
Abstract
Background:
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by the accumulation of amyloid-β (Aβ) plaques, neuroinflammation, and neuronal death. There are several well-established genetic and environmental factors hypothesized to contribute to AD progression including air pollution. However, the molecular mechanisms by which air pollution exacerbates AD are unclear.
Objective:
This study explored the effects of particulate matter exposure on AD-related brain changes using the APP/PS1 transgenic model of disease.
Methods:
Male C57BL/6;C3H wild type and APP/PS1 mice were exposed to either filtered air (FA) or particulate matter sized under 2.5μm (PM2.5) for 6 h/day, 5 days/week for 3 months and brains were collected. Immunohistochemistry for Aβ, GFAP, Iba1, and CD68 and western blot analysis for PS1, BACE, APP, GFAP, and Iba1 were performed. Aβ ELISAs and cytokine arrays were performed on frozen hippocampal and cortical lysates, respectively.
Results:
The Aβ plaque load was significantly increased in the hippocampus of PM2.5-exposed APP/PS1 mice compared to their respective FA controls. Additionally, in the PM2.5-exposed APP/PS1 group, increased astrocytosis and microgliosis were observed as indicated by elevated GFAP, Iba1, and CD68 immunoreactivities. PM2.5 exposure also led to an elevation in the levels of PS1 and BACE in APP/PS1 mice. The cytokines TNF-α, IL-6, IL-1β, IFN-γ, and MIP-3α were also elevated in the cortices of PM2.5-exposed APP/PS1 mice compared to FA controls.
Conclusion:
Our data suggest that chronic particulate matter exposure exacerbates AD by increasing Aβ plaque load, gliosis, and the brain inflammatory status.
INTRODUCTION
Alzheimer’s disease (AD) is a common age-related neurodegenerative disorder worldwide which has adverse long-term outcomes [1]. The pathological hallmarks of AD include amyloid-β (Aβ) plaques accumulating in the brain extracellular surface [2] and hyperphosphorylated tau containing neurofibrillary tangles depositing within neurons [3–5]. These pathologies are hypothesized to lead to neuronal damage, oxidative stress, mitochondrial dysfunction, synaptic collapse, gliosis, inflammation, and cognitive impairment [6, 7]. Worldwide, approximately 47 million people were reported to have dementia in 2015, and this number is anticipated to increase 3-fold by 2050 [8, 9]. AD is a leading cause of death in the United States with an estimated 5.8 million affected and, by mid-century, the number is expected to grow to 13.8 million [10].
The risks associated with developing AD are multifactorial with age being the greatest risk factor [11, 12]. Besides aging, various environmental factors also increase the risk of AD, including head trauma, obesity, diabetes, and air pollution [12–14]. Exposure to particulate matter is a major threat for various global diseases, including AD, potentially by inducing inflammation and oxidative stress [13, 15]. Worldwide, millions of people are exposed chronically to air pollutants with levels higher than recommended by the World Health Organization Air Quality Guidelines [16].
Air pollution is comprised of a heterogeneous mixture of particulate matter (PM), organic compounds (e.g., endotoxins and polycyclic aromatic hydrocarbons), gases (e.g., nitrogen oxides, carbon monoxide, sulfur oxides, and ground-level ozone), and metals (e.g., nickel, vanadium, iron, zinc, and manganese) present in indoor and outdoor air [13]. Particulate matter is predominantly categorized according to its size with coarse particles having an aerodynamic diameter of 2.5 to 10μm (PM10), fine particles less than 2.5μm (PM2.5), and ultrafine particles less than 0.1μm (PM0.1) [17, 18]. The National Ambient Air Quality Standards (NAAQS) recommend that for a healthy individual, annual exposure to PM2.5 should not surpass 15 mg/m3 [18, 19]. The size of PM is the basis of its adverse biological effects and PM2.5 may be especially harmful because it is able to traverse the endothelial lining of the lung tissue and enter the circulation. It is acutely toxic to lungs and cardiovascular tissues and crossing the pulmonary blood-air barrier to enter peripheral circulation and, ultimately, the brain [20–22]. Various studies have reported PM2.5 toxicity in both animal models and in humans [18, 23–26]. Several studies report a broad range of potential health complications associated with chronic exposure to PM2.5, including effects on the cardiovascular system [24, 26–29]. A few reports also suggest adverse effects of air pollution on cognitive function [30–32]. Specifically, PM2.5 induces neuroinflammation and causes cognitive deficits [13, 33–37]. Additionally, PM2.5 can infiltrate into the brain in vivo where it may contribute to neurodegeneration-related pathology [38–40]. Since neuroinflammation is a critical component of AD pathophysiology, it is feasible that PM2.5 exposure may exacerbate at least this aspect of AD development [41, 42]. Indeed PM exposure appears capable of potentiating Aβ-stimulated microglial changes in vitro [43] and long-term potentiation and memory performance in vivo [44, 45] through mechanisms involving both oxidative and inflammatory events. However, whether air pollution alters AD-related histopathology accumulation in the brain is not fully described.
To address this question, we used a chronic airborne PM2.5 exposure model in C57BL/6;C3H wild type and APP/PS1 AD mice to simulate exposure to PM2.5 levels close to the NAAQS recommendation. This study was designed in order to explore the risk of air PM2.5 exposure on brain changes related to AD by quantifying alterations in the brain inflammatory milieu and AD-related pathology.
MATERIALS AND METHODS
Particulate matter or filtered air exposure
Male wild type C57BL/6;C3H and AD transgenic mice, APPswe/PSEN1dE9 (APP/PS1) mice were obtained from The Jackson Laboratories (Bar Harbor, ME) at 8 weeks old and housed for 4 more weeks in the animal facility prior to exposure. The mice from both the strains were randomly divided into two groups for exposure to either filtered air (FA) or particulate matter (PM2.5) for 6 h/day, 5 days/week for 3 months. In total the mice were exposed for 55 days; however, PM2.5 values for 51 days were used for calculation of average exposure values due to monitor malfunction. No difference in the health of the mice was observed across FA- and PM2.5-exposed animals. The aerosol concentration system located at The Ohio State University was used for the concentrated PM2.5 exposure from the Columbus, OH region as described previously [18, 26]. The average daily PM2.5 concentration that the mice were exposed to was 25.8μg/m3. Since the animals were exposed for only 6 h/day, the concentration was equivalent to a 24 h period of 6.4μg/m3 per hour which is below the national air quality standard [18, 19]. The virtual impactor on the concentration system allowed any particle of size smaller <2.5μm to pass through, which also includes the ultra-fine fraction. Mice have no access to food or water during the duration of the exposure as they are whole body exposed. Mice were placed into SciReq’s, inExpose whole body chambers ‘pie slices’, with an airflow set to 2L of air/min. For the FA treatment, the same setup and model of SciReq inExpose chambers are used. The airflow for the FA chambers passes through a HEPA filter prior to chamber entry to remove all ambient particles, and then for precautionary redundancy, is run through another HEPA-VENT disc filter before flowing into each chamber at 2L of air/min. Both FA and PM airflow is channeled through desiccant beads to remove moisture. Animal use was approved by The Ohio State University and University of North Dakota Institutional Animal Care and Use Committees [18].
