
Abstract
Just as multiple sclerosis (MS) has long been primarily considered a white matter (WM) disease, Alzheimer’s disease (AD) has for decades been regarded only as a grey matter disorder. However, convergent evidences have suggested that WM abnormalities are also important components of AD, at the same extent as axonal and neuronal loss is critically involved in MS pathophysiology since early clinical stages. These observations have motivated a more thorough investigation about the possible mechanisms that could link neuroinflammation and neurodegeneration, focusing on amyloid-β (Aβ). Neuroimaging studies have found that patients with AD have widespread WM abnormalities already at the earliest disease stages and prior to the presence of Aβ plaques. Moreover, a correlation between cerebrospinal fluid (CSF) Aβ levels and WM lesion load was found. On the other hand, recent studies suggest a predictive role for CSF Aβ levels in MS, possibly due in the first instance to the reduced capacity for remyelination, consequently to a higher risk of WM damage progression, and ultimately to neuronal loss. We undertook a review of the recent findings concerning the involvement of CSF Aβ levels in the MS disease course and of the latest evidence of AD related WM abnormalities, with the aim to discuss the potential causes that may connect WM damage and amyloid pathology.
BIOLOGY OF AMYLOID: STRUCTURE, FUNCTION, AND REGULATION
Amyloid-β protein precursor (AβPP) is a single-pass transmembrane protein which is expressed at high levels in the brain. Its main biological function is likely to promote cell growth. AβPP is cleaved by two pathways. In the case of the nonamyloidogenic pathway, AβPP is first cleaved by α-secretase, generating membrane-tethered α-C terminal fragments (CTFs). The cleavage of AβPP by α-secretase releases sAβPPα from the cell surface and leaves an 83-amino-acid C-terminal AβPP fragment (C83). Further processing involves the intramembrane cleavage of α-CTFs by γ-secretase, which liberates the P3 (3 kDa) peptide. In the amyloidogenic pathway, cleavage via the β- and γ-secretases produces several species of Aβ fragments. β-site AβPP-cleaving enzyme 1 (BACE1) is the major β-secretase in the brain. Neurotoxic forms of Aβ created by cleavage of AβPP initially by BACE1 produce the 99-amino-acid C-terminal fragment of AβPP (C99) and sAβPPβ, and the C99 is then cleaved by γ-secretase to produce Aβ. The C99 fragment generated by β-secretase cleavage can be internalized and further processed by γ-secretase at multiple sites to produce cleavage fragments of 43, 45, 46, 48, 49, and 51 amino acids that are further cleaved to the main final Aβ forms, the 40-amino-acid Aβ40 (Aβ1–40) and the 42-amino-acid Aβ42 (Aβ1–42), in endocytic compartments [1]. The γ-secretase complex consists of four different proteins, presenilin 1, nicastrin, presenilin enhancer 2, and anterior pharynxdefective 1. Presenilin 1 (PSEN 1) is activated by auto-processing to generate N- and C-terminal cleavage products that both contain aspartyl protease sites that together are required for the activity of the mature γ-secretase. Moreover, both PSEN1 and presenilin 2 (PSEN2) regulate the proteolytic function of γ-secretase, and mutations in these proteins can change the activity of γ-secretase. Nicastrin, presenilin enhancer 2, and anterior pharynxdefective 1 are also critical components of γ-secretase and may modulate enzyme activity in response to physiological stimuli [2].
Aβ monomers aggregate into various types of assemblies, including oligomers, protofibrils, and amyloid fibrils. In their aggregated form, these peptides are able to induce neurotoxicity. In particular, amyloid fibrils are larger and insoluble, and they can further assemble into amyloid plaques, while amyloid oligomers are soluble and may spread throughout the brain. As defined 80 years ago, the amyloid fibrils must display the ‘cross-β’ diffraction pattern caused by the cross-β-sheet motif when irradiated with X-rays. The motif is composed of tightly interacting intermolecular β-sheets, and each β-sheet comprises thousands of identical copies of the same β-strand that stack through hydrogen bonding. Although the pathological properties of amyloid load and propagation remain unknown, the association of amyloid fibrils with Alzheimer-type pathology is strong.
