immunoreactivity in the dentate gyrus molecular layer of the hippocampus. The carboxyterminal globular domain of mammalian PrPC contains three alpha helical segments and a two-stranded antiparallel beta sheet [76], while the amino-terminal portion of PrPC appears intrinsically disordered [71]. Recombinant PrPC binds to soluble Aβ42 oligomers via two motifs, which span residues 23–27 and residues 95–110 [77–79] (Table 2).
A high-throughput screening panel can be utilized to discover anti-PrP antibodies for their ability to disrupt the Aβ-PrP interaction [80]. Two representative and extensively characterized monoclonal antibodies directed to these regions, ICSM-35 and ICSM-18, were shown to block the Aβ-mediated disruption of synaptic plasticity validating these antibodies as candidate therapeutics for AD either individually or in combination (Table 2).
Apart from antibodies, fragments of the PrPC protein are also effective therapeutic strategies to prevent deficits in synaptic plasticity and neuronal death induced by Aβ. Recombinant human prion protein (rPrP) and its N-terminal fragment N1 are strikingly potent inhibitors that block Aβ-induced inhibition of long-term potentiation (LTP) in hippocampal slices with Aβ cytotoxicity in primary hippocampal neurons [81] (Table 2).
OTHER THERAPEUTIC STRATEGIES
In addition to the receptors mentioned above, there are some other Aβ receptors can be recognized as possible therapeutic targets for AD. For these receptors, however, there were only some indirect therapies showing the mechanism of interfering with their upstream or downstream signaling pathways.
Immune-inhibitory receptor FcγRIIb is a receptor of Aβ [82]. The interaction between Aβ and FcγRIIb is largely increased in the brain of the patients with AD, and genetic ablation of FcγRIIb in PDAPP J20 mice reversed cognitive deficit, suggesting an essential role of FcγRIIb in Aβ toxicity. FcγRIIb also serves as a mechanistic link between Aβ and tau pathologies by modifying the pool of membrane-resident phosphatidylinositol derivatives [83]. KICG2576, an ATP competitive inhibitor of LYN, bl-ocked the phosphorylation of FcγRIIB2 resulting in the inhibition of tau hyperphosphorylation [84]. Additionally, the intracerebroventricular injection of KICG2576 into mice ameliorated Aβ-induced memory impairment.
The β-adrenergic receptor (β-AR) system can be considered a possible target that deserves further exp-loration in AD. The central noradrenergic system undergoes substantial changes during the course of AD and β-ARs have been implicated not only in amyloid formation in AD brain but also in amyloid-induced neurotoxicity [85]. However, the therapeutic effect of targeting β-AR remains controversial. Some β-ARs and antibodies have been reported to be able to slow cognitive decline in AD patients, but the underlying mechanism of action has not been explored [86, 87]. On the other hand, some animal studies reveled that blockage of β-AR exacerbated cognitive deficits. Administration of ICI 118551 [88], a selective β2 adrenergic receptor antagonist, increased Aβ levels, Aβ plaque burden, and tau phosphorylation. Given these contradicting reports, more studies are warranted to fully understand how β-Ars are involved in AD pathogenesis and how they could be modulated to produce most beneficial outcomes.
THERAPEUTIC STRATEGIES TARGETING Aβ TRANSPORTERS
Therapeutic strategies targeting RAGE
RAGE belongs to the immunoglobulin (Ig) superfamily [89] and binds a number of ligands including Aβ. Despite being recognized as a crucial regulator of the innate immune response, evidence suggests that RAGE signaling is involved in neurodegenerative diseases, especially AD [90].