Tissue collection
To collect tissue, animals were sacrificed and left hemispheres of the brains were fixed in 10% formalin with zinc for immunohistochemistry and the right hemispheres was flash frozen in liquid nitrogen for ELISAs, western blot, and cytokine analysis.
Antibodies and reagents
Antibodies were utilized for immunohistochemistry and western blot analyses. Primary antibodies against GFAP (D1F4Q) and Aβ (D54D2), PS1 (D39D1) and BACE (D10E5) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). The Iba1 antibody (019–19741) was purchased from Wako Chemicals USA, Inc. (Richmond, VA, USA). The CD68 (MCA1957) antibody was purchased from Bio-Rad Laboratories, Inc. (Hercules, CA, USA). The anti-APP, Y188 (ab32136) antibody was purchased from Abcam (Cambridge, MA, USA). The GAPDH (sc32233) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Elite Vectastain ABC reagents, Vector VIP, biotinylated anti-rabbit, and anti-mouse secondary antibodies were purchased from Vector Laboratories, Inc (Burlingame, CA, USA).
Immunohistochemistry
The fixed left-brain hemisphere from each animal was processed for histologic studies as previously described [46]. After collection, brains were fixed in 10% formalin with zinc. Following fixation, brains were cryoprotected in a gradient of 15% sucrose-PBS and 30% sucrose-PBS, for 72 h each. Formalin-fixed tissue was embedded in a 15% gelatin (in PBS) matrix and was submerged in 4% paraformaldehyde overnight, which enables complete polymerization. Thereafter, the gelatin brain blocks were cryoprotected by two changes of 30% sucrose-PBS, each for 72 h. The blocks were then flash frozen using dry ice and isomethyl pentane. Serial sections of 40μm were cut using a Leica freezing microtome and stored at 4°C in PBS with 0.1% sodium azide until immunostaining. For Iba1 and Aβ, antigen retrieval was required. Iba1 antigen retrieval was performed using Tris-EDTA at 95°C for 10 min and for Aβ, sections were incubated in 25% formic acid for 25 min, prior to blocking. Thereafter, sections were incubated with primary anti-Aβ (1:500), anti-GFAP (1:1000), anti-Iba1 (1:1000), and anti-CD68 (1:1000) antibodies with mild shaking overnight at 4°C. This was followed by washing and incubation with biotinylated secondary antibodies (1:2000 dilution) and an avidin/biotin complex (Vector ABC kit). Vector VIP was used as the chromogen to detect the immunoreactivity. The sections were then mounted onto subbed slides with subsequent standard dehydration and cover-slipped using VectaMount (Vector Laboratories). A Hamamatsu NanoZoomer 2.0HT Brightfield + Fluorescence Slide Scanning System was used to capture images and the quantitation of immunoreactivity was done using Adobe Photoshop™ as previously described [47, 48]. Immunohistochemistry quantitation was performed using 3 hippocampus containing coronal serial sections per mouse. Hippocampi were traced using a digital pen and WACOM tablet (model: CTE-430, WACOM Co., Ltd). All the hippocampal images of all the mice from all the groups were pasted into a single canvas and the processing of the individual images was identical and performed simultaneously [49]. The entire canvas was converted into greyscale followed by inversion using Adobe Photoshop CS3 software (Adobe Systems, San Jose, CA). Background was subtracted from the entire canvas simultaneously by selecting one region with only gelatin or no staining using the black dropper tool. After normalizing for background, the mean optical density value for each image was recorded by sequentially selecting individual hippocampi using the lasso tool in Adobe Photoshop CS3 software (Adobe Systems, San Jose, CA). Optical density values were averaged from all sections from each mouse per group.
Western blot analysis
A portion of the flash frozen hippocampus was prepared in RIPA buffer containing protease inhibitors then bullet blended (Bullet Blender Storm 24, Next Advance, Inc.) and centrifuged (21,000 g, 4°C, 10 min) to remove insoluble content. Protein content of the cell lysates was determined using BCA protein determination assay (Pierce Biotechnology, Rockford, IL, USA). The resultant supernatants were boiled in sodium dodecyl sulfate (SDS) containing gel-loading sample buffer for 5 min. 15μg of each total protein extract was resolved on a 10% SDS polyacrylamide gel. Separated proteins were transferred to Immobilin-FL PVDF membranes (Millipore). Blots were blocked in Odyssey Blocking Buffer (LI COR bioscience, Lincoln, NE) for 1 h at room temperature. The blocked membrane was incubated in blocking buffer containing 0.1% Tween 20 and the respective primary antibodies; anti-PS1 (1:1000), BACE (1:1000), GFAP (1:1000), Iba1 (1:500), APP (1:1000), and GAPDH (loading control), overnight at 4°C, followed by three 15 min washes. APP, BACE, GFAP, and Iba-1 were immunodetected from the same blot and normalized to the respective GAPDH. PS1 was immunodetected on a separate membrane with its respective GAPDH used for normalizing. Blots were then incubated in blocking buffer containing 0.1% Tween 20 (Sigma), 0.01% SDS, and IRDye®, 680RD or 800 CW labelled secondary antibody 1:15 000 (Li CORE Bioscience, Lincoln, NE, USA) for 1 h at RT. The blot was imaged using the LI COR Odyssey imaging system (Li CORE Bioscience, Lincoln, NE, USA) and quantified using IMAGE studios® software. Optical density values were normalized to the relevant loading control GAPDH optical density values from the same membrane [48].