The production of Aβ is normally counterbalanced by several processes, including proteolytic degradation, cell-mediated clearance, active transport out of the brain, and deposition into insoluble aggregates [2]. Several evidences suggest that proteolytic degradation is a particularly important determinant of cerebral Aβ levels and, by extension, of Aβ-associated pathology [3]. In addition to degradation, Aβ released into the extracellular space can be transported between different compartments, such as from the brain to the blood or from the blood to the brain, and can also be cleared by chaperones, such as apolipoprotein E (ApoE), which can affect Aβ metabolism after it is released by cells and influence Aβ aggregation, clearance, and transport. The carrier- and receptor-mediated transport of Aβ across the blood-brain barrier (BBB) regulates brain Aβ levels. The concentration of soluble Aβ in the central nervous system (CNS), which is central to the formation of neurotoxic oligomeric Aβ species and vascular aggregated forms of Aβ, is critically influenced by Aβ transport exchange across the BBB.
Alzheimer’s disease (AD) is the most common neurodegenerative disorder, and its devastating neuropathology is tightly linked to the cortical deposition of fibrillar Aβ plaques. The major component of these deposits is Aβ1–42 [4]. However, because of the weak correlation between plaque load and AD pathology, more recent findings indicate that Aβ dimers, small diffusible Aβ oligomers, are highly toxic and more strictly associated with memory dysfunction in the early stage of AD [5]. Although the toxic oligomers are enriched in β-sheet content, they are in a distinct conformation from amyloid fibrils. In vitro, Aβ has been described in many states including a membrane-bound monomer, a dimer, small oligomers, worm-like fibrils, ring-like oligomers, protofibrils, and dozens of polymorphic mature fibrils [5]. Although each of these various structural entities appear to be toxic, it remains unclear which of them plays the major role in AD pathophysiology and by which mechanism of toxicity. Moreover, the relationship between oligomers and fibrils still remains to be established.
The amyloid hypothesis proposes that the fundamental cause of AD lies in the deposits of extracellular Aβ peptides [6]. Mutations in the human APP gene cause the development of amyloid plaques and Alzheimer-like brain pathology, especially in early-onset familial cases of AD [7]. Mutations in the human APP gene are close to the γ-secretase site and can increase the Aβ1–42/Aβ1–40 ratio. It is reported that mutations that alter residues C-terminal to the Aβ1–42 site reduce cleavage efficiency and increase the Aβ1–42/Aβ1–40 ratio [8]. AD-causing mutations also occur in the PSEN1 and PSEN2 genes. Mutations in the human PSEN1 and PSEN2 genes affect γ-secretase activity and increase the Aβ1–42/Aβ1–40 ratio. Late-onset AD is characterized by a pattern of interwoven genetic and non-genetic factors. These genetic risk-factors may affect one or more pathogenic mechanisms, such as increased Aβ production and aggregation; decreased Aβ clearance and degradation; increased inflammation; and resistance to γ-secretase activity. All these mechanisms are believed to lead to neurodegeneration in AD brains [2].
ROLE OF WHITE MATTER ABNORMALITIES IN AD
In patients with AD, brain magnetic resonance imaging (MRI) detects frequently the presence of focal hyperintensities in the deep and subcortical white matter (WM) [9]. Their presence seems to increase the risk for conversion from mild cognitive impairment to AD, and to predict the progression of cognitive symptoms. Moreover, diffusion-weighted imaging studies have demonstrated the presence of WM microstructural changes in AD brains since the preclinical stages of disease [10]. Nevertheless, the nature of these WM hyperintensities (WMHs) remains largely unclear: traditionally, the main hypothesis considers them as chronic ischemic lesions caused by cerebral microangiopathy.