Increased RAGE expression was observed in the capillaries of the AD brain as compared to controls suggesting that RAGE is a crucial factor that infl-uences Aβ burden. RAGE interaction with Aβ me-diated surface binding and subsequent cellular processing of the ligand [91]. Aβ-RAGE interactions disrupt tight junction proteins and reduce breakage of BBB integrity via the Ca2 +-calcineurin pathway while disrupting microvessels that coexist with Aβ plaque-deposited areas, elevated RAGE expression and enhanced matrix metalloproteases secretion [92]. RAGE modulates β- and γ-secretase activity thus potentiating Aβ formation through the activation of GSK3β and p38 MAP kinase [93]. Compelling evidence from in vivo and in vitro studies indicated that RAGE signaling contributes to AD pathogenesis by activating neuro-inflammation. Tg mice expressing mAPP in neurons and RAGE in microglia showed increased production of IL-1β and TNF-α, enhanced infiltration of microglia and astrocytes, Aβ aggregation, decreased AChE activity, and rapid disruption of spatial learning/memory [94].
sRAGE, which is the secreted isoform of RAGE, inhibits the interaction between RAGE and Aβ by competing with cell-surface RAGE [95, 96]. To overcome its short life, a sRAGE-secreting umbilical cord derived mesenchymal stem cells (sRAGE-MSCs) were established to reduce Aβ deposition, RAGE dependent cell death and inflammation [97]. sRAGE-MSC treatment improved the viability of injected MSCs and enhanced the protective effects of sRAGE by inhibiting the binding of RAGE and RAGE ligands in vivo (Table 3).
The potential therapeutic strategies that increase Aβ transportation
Given the protective effects of sRAGE, peptide fragments from sRAGE were predicted to demonstrate the same biological activity. The extracellular domain of RAGE consists of three immunoglobulin-like regions, one V-type followed by two C-type, a transmembrane spanning domain and a 43 amino acid cytosolic tail [98, 99]. RAGE binds soluble Aβ in the nanomolar range (Kd 52.2±14.6 nM) with its V-type region. Using intrinsic RAGE tryptophan fluorescence and mass spectrometry of non-covalent protein–ligand complexes, RAGE was shown to be localized at the Aβ binding region to the 7 amino acid stretch of residues of the sequence LVFFAED [100] (Table 3).
Fragment peptides of RAGE that form exposed areas of the V-domains were synthesized and evaluated for their memory protection activity. The RAGE fragment sequence (60–76) under intranasal insert-ion was able to restore memory and improve morphological and biochemical state of neurons in the brain [101]. A further investigation showed that the shortened analogous peptide (60–70) and peptide (60–65) were able to bind Aβ in vitro and protect neuronal primary cultures from amyloid toxicity by preventing the caspase 3 activation [102, 103] (Table 3).
Other than design RAGE inhibition peptides by mimicking sRAGE, RP-1 was identified as a specific high-affinity RAGE inhibitor showing a high homology to the 16–23 (KLVFFAED) regions in the Aβ peptide [104]. RP-1 bound to RAGE and inhibited Aβ peptide-induced cellular stress in SH-SYSY cells in vitro likely by activation of the PI3K/AKT prosurvival pathway (Table 3).
Matrine (Mat) is a main component of traditional Chinese herbs used to treat dementia. Mat was reported to block the ligand binding site of RAGE and weakened the activation of the RAGE pathway resulting from Aβ stimulation [105]. Molecular docking analysis between Mat and RAGE V-domain showed a strong interaction in a hydrophobic region. In addition to the hydrogen bonding interaction between Mat and Lys37, Val35, Leu36, leu34, and Ile26 contribute to the stable interaction. Furthermore, Mat inhibited the Aβ aggregation and reduced in Aβ levels and chronic inflammation, which may also contribute to the reduced activation of the downstream RAGE signaling pathway (Table 3).
Azeliragon (TTP488), a small molecule RAGE antagonist, is currently in a phase III trial in patients with mild AD. The preclinical study in AD transgenic mice showed a reduction of amyloid load in the brain, improved performance on behavioral testing and normalization of electrophysiological recordings from hippocampal slices [106] (Table 3).