Enzyme linked immunosorbent assay (ELISA)
The flash frozen hippocampi were weighed and lysed in ice cold RIPA buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 1 mM Na3VO4, 10 mM NaF, 1 mM EDTA, 1 mM EGTA, 0.2 mM phenylmethylsulfonyl fluoride, 1% Triton, 0.1% SDS, and 0.5% deoxycholate) with protease inhibitors (AEBSF 1 mM, aprotinin 0.8μ M, leupeptin 21μM, bestatin 36 μM, pepstatin A 15μM, E-64 14μM). The tissue samples were homogenized using beads in a Bullet Blender and centrifuged to remove the insoluble content. The resulting supernatants were used to perform soluble Aβ1–40 and Aβ1–42 ELISAs. The insoluble content (pellet) was resuspended in 5M guanidine HCl/50 mM Tris HCl pH 8.0 and samples were again homogenized using the Bullet Blender and centrifuged (21,000 g, 4°C, 10 min) and the supernatants were used to perform insoluble Aβ1–40 and Aβ1–42 ELISAs. The levels of soluble and insoluble Aβ1–40 and Aβ1–42 were assessed using commercially available ELISA kits from Millipore Sigma. The levels of Aβ1–40 and Aβ1–42 (soluble and insoluble) were detected as pg/mL per mg protein derived from the standard curve for each protein. The protein concentrations of the sample lysates were measured using the bicinchoninic acid (BCA) protein determination assay (Thermo Fisher Scientific) [48].
Th1/Th2/Th17 cytokine assessment
RayBio Quantibody® Mouse TH17 Arrays (QAM-TH17–1, RayBio, Norcross, GA) were employed to assay lysates, following the manufacturer’s instructions. Due to limited sample amount available from the hippocampal lysates, temporal cortex lysates were used for cytokine analysis based upon the relevance of this region to AD and the fact that it is also a region with high plaque load and gliosis changes in AD mice. Eighteen different cytokines were evaluated: IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12p70, IL-13, IL-17, IL-17F, IL-21, IL-22, IL-23, IL28, IFN-γ, MIP-3α, TGF-β, and TNF-α. A GenePix 4400 scanner was used by RayBioTech for scanning the slide arrays using GenePix Pro software. Results were analyzed using the RayBio Analysis Tool. Using a standard curve, concentrations of each cytokine were determined.
Statistical analysis
Results are presented as mean values±standard error of the mean (SEM). Statistical analysis was performed by two-way ANOVA followed by Tukey’s multiple comparisons using GraphPad prism 8 software (GraphPad Software, Inc. La Jolla, CA). p < 0.05 was considered as statistically significant.
RESULTS
Particulate matter inhalation increased Aβ accumulation and APP processing enzymes in the hippocampus of APP/PS1 mice
Since accumulation of Aβ plaques is one of the pathological hallmarks of AD [2], and prior work has suggested that PM exposure may increase APP processing to Aβ [45], immunohistochemistry for Aβ was performed on the WT and APP/PS1 brains from the particulate matter and filtered air-exposed groups. As expected, no plaque immunoreactivity was detected in WT from either the FA or PM2.5-exposed groups. However, robust Aβ plaque deposition was seen in the FA APP/PS1 group, and int-erestingly, a significant increase in Aβ plaque density was observed in the hippocampus of their respective PM2.5 exposure group (p < 0.05) (Fig. 1A).
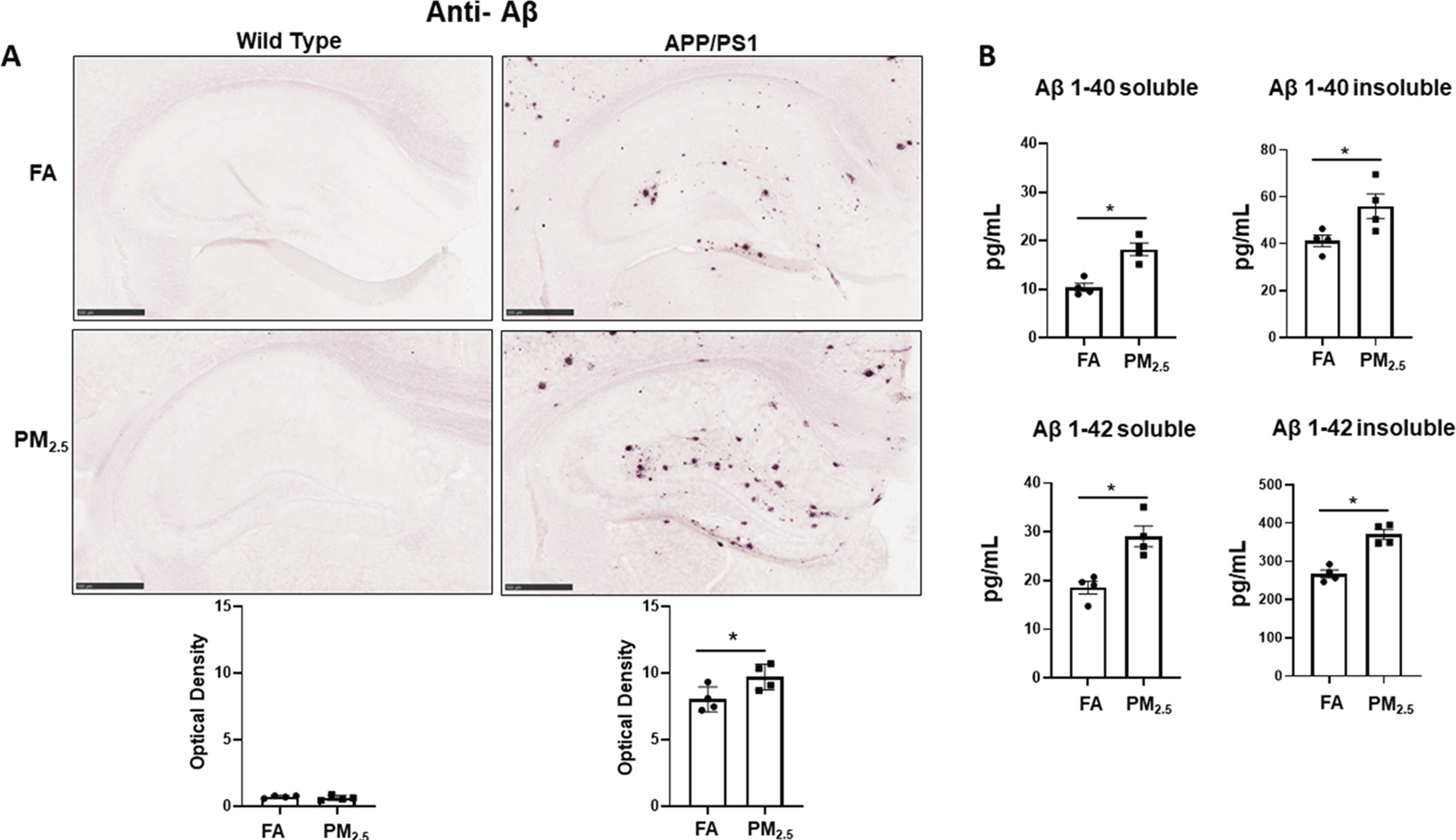
Particulate matter inhalation increased Aβ plaque load in APP/PS1 hippocampus. Male littermate control C57BL/6;C3H wild type and APP/PS1 mice were exposed to FA or PM2.5 for 3 months and brains were collected, sectioned, and immunostained with anti-Aβ antibody using Vector VIP as the chromogen. A) Representative immunohistochemical staining images (5x) of the hippocampus for Aβ are shown. Quantification of immunoreactivity was performed, and optical densities obtained were averaged and graphed as mean±SEM (n = 4), *p < 0.05, scale bar 500μm. B) The levels of both soluble and insoluble amyloid-beta 1–40 (Aβ 1–40) and 1–42 (Aβ 1–42), were measured in hippocampi from wild type and APP/PS1 mice using ELISAs. Results are shown as mean±SEM (n = 4), *p < 0.05.