Chronic hypoperfusion due to small vessel disease represents a potential cause for the development of WM degeneration in subjects with and without dementia. It is well known that several vascular changes reducing the cerebral blood flow, such as hypoxia-induced capillary loss, contribute to the formation of WMHs [11]. The crucial issue is exactly their relationship with aging and preexisting cardiovascular disorders, suggesting that vascular damage may be a predominant mechanism for their cause. Cerebral amyloid angiopathy (CAA) has been frequently observed in the brains of patients with AD at autopsy [12]. The vascular amyloid deposition of CAA on the walls of the cerebral blood vessels can produce luminal stenosis, endothelial damage, thrombosis, thickening of blood vessels, and vasospasm. This is the reason why CAA has been well accepted as the major cause of ischemic WM damage in AD brains. To minimize the risk of confounding variables associated with vascular comorbidities, several studies have selected patients with no remarkable history or risk factors for cardiovascular disease. They consistently showed that WMHs represent a crucial feature of AD pathology independently from patients’ vascular risk factors and disease stage.
At present, it remains unclear to what extent different factors may contribute in determining the accumulation of WMHs in AD brains. It is also unclear to what extent cerebral hypoperfusion exacerbates the progression of neurodegenerative disease processes such as the accumulation of Aβ [13]. This lack of information provides additional incentive on the importance of broadening knowledge about AD-related WM pathology, focusing for instance on the overlapping mechanisms linking together amyloid deposition and WM primary neuropathology.
Several studies suggest that Aβ is toxic to oligodendrocytes [14]. Although amyloid plaques are exceedingly rare in AD WM, the levels of soluble Aβ are elevated in the WM [15]. Thus, a direct exposure of WM oligodendrocytes to increased amounts of Aβ is likely to occur [16].
11C-Pittsburgh compound B (PiB) positron emission tomography (PET) is considered to be a useful tool for evaluating AD. PiB retention in the cortex is typically used to distinguish AD from other forms of dementia. Given the recent findings showing that WM changes are an important feature of AD, regional WM PiB retentions in AD patients were also investigated with conflicting results. Further study is required to clarify this issue.
Only few data until are available in literature on the relationship between measures of macrostructural and microstructural WM damage and cerebrospinal fluid (CSF) Aβ levels in AD. Kalheim and colleagues reported a remarkable extent of WM microstructural damage in patients with mild cognitive impairment who showed pathological CSF levels of Aβ1–42 [17]. Additionally, a paper by Dean III et al. [18] has contributed in clarifying the relationship between amyloid pathology and myelin alteration in preclinical AD. Measuring whole-brain longitudinal and transverse relaxation times and the myelin water fraction (MWF), a significantly negative association was observed between MWF and CSF Aβ levels [18]. Concerning inherited forms of AD, Lee and colleagues reported a correlation between WMHs and CSF Aβ levels [19]. To better understand the relationship between WMHs and amyloid pathology, it was investigated how CSF Aβ levels interact with measures of macrostructural and microstructural WM damage [20]. A group of patients with cognitive decline was recruited, classifying them as Aβ(+) and Aβ(–) based on their CSF Aβ levels. It was demonstrated that, even accounting for the ageing effect, CSF Aβ levels are the best predictor for the accumulation of WMHs: the lower the CSF Aβ levels, the higher the total WMHs (Fig. 1A). Consistently, Aβ(+) patients showed significantly higher WM lesion load when compared with Aβ(–) patients. Moreover, no significant difference in WM lesion load was observed when stratifying patients with AD for disease duration or global cognition. Against this background, it can be argued that WMHs are likely to occur at an early pathophysiological stage of AD. In this picture, these findings suggest that CSF Aβ reduction might be associated with the occurrence of WM metabolic damage due to Aβ deposition, possibly caused by impairment of pathways implicated in myelination and myelin repairing processes [21, 22].