Therapeutic strategies targeting LRP-1
LRP-1, also known as alpha-2-macroglobulin rec-eptor (A2 MR), apolipoprotein E receptor (APOER) or cluster of differentiation 91 (CD91), is a member of the low-density lipoprotein receptor family, which facilitates its multiple functions as a ligand receptor for endocytosis in the plasma membrane of cells [107, 108]. LRP-1 contributes to Aβ metabolism via different mechanisms. LRP-1 was initially studied for its effect of APP trafficking. As a direct receptor of Aβ, LRP-1 binds to Aβ40 immobilized with high affinity (Kd = 0.6–1.2 nM) compared to Aβ42 and mutant Aβ. This LRP-Aβ interaction mediated Aβ brain capillary binding, endocytosis, and transcytosis across the BBB. What’s more, it also reported that LRP-1 reduced Aβ production by competing with APP for BACE1 [109] and γ-secretase[110].
IgG1-iS18, an anti-LRP/LR specific antibody, has been recognized as a potential therapeutic strategy by blocking LRP-1 on Aβ formation and AD associated symptoms [111]. AD transgenic mice received IgG1-iS18 through intranasal administration showed improved short term memory and learning ability. IgG1-iS18 reduced the levels of soluble and insoluble Aβ in the whole brain and diminished accumulation of amyloid plaques in the hippocampus (Table 3).
Another potential therapeutic strategy of blocking the LPR-Aβ interaction is LRPIV-D3674G [112]. LRP-1 is a type I transmembrane protein, composed of a large 515 kDa N-terminal extracellular subunit and an 85 kDa C-terminal transmembrane subunit, which are noncovalently associated with one another. The extracellular domain of LRP-1 has four ligand binding domains (I–IV) with 2, 8, 10, and 11 cysteine-rich complement-type repeats, respectively. SPR analysis characterize Aβ interaction with the LRP-1 ligand binding domains revealed that Aβ binds to LRP-1 immobilized clusters II and IV [113]. As a modified sLRP-1 such as peptide, LRPIV-D3674G has a higher binding affinity to Aβ, but less binding affinity to other LRP-1 ligands than wild-type sLRP-1. Subcutaneous administration of LRPIV-D3674G reduced Aβ40 and Aβ42 levels in the hippo-campus, cortex, and CSF by 60–80%, and improved vascular and neuronal functions in Tg2576 mice (Table 3).
CONCLUSION
In this review, we present recent advances in the understanding of Aβ receptors and the related ther-apeutic strategies. Although the mechanism underlying the memory and cognitive defect in AD is hotly debated, Aβ has been identified as a major factor that participates in AD progression through its neurotoxic effects. The intracellular signaling in response to Aβ binding to neurons is highly variable between receptors. These receptors and transporters exert vital physiological effects such as synaptic plasticity, tissue patterning, angiogenesis, axon guidance, and anti-apoptosis. The binding of Aβ with these receptors and transporters may not only disturb these physiological functions but also induce neuronal toxicity, which is likely to present in the inhibition of LTP, dendritic spine loss, actin filament disassembly, neuron death and even the generation and clearance of Aβ.
For decades, great effort have been devoted to exploring AD therapeutics [114]; however, the high failure rate makes AD drug development one of the greatest challenges in modern medicine. Since 2003, hundreds of studies based on Aβ hypothesis have been processed, but none of them have been approved for AD treatment [115]. Most of them were terminated because of lack of efficiency or serious side effects.
It should be noticed that the majority Aβ-targeting agents aimed to alleviate Aβ burden by targeting Aβ aggregation [116], reducing the production of Aβ through inhibition of β- or γ-secretase [117–119], or reducing Aβ levels through immunotherapy [120, 121]. Very few of them have directly targeted the neurotoxic pathway of Aβ. The failures of clinical trials suggest that the current treatments might be too late for mild-to-moderate AD patients since irreversible damage to the brain may already have occurred. From that point of view, novel drug development based on blocking Aβ-induced neurotoxicity by inhibiting Aβ-cell interactions could present great promise to delay or even halt AD progression.
Footnotes
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (grant number 81903471 and 81971330), Key Program Natural Science Foundation of Shannxi province (Grant No. 2018ZDXM-SF-040), Natural Science Basic Research Plan in Shaanxi Province of China (grant number 2019JQ-202 and 2020JQ-877) and Xi’an Weiyang District Science and Technology Information Bureau (grant number 201933). Thank the support of The Youth Innovation Team of Shaanxi Universities.