In order to better quantify changes in Aβ levels, ELISA analyses were performed. The levels of both Aβ1–40 and Aβ1–42, soluble and insoluble, were measured from the hippocampus region of the APP/PS1 mice exposed to PM2.5 or FA. As predicted and consistent with the immunohistochemical staining, both soluble and insoluble Aβ1–40 and Aβ1–42 levels were elevated in APP/PS1 mouse brains. Particulate matter exposure also resulted in a significant increase in soluble and insoluble levels of both peptides (p < 0.05) (Fig. 1B). Only the APP/PS1 mice brains were assessed, as human specific Aβ ELISA was used.
Furthermore, to assess APP processing potential western blots were performed for APP, BACE, and PS1 proteins from the hippocampus of APP/PS1 mice. Interestingly, BACE and PS1 levels were significantly elevated in the mice exposed to PM2.5 in comparison to the FA controls (p < 0.05). This is consistent with prior work demonstrating elevated BACE1 levels in rodents following PM exposure [45]. Although APP showed an increasing trend in the PM2.5-exposed group, it was not significant from controls (Fig. 2). This data supports an observation of increased Aβ production along with elevated Aβ plaque load in the hippocampus of APP/PS1 mice upon PM2.5 exposure.
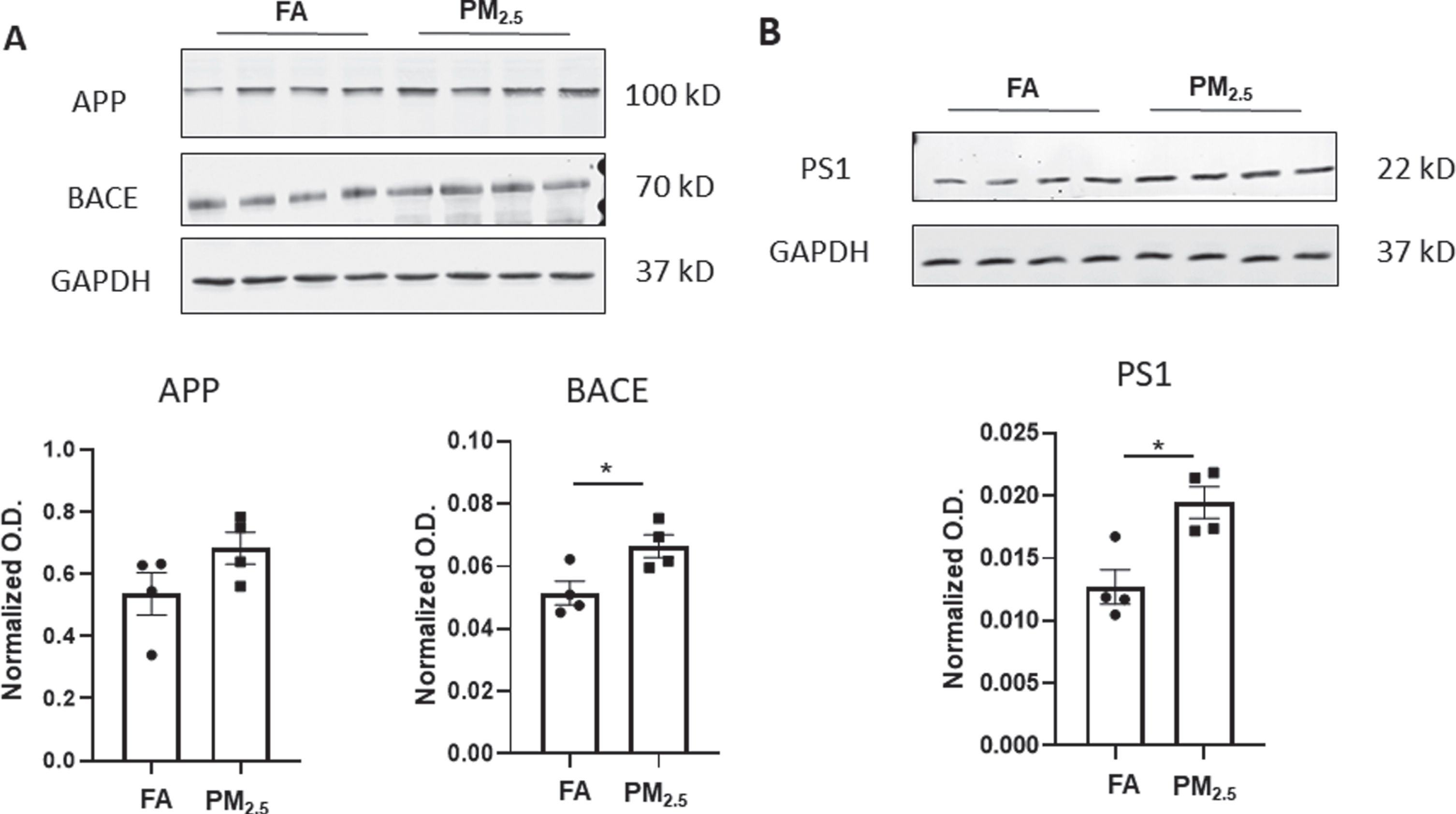
Particulate matter inhalation increased APP processing enzyme levels in APP/PS1 hippocampus. Frozen hippocampi from the APP/PS1 mice exposed to FA or PM2.5 were lysed and resolved via SDS-PAGE and western blots performed using (A) anti-APP, anti-BACE, and (B) anti-PS1 antibodies. Anti-GAPDH antibodies were used to probe the respective blots as a loading control. Quantification shows arbitrary densitometry units of each protein signal normalized to its respective GAPDH loading control. Results are shown as mean±SEM (n = 4), *p < 0.05.