A) Linear regression of total white matter (WM) lesion load in function of CSF amyloid-β (Aβ) levels in both AD and non-AD patients. Scattered line shows CSF Aβ threshold for AD diagnosis, which was set at the value of 600 pg/ml (adapted from Pietroboni et al., 2018 [20]). B) Comparison of CSF Aβ levels in MS patients stratified according to Expanded Disability Status Scale (EDSS) score (<3 or ≥3) at 5-year follow-up. Threshold was set at the value of 813 pg/ml. C) Multiple regression analysis of EDSS score at 5-year follow-up in function of CSF Aβ levels (B and C adapted from Pietroboni et al., 2019 [26]). D) Comparison of CSF Aβ levels in MS active patients according to their normal-appearing white matter (NAWM) amyloid tracer mean standard uptake value (SUV) (adapted from Pietroboni et al., 2019 [37]).
In conclusion, all these studies suggest that WM lesions and their microstructural substrate represent a crucial feature of AD, independent of vascular risk factors and disease stage.
ROLE OF AMYLOID-β IN MS
When taking into account the hypothesis of neurodegeneration as a major contributor to MS disability, Aβ1–42 has recently become an interesting candidate for its putative role in this process. AβPP has been detected in MS plaques with a higher AβPP immunoreactivity in actively demyelinating than in chronic lesions, thus indicating a modification of AβPP metabolism across disease stages [23]. Moreover, AβPP was found upregulated in both acute and chronic MS lesions and has been regarded as a sensitive marker of axonal damage [24]. Reduced CSF Aβ levels have already been reported in MS patients [22, 25], although the interpretation of these findings remains controversial. A recent study in a relatively small group of MS patients revealed that lower baseline levels of CSF Aβ are predictive of a more severe disease progression over 3-year clinical follow-up [22]. Following this preliminary study, a larger cohort of MS patients with a longer follow-up was recruited, classifying them as those Aβlow and those Aβhigh based on their CSF Aβ levels. With respect to the clinical outcome, the Aβlow status was associated with a higher risk of disease progression [26] (Fig. 1B, C). As a confirmation of this, CSF Aβ levels came out as a predictor of clinical disability.
With respect to the underlying processes that might explain why reduced CSF Aβ levels associate to a worse clinical outcome, the spectrum of hypotheses appears multifaceted. There is the myelin model that combines the occurrence of WM metabolic damage with Aβ deposition [21]. On the one hand, the inflammation increases BACE1 activity, which was previously found increased in patients with reduced CSF Aβ levels [27]. BACE1 is also involved in the cleavage of neuregulin 1 (NRG1) [28], a protein that plays a crucial role in myelin repair. On the other hand, CSF Aβ reduction could depend on AβPP deposition around injured axons, although there is no evidence of Aβ deposition in MS plaques. Nevertheless, these studies suggest that CSF Aβ levels may represent a significant feature in MS, as they resulted being predictive biomarkers of disease progression [22, 26]. As part of a fascinating hypothesis, it can be speculated that lower CSF Aβ levels may be associated with a decreased ability to remyelinate CNS axons, with an early WM and GM damage, and with a higher probability of clinical disease progression. Considering the relationship existing between some genetic factors (i.e., apolipoprotein polymorphisms), myelin repair efficiency, and the risk of amyloidosis, it is possible that genetic modulations, whose effect is typically observed in old age, may have an impact in younger ages only when individuals are affected by a highly impacting condition such as MS. A relationship between APOE genotyping and MS severity, however, remains poorly investigated and controversial [29, 30]. Similarly, the exact role played by Aβ in MS pathophysiology remains to be fully understood.
PET with amyloid tracers (e.g., PiB, florbetapir, florbetaben, flutemetamol) was originally developed for imaging of amyloid deposition in neurodegenerative disorders and dementia [31]. Only recently it has been proposed as an imaging marker for quantification of myelin loss and repair in MS [32–35]. Amyloid tracers bind extensively to WM, and its uptake decreases in the presence of demyelination [32] (Fig. 2). The usefulness of amyloid tracers in MS is traditionally considered secondary to their nonspecific binding to WM, possibly being trapped in β-sheet structures of myelin proteins or being highly soluble in the myelin-associated lipid bilayer [36]. Acute WM lesions show reduced amyloid tracer uptake compared with the normal-appearing WM (NAWM), thus reflecting a more extensive myelin loss within the lesions than in the NAWM [34]. In light of these data, amyloid PET tracer WM uptake may be considered as a biomarker of demyelination.