Particulate matter inhalation increased astrocytosis in the hippocampus of APP/PS1 mice
It is well-known that reactive glia, including astrocytes and microglia, and associated inflammatory changes are associated with various neurodegenerative diseases including AD [50]. In the central nervous system, astrocytes are the most abundant glial subtype, and play a vital role in the regulation of neuroinflammation [51]. Thus, to assess astrogliosis in response to exposure to PM2.5, anti-GFAP immunostaining of the brain was performed. In both WT and APP/PS1 FA-exposed brains, a basal level of GFAP immunoreactivity was observed (Fig. 3A). Although PM2.5 exposure did not alter WT hippocampal GFAP immunoreactivity, it produced a significant increase in APP/PS1 mice compared to their respective FA controls (p < 0.05) (Fig. 3A). The protein levels of GFAP in the hippocampus of APP/PS1 mice were further quantified using western blot analysis to demonstrate a significant increase in the PM2.5-exposed group with respect to their respective FA controls (Fig. 3B). This indicated a susceptibility to astrogliosis in the APP/PS1 mice, in particular, upon exposure to particulate matter.
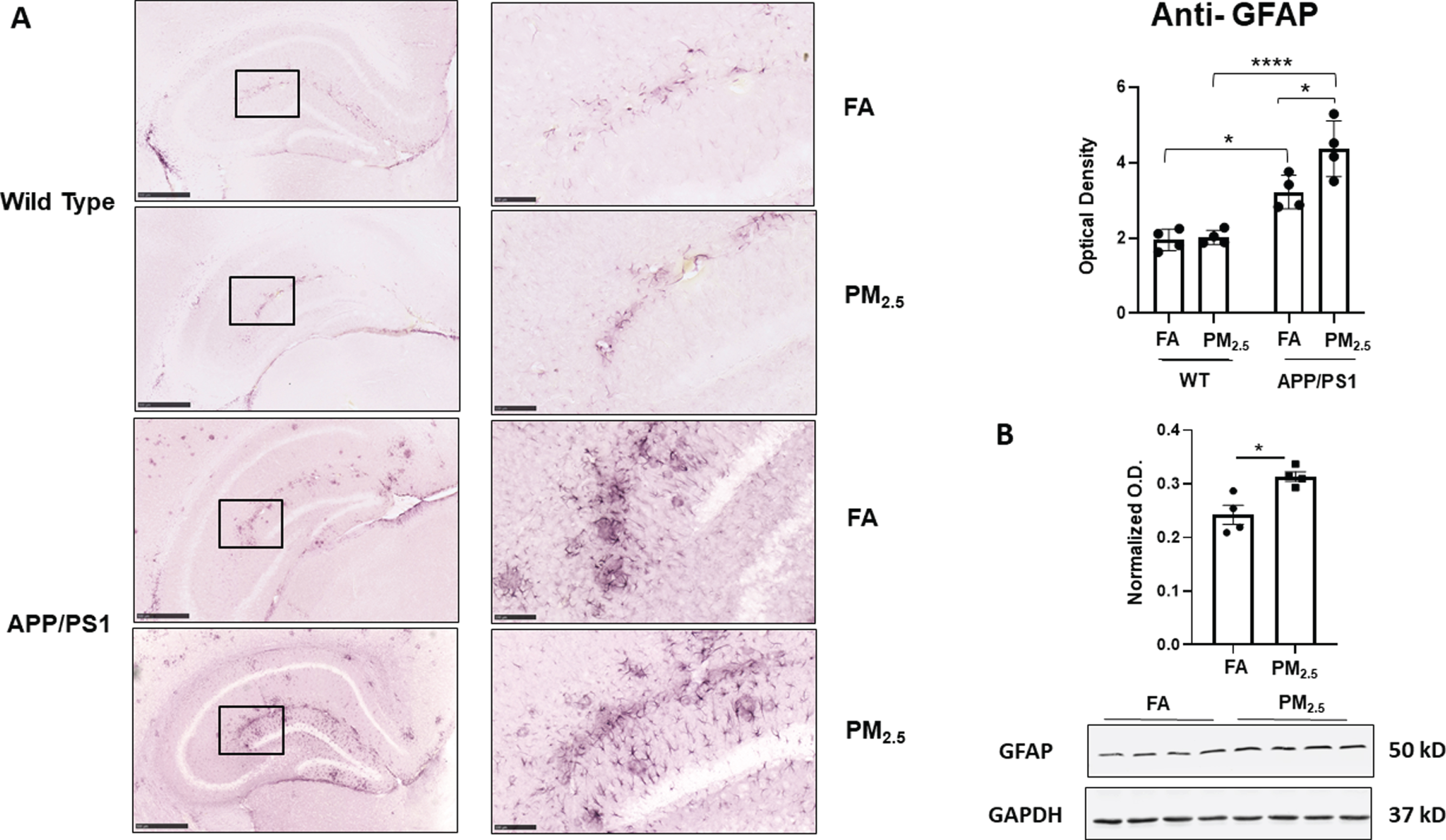
Particulate matter inhalation increased astrogliosis in APP/PS1 hippocampus. Male littermate control C57BL/6;C3H wild type and APP/PS1 mice were exposed to FA or PM2.5 for 3 months and brains were collected, sectioned and immunostained with anti-GFAP antibody using Vector VIP as the chromogen. Representative immunohistochemical staining images (5×and 20×) of the hippocampus are shown (A). Quantification of immunoreactivity was performed, and optical densities obtained were averaged and graphed as mean±SEM (n = 4), *p < 0.05 and ****p < <0.0001; scale bar 500μm. (B) Frozen hippocampi from the mice were lysed and resolved via SDS-PAGE and western blots performed using anti-GFAP and anti-GAPDH antibodies. Quantification shows arbitrary densitometry units of protein signal normalized to its respective GAPDH loading control. Results are shown as mean±SEM (n = 4), *p < 0.05.
Particulate matter inhalation increased microgliosis in the hippocampus of APP/PS1 mice
Microglial activation is also hypothesized to play an important role in AD pathophysiology [52]. To quantify microglial activation in response to PM2.5 exposure, anti-Iba1 immunostaining was performed. In WT mice, diffuse microglial staining was observed in both the FA and PM2.5 groups with no significant differences in hippocampal staining intensity (Fig. 4A). Robust plaque-associated microglial immunoreactivity was observed in the FA APP/PS1 mice with a significant increase in the PM2.5-exposed group (p < 0.05) (Fig. 4A). This increased microglial immunoreactivity was confirmed by assessing Iba1 protein levels in the hippocampus of APP/PS1 mice by western blot. This demonstrated a significant increase with PM2.5 exposure (Fig. 4B). Similar to the astrocyte assessment, this indicated an effect of PM2.5 to increase microgliosis in the APP/PS1 mice. As another means of assessing microglial activation, anti-CD68 immunoreactivity was examined as a marker of a reactive, phagocytic phenotype. Diffuse, limited staining was observed in both the FA and PM2.5 treated WT groups (Fig. 5). However, as anticipated, FA APP/PS1 mice showed a dramatic plaque-associated immunoreactivity with a significant increase in the PM2.5-exposed mice (p < 0.05) (Fig. 5). This provided further support the PM2.5 exposure significantly increases microglial activation in the APP/PS1 line.

Particulate matter inhalation increased microgliosis in APP/PS1 hippocampus. Male littermate control C57BL/6;C3H wild type and APP/PS1 mice were exposed to FA or PM2.5 for 3 months and brains were collected, sectioned and immunostained with anti-Iba1 antibody using Vector VIP as the chromogen. Representative immunohistochemical staining images (5x) of the hippocampus are shown (A). Quantification of immunoreactivity was performed and optical densities obtained were averaged and graphed as mean±SEM (n = 4), *p < 0.05 and ****p < 0.0001; scale bar 500μm. B) Frozen hippocampi from the mice were lysed and resolved via SDS-PAGE and western blots performed using anti-Iba1 and anti-GAPDH antibodies. Quantification shows arbitrary densitometry units of protein signal normalized to its respective GAPDH loading control. Results are shown as mean±SEM (n = 4), *p < <0.05.
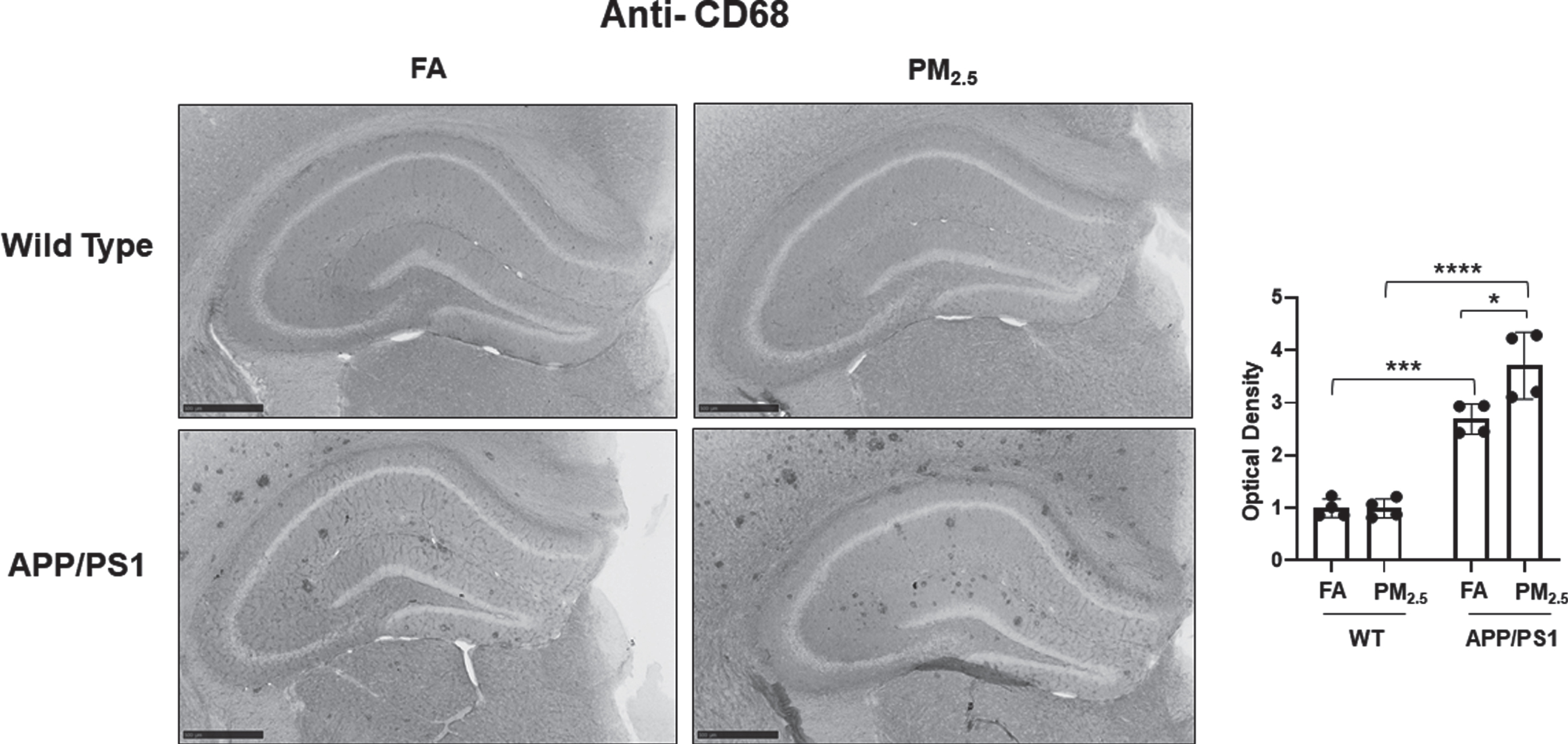
Particulate matter inhalation increased microglial phagocytic phenotype in APP/PS1 hippocampus. Male littermate control C57BL/6;C3H wild type and APP/PS1 mice were exposed to FA or PM2.5 for 3 months and brains were collected, sectioned and immunostained with anti-CD68 antibody using Vector VIP as the chromogen. Representative immunohistochemical staining images (5x) are shown. Quantification of immunoreactivity was performed and optical densities obtained were averaged and graphed as mean±SEM (n = 4), *p < 0.05, ***p < 0.001 and ****p < 0.0001; scale bar 500μm.