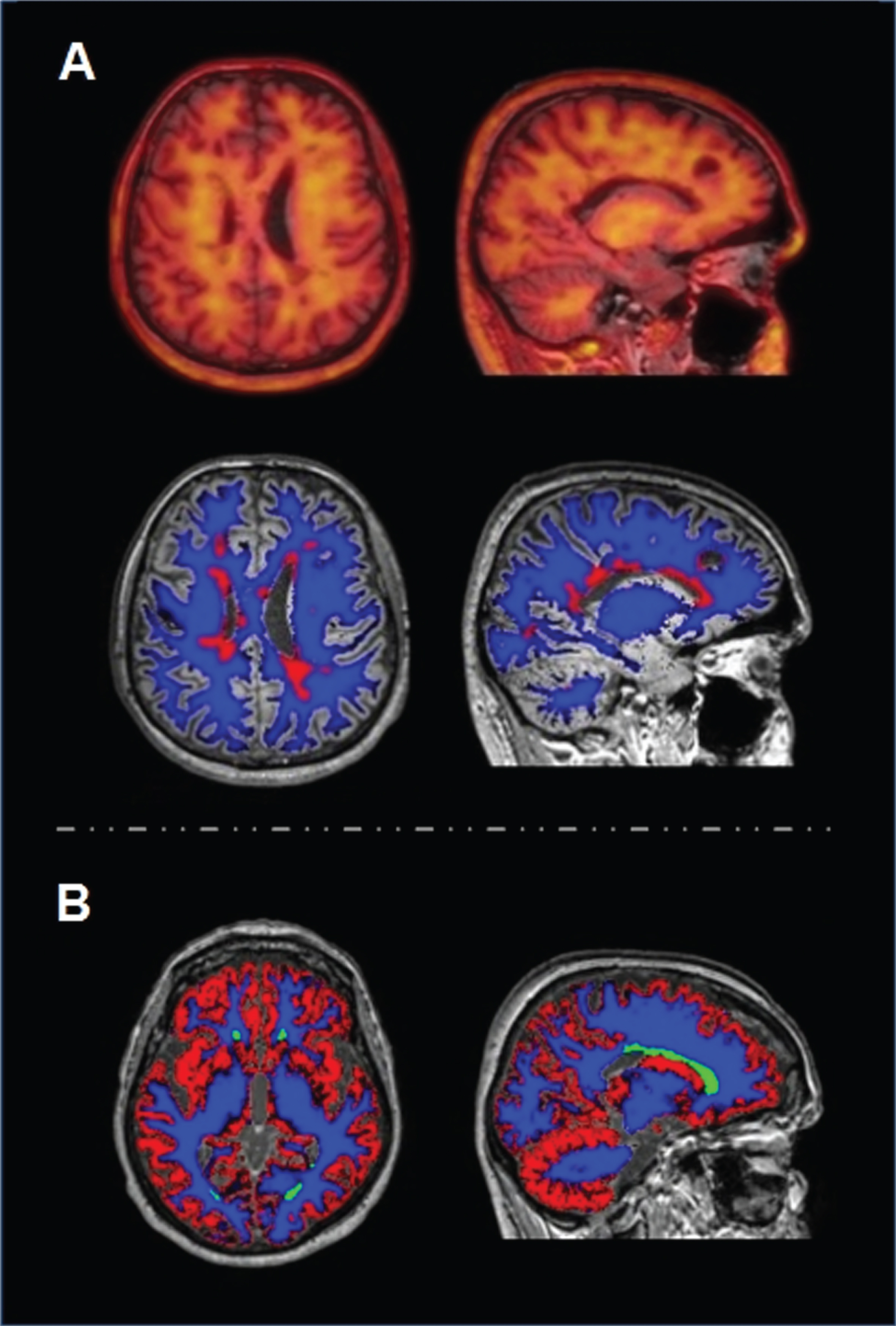
Brain MRI and amyloid PET imaging in MS (A) and AD (B). A) T1-weighted MRI/PET fusion images. Right: Normal appearing white matter (blue) and damaged white matter (red) segmentation on T2-weighted FLAIR MRI. B) Grey matter (red), normal appearing white matter (blue) and damaged white matter (green) segmentation on T2-weighted FLAIR MRI.