Particulate matter inhalation altered cytokine levels in the brains of both WT and APP/PS1 mice
The changes in astrocyte and microglial marker im-munoreactivity following PM2.5 exposure suggested inflammatory changes were potentiated. To quantify a change in the brain immune environment, cytokine levels were quantified from cortices in each group. In wild type mice, negligible changes were seen in cytokine levels in the PM2.5-exposed group compared to the FA controls (p < 0.05) (Fig. 6). However, in the APP/PS1 mice the pro-inflammatory cytokines TNF-α, IL-1β, IL-6, MIP-3α, and IFN-γ were elevated in the PM2.5-exposed compared to FA controls (IL-1β, p < 0.001; TNF-α and IFN-γ, p < 0.01; IL-6 and MIP-3α, p < 0.05) (Fig. 6). Conversely, levels of two anti-inflammatory cytokines, TGF-β (p < 0.05) and IL-10 (p < 0.01) (Fig. 6), were significantly reduced in the PM2.5-exposed compared to the FA control group. In addition, other cytokines IL-2, IL-5, IL-12, IL-13, IL17, IL-17F, IL-22 and IL-23; did not show any changes (Supplementary Figure 1).
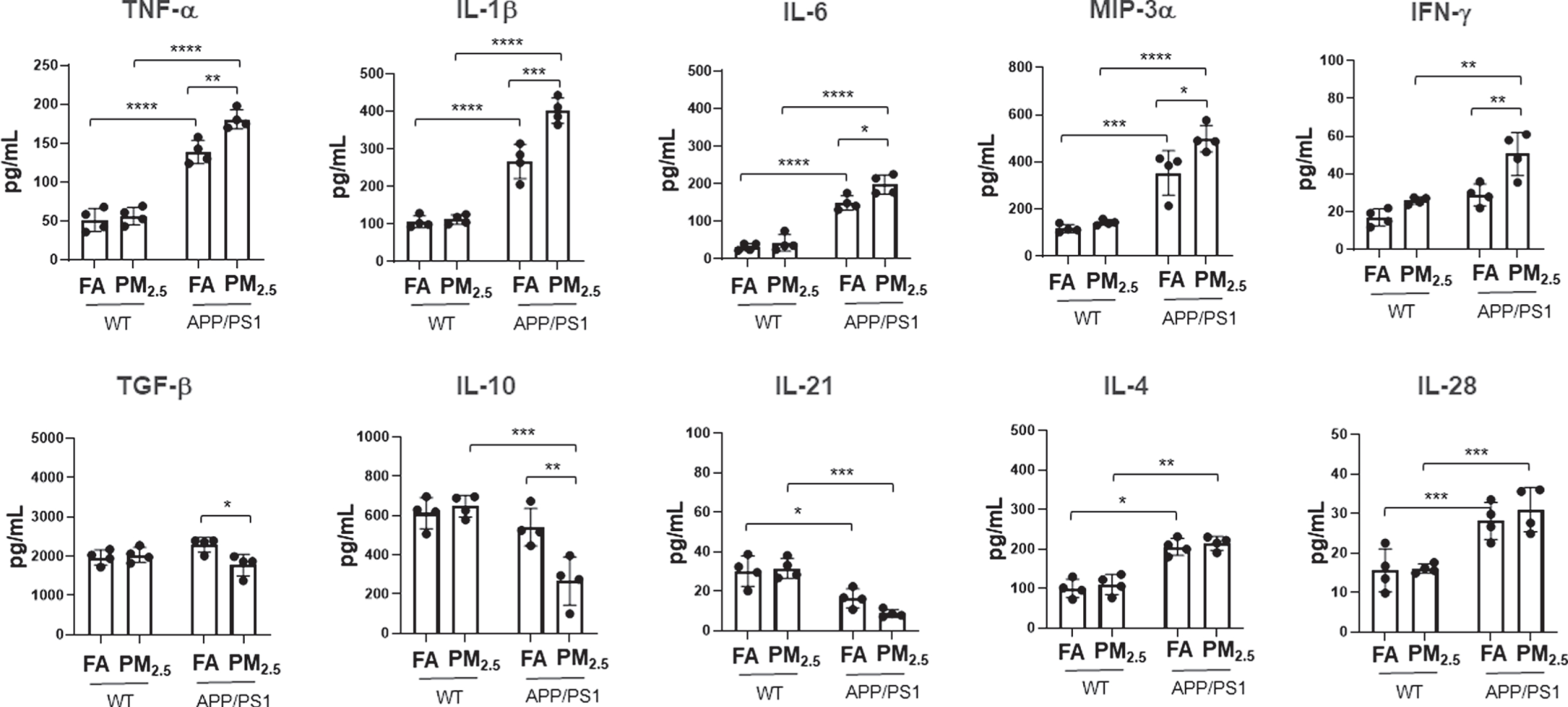
Particulate matter inhalation minimally affected brain cytokines in wild type mice. Male littermate control C57BL/6;C3H wild type and APP/PS1 mice were exposed to FA or PM2.5 for 3 months and brains were collected. Temporal cortices of wild type and APP/PS1 mice were lysed and used for a slide-based mouse cytokine array. Results from wild type and APP/PS1 brains are shown as mean±SEM (n = 4), *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
DISCUSSION
Our data suggest that chronic exposure to PM2.5 for three months exacerbates AD pathology indicated by an increase in Aβ levels, plaque load, reactive gliosis, and alteration of the brain inflammatory status indicated by elevated levels of pro-inflammatory cytokines. In our experimental paradigm, wild type and APP/PS1 mice were exposed to airborne PM2.5 from Columbus, Ohio using the modified SciReq inExpose system that concentrates airborne PM2.5 and exposes it to the animals. This exposure scheme has been explained in detail and has been employed for a variety of experiments [18, 53–55]. Concentrated PM2.5 from the Columbus, Ohio region was characterized previously and was found to be comprised of fairly high levels of elemental sulfur along with a variety of harmful metals, such as lead, zinc, and iron [18, 57]. The dose of exposure used in this study is approximately similar to the PM2.5 levels detected in several cities and industrial areas [18]. Mice were exposed for three months based upon prior work demonstrating a similar exposure induces an early cardiovascular phenotype [58].
PM2.5 includes both organic and inorganic comp-ounds produced mainly from industrial condensation of high-temperature vapor that include ammonium, hydrogen ions, sulfates, nitrates, carbon, hazardous metals, lipopolysaccharides, and water [13, 59]. It has been reported that lungs are not the only site for the toxicity of PM2.5. Depending on particulate characteristics and the duration of exposure, PM2.5 can enter the circulation and induce detrimental effects on other tissues/organs [60]. A report by Ku et al. showed that PM2.5 exposure results in deposition of metals within various tissues/organs, including the brain [61]. In addition, there are several other studies in humans [33, 40] and dogs [33, 63], which illustrate the role of coarse or fine particulate matter exposure on inflammatory status on the presentation of AD pathology. Additionally, our group has previously reported that chronic exposure to PM2.5 in utero alters neuroimmune phenotype [18] and also induces early AD-like pathological changes in the mouse brain [23].
The Aβ plaque load is a primary pathological hallmark of AD [2]. In our study, the amyloid-associated pathology was enhanced by chronic inhalation of airborne PM2.5. Also, it was associated with significantly elevated levels of both soluble and insoluble Aβ1–40 and Aβ1–42. Additionally, protein levels of BACE and the γ-secretase enzyme, PS1, were elevated correlating with the elevated Aβ plaque load and ELISAs suggesting Aβ production might be increasing. These changes support our hypothesis that chronic exposure to NAAQS levels of airborne PM2.5 exacerbate AD progression. At this time, it is unclear why Aβ levels and plaque load are increased in the PM2.5-exposed mice. It is possible that altered phagocytic clearance, increased production, or reduced blood clearance from the brain are all contributing to the changes we observed. Future work will need to assess these possible mechanisms. We elected to focus our Aβ analysis on the hippocampus as this is one of the earlier regions hypothesized to be affected during disease [64–67]. In addition, hippocampal function is associated with learning and memory and problems in these cognitive aspects are also associated with early AD [68, 69]. Finally, the hippocampus is particularly vulnerable to injury, inflammatory, or traumatic insult [70, 71].
Neuroimmune changes are another critical change associated with AD in which pro-inflammatory activities are hypothesized to exacerbate neuronal loss and cognitive decline [41, 73]. Some studies have shown that PM2.5 itself, or immune mediators, either through the circulation or via the olfactory nerve, infiltrate into the brain which in turn leads to inflammation and gliosis [13, 74]. In our study, long term PM2.5 exposure resulted in increased immunoreactivity of astroglia and microglia, indicated by the elevated levels of GFAP and Iba1/CD68, respectively. This demonstrated that the increased reactive gliosis normally associated with AD was potentiated by PM2.5 exposure. Indeed, the lack of any significant change in glial makers in wild type mice suggests that the basal gliosis of the APP/PS1 mice may have primed them selectively to have increased vulnerability to the PM2.5 insult.
Although reactive microglia and astroglia are not exclusive to AD, their reactive phenotype has a clear relationship with amyloid plaques [75–77]. Microglia become activated and engaged at the site of any trauma during an inflammatory response in the brain. Many reports in both animal models of AD [47, 78–81] and human disease [77, 82–86] have revealed activated microglia around Aβ plaques demonstrating a tangible interaction with Aβ [72, 73]. Similar to microglia, astrocytes also play a crucial role in regulating neuroinflammation [51]. Reactive astrocytes also localize nearby amyloid plaques, surrounding Aβ deposits leading to glial scarring [42, 87] in AD mouse models [88] as well as in AD patients [89]. Although the exact cause of increased glial immunoreactivity in the PM2.5-exposed APP/PS1 mouse brains is not certain, it is reasonable to expect that the glial changes are a direct consequence of the increased Aβ plaque load observed.
The notion that reactive, secretory gliosis is associated with elevated Aβ levels is supported by the fact that microglia and astrocyte activation by Aβ or other trauma correlates with increased levels of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, and IFN-γ all hypothesized to aggravate an inflammatory response and promote neuronal loss [47, 90–101]. In support of this idea, we observed elevated levels of the aforementioned pro-inflammatory cytokines in response to PM2.5 exposure in APP/PS1 mice. Another pro-inflammatory chemokine, MIP-3α, was also elevated in PM2.5-exposed brains and is known to be released by activated astrocytes [92]. A study by Campbell and colleagues reported similar findings that PM2.5 exposure for only two weeks was sufficient to potentiate brain TNF-α and IL-1α levels and increase activation of NF-κB in mice [102]. In our study, a reduction in the anti-inflammatory cytokines, IL-10 and TGF-β, was also observed. This suggests that the brain immune environment in the APP/PS1 mice was further impaired by exacerbation of a pro-inflammatory condition. Although optimal glial cell activation may provide protective effects by aiding in clearance of Aβ from the brain, PM2.5-dependent phenotype changes in these cells due to elevated Aβ or an altered brain immune environment could render an exaggerated inflammatory response that may worsen the neurodegenerative processes in AD [103]. It is likely that Aβ plaque formation and neuroinflammation in the APP/PS1 brains exhibit a dependent relationship. Aβ plaque deposition may result in activation of microglia and astroglia and subsequent pro-inflammatory secretion [103]; however, both microglia and astroglia activation can potentiate Aβ deposition under certain inflammatory conditions. For instance, TNF-α with IFN-γ [104, 105] or IL-1β [104] stimulation reportedly increases the production of Aβ by astrocytes. Microglial activation leads to the upregulation of pro-inflammatory factors, including IL-1β, TNF-α, and IFN-γ, which induce nearby astrocyte-neuron activation and subsequently increased production of Aβ42 [106].
There are several studies which have reported an increase in neuroinflammation in wild type mice with exposure to particulate matter [102, 108]. However, in our study we did not find any significant alteration in the neuroinflammatory assessments. A probable reason for this contradiction might be a difference in the composition of particulate matter and dosage or duration of exposure. For example, a recent study by Shou et al. revealed that PM2.5 exposure for 9–12 months, but not 3–6 months exposure, was required for a significant increase in the levels of proinflammatory factors TNF-α, IL-1β, and IL-6 in the brain of wild type mice. Our data are quite consistent with this study. Also, the study showed that PM2.5 exposure for at least 9 months was needed for an impairment of cognitive function [109].
In conclusion, our findings demonstrate that PM2.5 exposure for three months is associated with exacerbation of AD pathophysiology in mice, potentially by the microglial and astrocyte activation, correlating with upregulation of pro-inflammatory mediators including TNF-α, IL-1β, IL-6, MIP-3α, and IFN-γ in the brain. Although this study provided significant insight into a selective vulnerability of AD mice to effects of PM2.5 exposure, further work including neurobehavioral studies, collection at different/longer exposure times, and further analysis of epigenomic/transcriptomic changes could improve understanding of the risk of particulate matter exposure to progression of AD.
Footnotes
ACKNOWLEDGMENTS
This work was supported by funding from R01 R01AG057046, P20GM103442, U54GM128729, and funds from the UND VPR Office and UNDSMHS. Imaging studies were conducted in the UND Histology Core Facility supported by NIH grant P20GM113123, DaCCoTA CTR NIH grant U54GM128729, and UNDSMHS funds. The authors also thank Jeremy Adelstein, Jacob Grimmer, Neill Schwieterman, Roy (Drew) Miller, Dr. Harpreet Kaur, and Dr. Mona Sohrabi, for their assistance.