Given these premises, amyloid tracer uptake in damaged WM (DWM) and NAWM of MS patients divided according to their disease activity was recently studied [37]. It was also investigated possible correlations between amyloid tracer uptake and CSF Aβ levels. In line with previous studies [34, 36], first it was found that the amyloid tracer uptake in larger WM lesions is reduced compared to that in the NAWM, thus confirming the role of amyloid PET as a biomarker of myelin loss. Moreover, it was showed that 18F-florbetapir uptake in patients with active disease is lower than that in non-active patients, suggesting an interesting link between early WM damage and disease activity [37]. The most interesting finding of this study is that the tracer uptake in the NAWM correlates with CSF Aβ levels: the lower the uptake, the lower the CSF Aβ concentration [37] (Fig. 1D). It can be hypothesized that active patients, i.e., those with lower uptake in both DWM and NAWM and with lower CSF Aβ levels, may have a reduced capacity for remyelination and consequently a higher risk of disease progression. Strengthening this hypothesis, it was also found lower uptake in the NAWM and lower CSF Aβ levels in those patients with smaller NAWM volume. In line with these findings and the aforementioned hypotheses, the correlation described between amyloid tracer uptake and CSF Aβ concentration seems to confirm that amyloid plays a role in the progression of WM damage in MS. As part of this speculation, lower CSF Aβ levels could be associated with a decreased ability for remyelination of CNS axons, with early WM and GM damage, resulting in a higher probability of disease progression.
AMYLOID-β AND WM DAMAGE
A big issue that still needs addressing is why CSF Aβ levels correlate with macro and/or microstructural WM damage in both conditions, AD and MS. At first sight, it seems complicated to identify a single mechanism for these two different conditions. With regards to MS, the most convincing explanation is that a precocious micro-inflammatory milieu increases Aβ secretion. However, both these processes (the persistence of inflammation and the Aβ production) exercise mutual influence: just as brain’s inflammatory response can increase Aβ secretion (i.e., reactive astrocytes increase the expression of BACE1 and, therefore, Aβ production), Aβ itself can be considered as a proinflammatory mediator due to its ability to induce inflammation [38]. Furthermore, Aβ can stimulate proinflammatory cytokine release from astrocytes. The same applies to AD, even if reversed. Increased amounts of Aβ trigger a cascade of neuropathological events, including demyelination, death of oligodendroglial cells and mild glial reaction [39], as well as neuronal dysfunction and alterations in the neurotransmission [40]. As mentioned above, Aβ peptides exert a cytotoxic effect on the oligodendrocyte. As the oligodendrocyte is indispensable for the maintenance of the myelin sheath, Aβ-induced oligodendrocyte death can subsequently disrupt its integrity. Aβ peptides induce, in turn, a micro-inflammatory milieu, which maintains and often further affects the WM damage (Fig. 3). In any case, it remains to be clarified whether Aβ plays a causal role in the injury of the WM or represents only an epiphenomenon of the failure of its reparative processes.
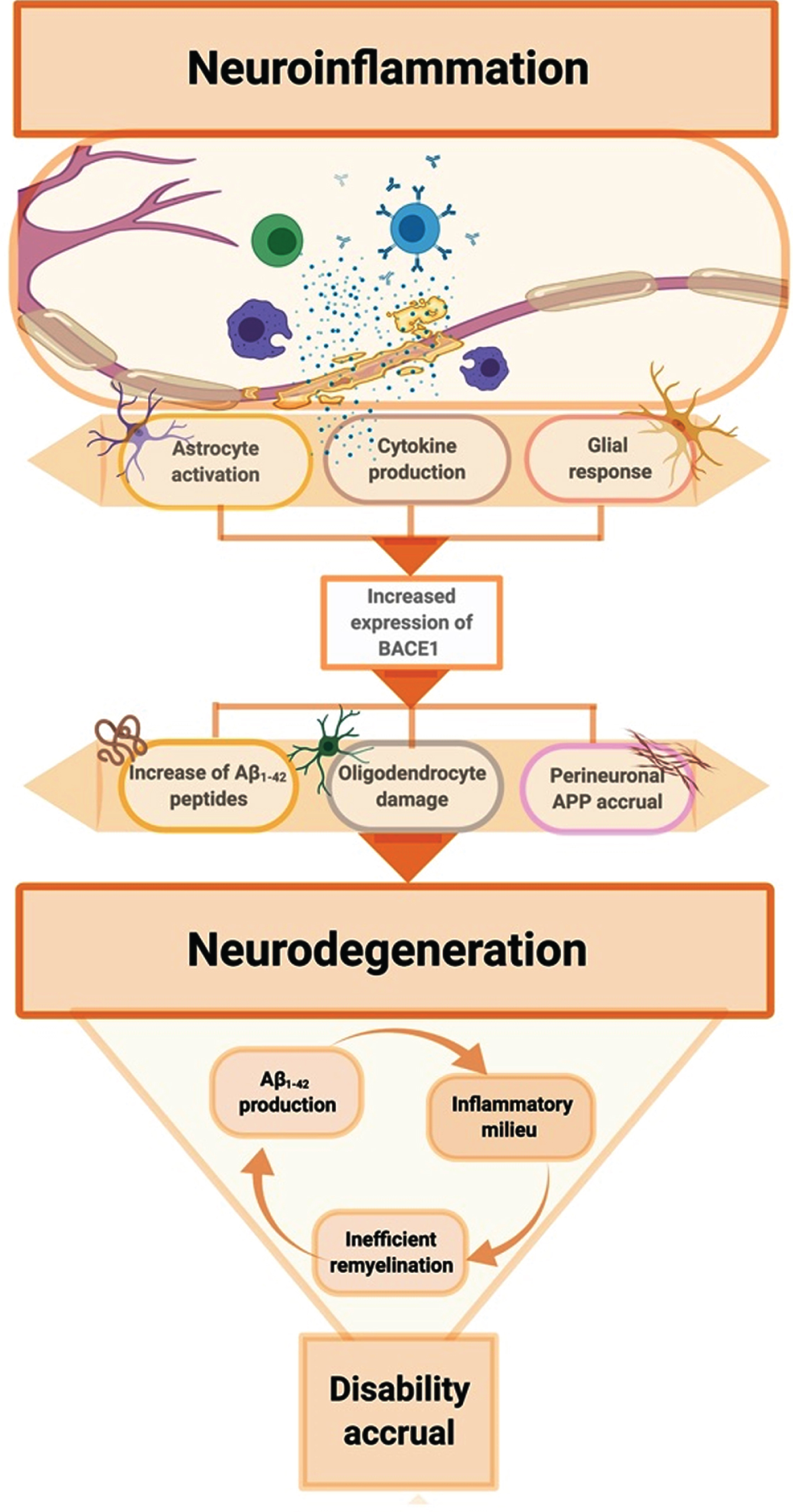
Flowchart showing the putative biological mechanism coupling neuroinflammation and neurodegeneration through amyloid-related pathways. Neuroinflammation is considered the mainstay of multiple sclerosis pathogenesis, although it is reported to occur in neurodegenerative diseases as well such as Alzheimer’s disease. In this scenario, the activation of immune response cascade promotes the expression of BACE1, which is an inflammatory-induced protein. By means of preferential activation of the amyloidogenic way, an increased production of Aβ1–42 peptides occurs, which is assumed to exert a direct toxic effect on oligodendrocytes. In addition of that, inflammation promotes the overexpression of AβPP itself and reduces the clearance of Aβ1–42 peptides, overall concurring to sustain amyloid pathology accrual. Noteworthy, BACE1 is also involved in Neuregulin1 cleavage, presumed to be fundamental for regulation of myelination and synaptic plasticity. As a whole, these mechanisms are likely to affect myelin dynamics, without regard to the original biological insult that arouse the inflammatory response. Ultimately, the inefficiency of damage repair system promotes the persistency of such an inflammatory milieu, further enhancing the secretion of the abovementioned toxic amyloid peptides. All these interconnected conditions concur to the establishment of a disruptive self-maintaining mechanism of white matter damage progression, that ultimately leads to neurodegeneration and clinical disability accrual.
As speculation, one single model that could unify these two points of view may be the metabolic one, which associates the Aβ deposition with the occurrence of WM metabolic damage due to several, partially still unknown factors. In line with this hypothesis, the myelin vulnerability could be the key element. The expansion of brain volume and connectivity seem largely myelin-driven, over the lifespan as in the evolutionary trajectory. Human cognitive functions and behaviors are highly dependent on later-myelinating oligodendrocytes, cells that continue myelinating the circuitry of neural networks [41]. These later-myelinating oligodendrocytes and their myelin are structurally more complex and metabolically overextended. Thus, they seem to be especially vulnerable to neuroinflammatory and neurodegenerative processes [42]. As proposed by Haroutunian and colleagues [41], the human species’ exceptional myelination is supported by very recent evolutionary changes involving lactate dehydrogenase to augment energy production, peroxisome organelle function to augment lipid production, and ApoE to augment lipid transport/recycling. The authors explained that these adaptations may have evolved in part to support the extremely metabolically expensive processes of creating and maintaining a highly myelinated CNS [41]. In this vision, multiple insults to myelin (i.e., inflammatory attacks during MS relapses or metabolic overload during neurodegenerative processes) could be directly responsible for Aβ secretion and deposition, or indirectly linked to amyloid pathology on this as an epiphenomenon of the failure of myelin repairing processes. On the other hand, the regional vulnerability to Aβ deposition could explain why the very early GM atrophy patterns observed in both AD and MS seem to be consistent and involve the same neocortical areas [43]. In other words, cortical atrophy could be increased in those areas known to have the highest early Aβ load, such as the precuneus and the anterior and posterior cingulate areas also in MS, suggesting a direct relationship between GM atrophy and Aβ deposition in these regions. Moreover, a recent study identified a specific set of early Aβ accumulating regions, which largely correspond to the default-mode network (DMN), revealing that the hypoconnectivity within the DMN and between DMN and the frontoparietal network is associated with low CSF Aβ levels [44]. Interesting results were also obtained with transcranial magnetic stimulation protocols, showing in early AD not only the involvement of the hippocampus but also of the associative cerebral cortices, suggesting that Aβ-related pathology would derive from the disturbance of the brain’s effective connectivity implying abnormal interactions between neuronal systems [45]. Taken together, all these findings seem to confirm that selective vulnerability of myelin or neurons may be related to an increased amount of metabolic and/or plastic requirements and not be associated with disease-specific pathological processes.
CONCLUSIONS AND FUTURE DIRECTIONS
Although further research on amyloid pathology in WM damage is necessary, recent studies have demonstrated that 1) Aβ plays a role in MS; 2) WMHs represent an early feature of AD; 3) neuroinflammation and neurodegeneration often have overlapping and uncertain boundaries; and 4) CSF Aβ levels are predictors for WM micro- and macrostructural damage in both AD and MS. There are additional biomarkers that can be obtained in vivo and could, in combination with wet biomarkers, help clarify the relationship between inflammation and neurodegeneration. Future studies, possibly with a longitudinal design, should include multiparametric measures targeting various aspects of inflammation and neurodegeneration.